1.本发明提供单一生物单元的序列信息的新型处理方法、系统以及相关技术。更具体而言,提供微生物基因组数据的自动构建和提供系统。
背景技术:
2.虽然微生物基因组数据的构建正在取得进展,但目前的数据大多基于宏基因组信息,当将复杂的菌群(bacterial flora)作为分析对象时,信息的质和量均不足。
3.尽管已经部分地取得了每个单一生物单元的遗传信息(基因组信息等),但并未提供对其的高品质的信息处理。
技术实现要素:
4.用于解决课题的手段
5.本发明人进行了锐意研究,结果完成了下述系统:累积单一生物单元水平上的单一生物单元的序列信息,并从其中自动构建和提供高精度的微生物基因组数据。
6.作为本发明的实施方式的示例,列举如下。
7.项目1
8.一种方法,其是处理单一(single)生物单元的序列信息的方法,其中,该方法包括如下步骤:
9.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
10.(b)根据需要,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及
11.(c)使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
12.项目2
13.根据项目1所述的方法,其中,所述方法还包括:当进行所述(b)时,利用数据库。
14.项目3
15.一种方法,其是处理单一(single)生物单元的序列信息的方法,其中,该方法包括如下步骤:
16.a)从数据库提取草图中无重复的基因;
17.b)计算对应于每个该基因的草图的个数或比例;以及
18.c)选择该对应草图的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
19.项目3a
20.一种方法,其是处理单一(single)生物单元的序列信息的方法,其中,该方法包括如下步骤:
21.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
22.(b)将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。
23.项目4
24.一种方法,其是处理单一生物单元的序列信息的方法,其中,该方法包括如下步骤:
25.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
26.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
27.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
28.项目5
29.根据项目4所述的方法,其中,该方法包括如下步骤:
30.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
31.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
32.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
33.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目4中所述的步骤。
34.项目6
35.一种方法,其是处理单一生物单元的序列信息的方法,其中,该方法包括如下步骤:
36.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
37.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
38.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
39.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
40.项目7
41.根据项目6所述的方法,其中,所述再次聚类化通过网络分析和社群检测来进行。
42.项目8
43.一种方法,其是处理单一生物单元的序列信息的方法,其中,该方法包括如下步骤:
44.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
45.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
46.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
47.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
48.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
49.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
50.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
51.项目9
52.一种程序,其将处理单一(single)生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
53.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
54.(b)根据需要,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及
55.(c)使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
56.项目10
57.根据项目9所述的程序,其中,所述方法还包括:当进行所述(b)时,利用数据库。
58.项目11
59.一种程序,其将处理单一(single)生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
60.a)从数据库提取草图中无重复的基因;
61.b)计算对应于每个该基因的草图的个数或比例;以及
62.c)选择该对应草图的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
63.项目11a
64.一种程序,其将处理单一(single)生物单元的序列信息的方法安装在计算机中,
其中,该方法包括如下步骤:
65.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
66.(b)将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。
67.项目12
68.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
69.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
70.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
71.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
72.项目13
73.根据项目12所述的程序,其中,该方法包括如下步骤:
74.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
75.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
76.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
77.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目12中所述的步骤。
78.项目14
79.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
80.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
81.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
82.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
83.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
84.项目15
85.根据项目14所述的程序,其中,所述再次聚类化通过网络分析和社群检测来进行。
86.项目16
87.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
88.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
89.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
90.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
91.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
92.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
93.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
94.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
95.项目17
96.一种记录介质,其储存有将处理单一(single)生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
97.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
98.(b)根据需要,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及
99.(c)使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
100.项目18
101.根据项目17所述的记录介质,其中,所述方法还包括:当进行所述(b)时,利用数据库。
102.项目19
103.一种记录介质,其储存有将处理单一(single)生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
104.a)从数据库提取草图中无重复的基因;
105.b)计算对应于每个该基因的草图的个数或比例;以及
106.c)选择该对应草图的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
107.项目19a
108.一种记录介质,其储存有将处理单一(single)生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
109.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
110.(b)将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。
111.项目20
112.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
113.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
114.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
115.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
116.项目21
117.根据项目20所述的记录介质,其中,该方法包括如下步骤:
118.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
119.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
120.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
121.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目20中所述的步骤。
122.项目22
123.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
124.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
125.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
126.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
127.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
128.项目23
129.根据项目22所述的记录介质,其中,所述再次聚类化通过网络分析和社群检测来进行。
130.项目24
131.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
132.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
133.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
134.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
135.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
136.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
137.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;
138.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
139.项目25
140.一种系统,其用于处理单一(single)生物单元的序列信息,该系统包括:
141.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
142.(b)追加信息追加部,其根据需要而将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及
143.(c)草图创建部,其使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
144.项目26
145.根据项目25所述的系统,其中,当所述系统包括所述(b)追加信息追加部时,其还包括利用数据库的数据库利用部。
146.项目27
147.一种系统,其用于处理单一(single)生物单元的序列信息,该系统包括:
148.a)提取部,其从数据库提取草图中无重复的基因;
149.b)计算部,其计算对应于每个所述基因的草图的个数或比例;以及
150.c)选择部,其选择所述对应草图的个数或比例为规定值以上的基因作为生物谱系
鉴定用序列候选。
151.项目27a
152.一种系统,其用于处理单一(single)生物单元的序列信息,该系统包括:
153.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
154.(b)鉴定部,其将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。
155.项目28
156.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
157.(d)排序部,其基于规定的判断基准而将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
158.(e)草图构建部,其基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
159.(e’)选择部,其选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
160.项目29
161.根据项目28所述的系统,其中,该系统包括:
162.(f)选择部,其将所述所选择的草图与所述(e)草图构建部和(e’)选择部中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
163.(g)草图改善部,其使用(f)选择部中所选择的序列信息和该所选择的草图,生成更长的草图;
164.(g’)草图构建部,其根据需要使(g)草图改善部重复进行草图生成,直至该更长的草图达到序列信息的全长为止;
165.(g”)机构,其根据需要而在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,使项目28中所述的(d)排序部、(e)草图构建部以及(e’)选择部重复进行排序、草图构建以及选择。
166.项目30
167.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
168.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
169.(h)再次聚类部,其在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
170.(h’)比较部,其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
171.(i)注册部,其对于比较结果,基于规定的判断基准来判断(h)再次聚类部中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
172.项目31
173.根据项目30所述的系统,其中,所述再次聚类部通过网络分析和社群检测来进行再次聚类化。
174.项目32
175.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
176.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
177.(d)排序部,其基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
178.(e),其基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
179.(e”)草图构建部,其选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
180.(h)再次聚类部,其在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
181.(h’)比较部,其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
182.(j)机构,其对于比较结果,基于规定的判断基准来判断(h)再次聚类部中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
183.项目a1
184.一种方法,其发出指令使计算机执行单一(single)生物单元的序列信息的处理,其中,接收该指令的该计算机执行如下步骤:
185.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
186.(c)使用该单一生物单元的序列信息的部分序列信息和独立于该聚类化而生成的数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
187.项目a2
188.根据上述项目所述的方法,其中,所述方法还包括如下步骤:(b)将所述数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。
189.项目a3
190.根据上述项目中任一项所述的方法,其中,所述(c)包括:除去一定量的包含确认到多个重复的序列部位的部分序列信息,并校正序列读段的偏差。
191.项目a4
192.一种方法,其发出指令使计算机执行生物谱系鉴定用序列候选的筛选,其中,接收该指令的该计算机执行如下步骤:
193.a)从数据库提取草图中无重复的基因;
194.b)计算每个该基因的单拷贝基因的个数或比例;以及
195.c)选择该单拷贝基因的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
196.项目a5
197.一种方法,其发出指令使计算机执行单一生物单元的序列信息的处理,其中,接收该指令的该计算机执行如下步骤:
198.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
199.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
200.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
201.项目a6
202.根据上述项目中任一项所述的方法,其中,接收该指令的该计算机执行如下步骤:
203.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
204.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
205.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
206.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目5中所述的步骤。
207.项目a7
208.一种方法,其发出指令使计算机执行单一生物单元的序列信息的处理,其中,接收该指令的该计算机执行如下步骤:
209.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
210.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
211.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
212.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
213.项目a8
214.根据上述项目中任一项所述的方法,其中,所述再次聚类化通过网络分析和社群
检测来进行。
215.项目a9
216.一种方法,其发出指令使计算机执行单一生物单元的序列信息的处理,其中,接收该指令的该计算机执行如下步骤:
217.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
218.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
219.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
220.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
221.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
222.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
223.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
224.项目a10
225.根据上述项目中任一项所述的方法,其中,所述部分序列信息由长读段序列来确定。
226.项目a11
227.一种程序,其将处理单一(single)生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
228.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
229.(c)使用该单一生物单元的序列信息的部分序列信息和独立于该聚类化而生成的数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
230.项目a12
231.根据上述项目所述的程序,其中,所述方法还包括如下步骤:(b)将所述数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。
232.项目a13
233.根据上述项目中任一项所述的程序,其中,所述(c)包括:除去一定量的包含确认到多个重复的序列部位的部分序列信息,并校正序列读段的偏差。
234.项目a14
235.一种程序,其将筛选生物谱系鉴定用序列候选的方法安装在计算机中,其中,该方
法包括如下步骤:
236.a)从数据库提取草图中无重复的基因;
237.b)计算每个该基因的单拷贝基因的个数或比例;以及
238.c)选择该单拷贝基因的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
239.项目a15
240.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
241.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
242.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
243.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
244.项目a16
245.根据上述项目中任一项所述的程序,其中,该方法包括如下步骤:
246.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
247.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
248.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
249.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目15中所述的步骤。
250.项目a17
251.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
252.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
253.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
254.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
255.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
256.项目a18
257.根据上述项目中任一项所述的程序,其中,所述再次聚类化通过网络分析和社群检测来进行。
258.项目a19
259.一种程序,其将处理单一生物单元的序列信息的方法安装在计算机中,其中,该方法包括如下步骤:
260.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
261.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
262.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
263.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
264.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
265.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
266.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
267.项目a20
268.根据上述项目中任一项所述的程序,其中,所述部分序列信息由长读段序列来确定。
269.项目a21
270.一种记录介质,其储存有将处理单一(single)生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
271.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
272.(c)使用该单一生物单元的序列信息的部分序列信息和独立于该聚类化而生成的数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
273.项目a22
274.根据上述项目所述的记录介质,其中,所述方法还包括如下步骤:(b)将所述数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。
275.项目a23
276.根据上述项目中任一项所述的方法,其中,所述(c)包括:除去一定量的包含确认到多个重复的序列部位的部分序列信息,并校正序列读段的偏差。
277.项目a24
278.一种记录介质,其储存有将筛选生物谱系鉴定用序列候选的方法安装在计算机中的程序,其中,该方法包括如下步骤:
279.a)从数据库提取草图中无重复的基因;
280.b)计算每个该基因的单拷贝基因的个数或比例;以及
281.c)选择该单拷贝基因的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
282.项目a25
283.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
284.(d)基于规定的判断基准,将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
285.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
286.(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
287.项目a26
288.根据上述项目中任一项所述的记录介质,其中,该方法包括如下步骤:
289.(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
290.(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;
291.(g)根据需要,重复(g),重复进行至该更长的草图达到序列信息的全长为止;以及
292.(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复项目25中所述的步骤。
293.项目a27
294.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
295.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
296.(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
297.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
298.(i)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
299.项目a28
300.根据上述项目中任一项所述的记录介质,其中,所述再次聚类化通过网络分析和社群检测来进行。
301.项目a29
302.一种记录介质,其储存有将处理单一生物单元的序列信息的方法安装在计算机中的程序,其中,该方法包括如下步骤:
303.(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
304.(d)基于规定的判断基准,将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
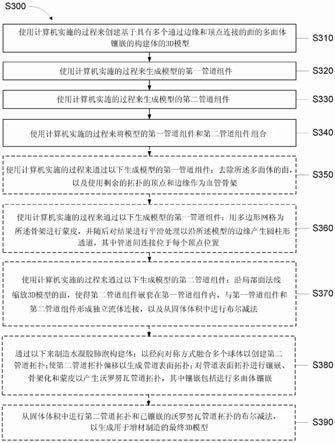
305.(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
306.(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
307.(h)在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
308.(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;
309.(j)对于比较结果,基于规定的判断基准来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
310.项目a30
311.根据上述项目中任一项所述的记录介质,其中,所述部分序列信息由长读段序列来确定。
312.项目a31
313.一种系统,其用于处理单一(single)生物单元的序列信息,该系统包括:
314.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及
315.(c)草图创建部,其使用该单一生物单元的序列信息的部分序列信息和独立于由该(a)聚类部进行的聚类化而生成的数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。
316.项目a32
317.根据上述项目所述的系统,其中,所述系统还包括:(b)追加信息追加部,其将所述数据库中的对应于该聚类的该单一生物单元的部分序列信息追加至该聚类中。
318.项目a33
319.根据上述项目中任一项所述的系统,其中,所述(c)草图创建部包括均质化处理部,所述均质化处理部除去一定量的包含确认到多个重复的序列部位的部分序列信息,并校正序列读段的偏差。
320.项目a34
321.一种系统,其用于筛选生物谱系鉴定用序列候选,该系统包括:
322.a)提取部,其从数据库提取草图中无重复的基因;
323.b)计算部,其计算每个所述基因的单拷贝基因的个数或比例;
324.c)选择部,其选择所述单拷贝基因的个数或比例为规定值以上的基因作为生物谱系鉴定用序列候选。
325.项目a35
326.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
327.(d)排序部,其基于规定的判断基准而将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
328.(e)草图构建部,其基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;以及
329.(e’)选择部,其选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行选择。
330.项目a36
331.根据上述项目中任一项所述的系统,其中,该系统包括:
332.(f)选择部,其将所述所选择的草图与所述(e)草图构建部和(e’)选择部中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;
333.(g)草图改善部,其使用(f)选择部中所选择的序列信息和该所选择的草图,生成更长的草图;
334.(g’)草图构建部,其根据需要使(g)草图改善部重复进行草图生成,直至该更长的草图达到序列信息的全长为止;
335.(g”)机构,其根据需要而在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,使项目35中所述的(d)排序部、(e)草图构建部以及(e’)选择部重复进行排序、草图构建以及选择。
336.项目a37
337.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
338.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
339.(h)再次聚类部,其在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
340.(h’)比较部,其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
341.(i)注册部,对于比较结果,基于规定的判断基准来判断(h)再次聚类部中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
342.项目a38
343.根据上述项目中任一项所述的系统,其中,所述再次聚类部通过网络分析和社群检测来进行再次聚类化。
344.项目a39
345.一种系统,其用于处理单一生物单元的序列信息,该系统包括:
346.(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;
347.(d)排序部,其基于规定的判断基准而将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;
348.(e),其基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息的草图;
349.(e”)草图构建部,其选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准,对至此所生成的草图进行评价;
350.(h)再次聚类部,其在草图的评价未因序列信息的组的集合数量增大而发生变动的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;
351.(h’)比较部,其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及
352.(j)机构,其对于比较结果,基于规定的判断基准来判断(h)再次聚类部中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
353.项目a40
354.根据上述项目中任一项所述的系统,其中,所述部分序列信息由长读段序列来确定。
355.项目b1
356.一种数据结构,包括经如下处理的多个单一生物单元的序列信息的部分序列信息,所述处理为:对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化。
357.项目b2
358.根据上述项目中任一项所述的数据结构,其中,所述数据结构中包含的部分序列信息来源于两个以上的经独立地聚类化而生成的数据库。
359.项目b3
360.根据上述项目中任一项所述的数据结构,其中,所述经独立地聚类化的相关信息与所述部分序列信息关联地储存。
361.项目b4
362.根据上述项目中任一项所述的数据结构,其中,当将所述部分序列信息整合时,构成基因组信息。
363.项目b5
364.根据上述项目中任一项所述的数据结构,其中,所述部分序列信息为按照每个单一生物单元收集的信息。
365.项目b6
366.根据上述项目中任一项所述的数据结构,其中,所述部分序列信息与其来源的单一生物单元的识别信息(id信息)关联地储存。
367.项目b7
368.一种数据结构,其是将多个包括经如下处理的多个单一生物单元的序列信息的部分序列信息的数据结构整合而成的单一生物单元的数据结构,所述处理为:对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化。
369.项目b8
370.根据项目b7所述的数据结构,其中,所述数据结构还包括上述项目中任一项或多项所述的一个以上的特征。
371.本发明旨在上述的一个或多个特征除了以明确示出的组合来提供之外还可以进一步组合来提供。本领域技术人员可根据需要阅读以下的详细说明并加以理解而认识到本发明的更多的实施方式和优点。
372.发明的效果
373.根据本发明,能够以更高精度提供单一生物单元水平上的单一生物单元序列信息。通过使用本发明,能够阐明不可培养的微生物的几乎完整的基因组序列或分析同一株微生物之间的遗传异质性。
附图说明
374.图1为本发明的整体示意图。在本发明中,使用注册在本系统中的生物谱系鉴定用序列,将新获得的单一生物单元的部分序列信息聚类化为同一谱系,并整合所聚类化的多个单一生物单元的部分序列信息,构建最佳的草图基因组序列。注册在微生物基因组数据库中的草图基因组序列在每次测定和注册新的单一生物单元时进行更新,并且品质逐渐提高。
375.图2为本系统中使用的微生物数据库结构。微生物基因组数据库由单一生物单元基因组信息和将该信息整合后创建的草图基因组信息构建而成。草图基因组信息中记录基因组序列所附带的数据,例如,暂定谱系分类、完整率(complete rate)、污染率(contamination rate)、品质类别(quality category)、重叠群(contig)数量、n50统计值、gc含量等。一个草图基因组信息中对应有多个组装碱基序列和基因信息。在基因信息中记录基因所附带的数据,例如,基因名称、基因长度、蛋白质家族、gc含量、标记物类型、单拷贝等。一个基因信息中对应有一个基因碱基序列。单一生物单元基因组信息也附带有与草图基因组信息同等的数据。与草图基因组信息同样地,一个单一生物单元基因组信息与多个组装碱基序列、基因信息相对应,一个基因信息与一个基因碱基序列相对应。并且,一个单一生物单元基因组信息与多个部分碱基序列相对应。
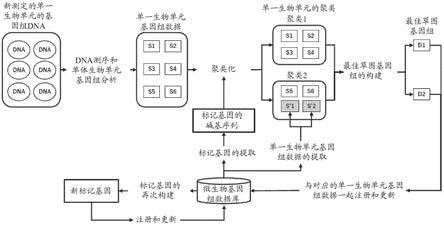
376.图3为通过微生物数据库将新测定的单一生物单元聚类化为同一谱系的方法。用dna测序仪测定单一生物单元的基因组dna,获得部分碱基序列。对部分碱基序列进行组装,获得组装碱基序列。对组装碱基序列进行基因鉴定,获得基因碱基序列。使用蛋白质数据库等,对各基因碱基序列进行功能推定,获得基因信息。同时,对组装碱基序列进行谱系分类,
获得暂定谱系分类信息。并且,评价组装碱基序列的品质,获得基因组信息。将分析单一生物单元而获得的部分碱基序列、基因组信息、组装碱基序列、谱系分类信息、基因信息、基因碱基序列作为单一生物单元基因组数据。使用谱系分类信息,从微生物基因组数据库检索同一谱系的草图基因组信息。进而,基于与草图基因组信息对应的基因信息,获得生物谱系鉴定用序列和其蛋白质家族。从单一生物单元基因组数据中提取具有与所获得的蛋白质家族相同蛋白质家族的基因信息和基因碱基序列,并通过草图基因组的生物谱系鉴定用序列与单一生物单元基因组的对应基因碱基序列的组合来计算同源性。从后述的处理排除具有某预定的同源性以下的组合。将在生物谱系鉴定用序列中同源性最高的基因鉴定为单一生物单元的生物谱系鉴定用序列。通过与同源性匹配的碱基序列长度的加权平均等来评价草图基因组与单一生物单元的相似度。将评价最高的草图基因组作为单一生物单元所属的聚类。万一存在多个同一评价值的草图基因组,则使用所有组装碱基序列而不是生物谱系鉴定用序列来进行同样的评价并加以判定。
377.图4为在不使用微生物数据库的情况下将新测定的单一生物单元聚类化为同一谱系的方法。关于不存在对应于微生物基因组数据库的草图基因组的单一生物单元基因组数据,通过暂定谱系分类来构建暂定的聚类。各暂定的聚类所属的单一生物单元基因组数据使用微生物基因组数据库的生物谱系鉴定用序列,因而判断是否应该进一步分割。分割的方法示于图6中。
378.图5为使用注册在微生物数据库中的基因信息来重新鉴定生物谱系鉴定用序列的方法。随着微生物基因组数据库的数据的积累,草图基因组信息或对应的基因信息的品质逐渐提高。因此,在本系统中,根据高品质草图基因组信息和基因信息来安装新的生物谱系鉴定用序列的再鉴定方法。从微生物基因组数据库提取与已注册的每个草图基因组对应的蛋白质家族并计算其出现频率,创建出现频率矩阵。此时,可以排除品质差的草图基因组。在出现频率矩阵中,对于一个草图基因组以多个频率出现的蛋白质家族因有污染的可能性而被排除在外。当仅有一个蛋白质家族对应时,其蛋白质家族鉴定为单拷贝基因。当蛋白质家族的单拷贝基因以预定以上的比例存在于全部草图基因组内时,该蛋白质家族所对应的基因为生物谱系鉴定用序列候选。按照比例从多至少的顺序排序,采用某基准值以上(例如90%以上)或排前列的几件作为新的生物谱系鉴定用序列。
379.图6为将设为同一谱系的聚类内的新测定单一生物单元进行细分化的方法。在新测定的单一生物单元中被判定为同一聚类的单一生物单元中,通过从微生物基因组数据库提取的生物谱系鉴定用序列,以循环方式评价各单一生物单元的相似度(距离)。使用相似度(距离)矩阵来进行网络分析或聚类分析,将单一生物单元细分化。
380.图7为通过设为同一谱系的聚类内的单一生物单元构建最佳草图基因组的方法。提取设为同一谱系的新型单一生物单元基因组数据,且若存在则从微生物基因组数据库提取同一谱系的单元基因组数据,设为聚类。聚类内的单一生物单元基因组数据基于规定的判断基准(例如,完整率、污染率)进行重排。从以后的处理排除未达到某预定水平的单元基因组数据。将重排的单一生物单元基因组数据按照等级由高到低的顺序选择两个,设为暂定草图基因组构建用单一生物单元基因组数据组。同样地,构建多个按照等级由高到低的顺序追加了单一生物单元基因组数据的暂定草图基因组构建用单一生物单元基因组数据组。对于这些暂定草图基因组构建用单一生物单元基因组数据组,使用ccsag法构建暂定草
图基因组。基于规定的判断基准(例如,完整率、污染率),选择基准最高的暂定草图基因组作为最佳草图基因组。当基准高于微生物基因组数据库中存在的现有的草图基因组时,将新创建的草图基因组注册在微生物基因组数据库中并进行更新。另外,将构建暂定草图基因组的单一生物单元基因组数据数作为说明变量,将基准值(例如,完整率、污染率)作为目标变量,创建暂定草图基因组的评价数据。这在判断为草图基因组的品质收敛而即使追加单一生物单元基因组数据也无法预期进一步的改进等时发挥作用。
381.图8为就注册在微生物数据库中的草图基因组而言构建更高品质的草图基因组的方法。在微生物基因组数据库内的草图基因组中,提取推定为品质收敛的草图基因组作为后处理对象(finishing targets)。所提取的单一生物单元基因组数据基于规定的判断基准(例如,完整率、污染率)进行重排。从以后的处理排除未达到某预定水平的单一生物单元基因组数据。对于这些所提取的单一生物单元,设定与通常进行的草图基因组构建相比容许度更高的参数来构建草图基因组。另一方面,对草图基因组再构建用的单一生物单元基因组数据的组装碱基序列和草图基因组的组装碱基序列进行同源性检索,并检测用于结合草图基因组的组装碱基序列的桥接组装碱基序列。当能够检测到桥接组装碱基序列时,使用其来结合草图基因组的组装碱基序列。对如此构建的两个草图基因组数据和已经在微生物基因组数据库注册的草图基因组数据进行比较评价,并选择基准值高的一方。当选择新的草图基因组数据时,将其注册在微生物基因组数据库中并进行更新。
382.图9为就注册在微生物数据库中的草图基因组而言用于进行进一步的细分化的方法。在微生物基因组数据库内的草图基因组中,提取推定为品质收敛的草图基因组作为细分化对象。所提取的单一生物单元基因组数据基于规定的判断基准(例如,完整率、污染率)进行重排。从以后的处理排除未达到某预定水平的单一生物单元基因组数据。对这些所提取的单一生物单元进行图6的细分化处理。针对所细分化的各聚类的单一生物单元基因组数据,分别构建草图基因组。将由细分化所构建的草图基因组数据与已注册在微生物基因组数据库中的草图基因组数据进行比较,选择基准值高的一方。当选择新的草图基因组数据时,注册在微生物基因组数据库中并进行更新。
383.图10为表示独立地进行分析时的系统结构的图。从dna碱基序列输出的单一生物单元的部分碱基序列数据通过便携式hdd(hard disk drive)等外部存储装置而记录在分析用计算机的辅助存储装置中。另外,在辅助存储装置中记录有用于进行序列处理的程序群、微生物基因组数据库。程序和部分碱基序列数据从辅助存储装置加载到主存储装置中,并通过中央运算处理装置来执行处理。一系列处理通过键盘、鼠标等输入装置来进行,处理结果输出在监视器等输出装置和辅助存储装置中。
384.图11为表示通过云端分析等互联网进行处理时的系统结构的图。从dna碱基序列输出的单一生物单元的部分碱基序列数据可记录在ftp服务器等中并通过互联网下载。将部分碱基序列数据上传到hpc(high-performance computing,高性能计算)系统,并在hpc系统上进行处理。微生物基因组数据库可通过数据库服务器进行访问,也可以将数据库本身下载到hpc系统上来使用。这一系列处理通过连接到互联网的分析用终端来进行。
385.图12为表示可通过将外部数据库中的序列追加到聚类中来构建高品质的基因组序列的图。
386.图13为偏差均质化处理的示意图。
387.图14为表示偏差均质化处理之前的大肠杆菌sag序列数据的偏差评价和获取基因组序列的图。
388.图15为表示偏差均质化处理之后的大肠杆菌sag序列数据的偏差评价和获得基因组序列的图。
具体实施方式
389.以下,示出最佳方式来说明本发明。应当理解的是,在整个说明书中,除非另有说明,单数形式的表述还包括其复数概念。因此,应当理解的是,除非另有说明,单数形式的冠词(例如,英语中的“a”、“an”、“the”等)还包括其复数概念。另外,应当理解的是,除非另有说明,本说明书中使用的术语以本领域通常使用的含义来使用。因此,除非另有定义,本说明书中使用的所有专业术语和科技术语具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。如有冲突,以本说明书(包括定义)为准。
390.(定义等)
391.下面适当说明本说明书中具体使用的术语的定义和/或基本的技术内容。
392.在本说明书中,“单一生物单元”是指具有遗传信息或其他生体分子的信息的单元。单一生物单元中可以包括细胞、细胞样结构物等,但不限定于此,还可包含人工生产的细胞(所谓人工细胞)或数字化细胞(作为信息提供)等。
393.在本说明书中,“细胞”是内部包含具有遗传信息的分子的粒子,是指能够被复制(无关于是否单独进行)的任意粒子。作为本说明书中的“细胞”,包含单细胞生物的细胞、细菌、源自多细胞生物的细胞、真菌等。
394.在本说明书中,“细胞样结构物”是指内部包含具有遗传信息的分子的任意粒子。作为本说明书中的“细胞样结构物”,包含细胞器,例如,线粒体、细胞核和叶绿体、以及病毒等。
395.在本说明书中,“遗传信息或其他的生体分子的信息”是指规定生体分子或其类似物的信息。遗传信息或其他生体分子的信息可包括核酸、氨基酸、脂质或糖链、或者它们的类似物的结构信息等,但不限定于此,还可包含代谢物质等生体内分子或其类似物的相互作用的多样性信息等。“遗传信息”还被称为“核酸信息”,两者的含义相同。
396.在本说明书中,“生体分子”是指任意的生物或病毒所具有的分子。生体内分子可包含核酸、蛋白质、糖链或脂质等。在本说明书中,“生体分子的类似物”是指生体分子的天然或非天然的变体。生体内分子的类似物可包含修饰核酸、修饰氨基酸、修饰脂质或修饰糖链等。
397.在本说明书中,“集合”是指包含两个以上单一生物单元、细胞或细胞用结构物的汇集。
398.在本说明书中,“子集”是指当与“集合”一同使用时具有比集合更少数量的单一生物单元、细胞或细胞用结构的集合的一部分。
399.在本说明书中,“凝胶”是指:在胶体溶液(溶胶)中,高分子物质或胶体粒子在其相互作用下整体形成网络结构,包含大量作为溶剂或分散介质的液相的同时失去流动性的状态。在本说明书中,“凝胶化”是指使溶液变为“凝胶”的状态。
400.在本说明书中,“胶囊”是指具有能够在其中保持细胞或细胞样结构物的形状的物
质。在本说明书中,“凝胶胶囊”是指可在其中保持细胞或细胞样结构物的凝胶状的微粒子状结构体。
401.在本说明书中,“基因分析”是指研究生物样品中的核酸(dna、rna等)的状态。在一个实施方式中,基因分析可列举利用核酸扩增反应的基因分析。作为包括它们在内的基因分析的示例,可列举序列确定、基因型判定/多态性分析(snp分析、拷贝数多态性、限制性酶片段长度多态性、重复数多态性)、表达分析、荧光猝灭探针(quenching probe:q-probe)、sybr green法、熔解曲线分析、实时pcr、定量rt-pcr、数字pcr等。
402.在本说明书中,“单一生物单元水平”是指:针对一个单一生物单元中包含的遗传信息或其他的生体分子的信息,以能够与其他单一生物单元中包含的遗传信息或其他的生体分子的信息加以区别的状态进行处理。
403.在本说明书中,“单细胞水平”是指;针对一个细胞或细胞样结构物中包含的遗传信息或其他的生体分子的信息,以能够与其他细胞或细胞样结构物中包含的遗传信息或其他的生体分子的信息加以区别的状态进行处理。例如,当将“单一生物单元水平”或“单细胞水平”上的多核苷酸扩增时,分别以某单一生物单元或者某细胞或细胞样结构物中的多核苷酸与其他单一生物单元或者其他细胞或细胞样结构物中的多核苷酸能够加以区别的状态,进行各自的扩增。在本发明的一个实施方式中,使该多核苷酸与扩增用试剂接触并在凝胶胶囊中扩增该多核苷酸的工序也可以在凝胶胶囊中保持凝胶状态的同时扩增该多核苷酸。
404.在本说明书中,“单一生物单元分析”是指以将一个单一生物单元(例如,细胞或细胞样结构物)中包含的遗传信息或其他的生体分子的信息与其他单一生物单元(例如,细胞或细胞样结构物)中包含的遗传信息或其他的生体分子的信息加以区别的状态进行分析。
405.在本说明书中,“单细胞分析”是指以将一个细胞或细胞样结构物中包含的遗传信息或其他的生体分子的信息与其他细胞或细胞样结构物中包含的遗传信息或其他的生体分子的信息加以区别的状态进行分析。
406.在本说明书中,“遗传信息”是指编码一个细胞或细胞样结构物中包含的基因等信息的核酸信息,包括特定基因序列的有无、特定基因的产量或总核酸产量。
407.在本说明书中,“生体分子的信息”是指一个细胞或细胞样结构物中包含的生体分子(核酸等、除核酸以外还包含蛋白质、糖、脂质等)或其类似物的信息,包括特定生体分子的结构或序列的有无、结构或序列的一致性、特定生体分子的产量以及总体生体分子的产量。
408.在本说明书中,“核酸信息”是指一个细胞或细胞样结构物中包含的核酸信息,并包括特定基因序列的有无、特定基因的产量或总核酸产量。
409.在本说明书中,“一致性”是指两个生体分子之间的结构或序列的相似性。当对象为序列时,一致性还可以通过将为了比较而能够比对的各序列中的位置进行比较来确定。
410.在本说明书中,“长读段序列”是指使用长读段(为了分析而片段化的核苷酸链)进行全部序列的测序的方法。通常,长读段序列用400个碱基以上的长度的读段来进行解读。
411.(优选的实施方式)
412.以下,记载优选实施方式的说明,但应当理解的是,该实施方式为本发明的示例,本发明的范围不限定于这样的优选实施方式。应当理解的是,本领域技术人员还可以参考
以下所述的优选实施例在本发明的范围内容易地进行修改、变更等。对于这些实施方式,本领域技术人员可适当组合一个或多个任意的实施方式。
413.(序列信息处理)
414.在一个方案中,本发明提供处理单一(single)生物单元(例如,细胞或细胞结构物)的序列信息的方法。该方法包括如下步骤:(a)对多个单一生物单元的序列信息(例如,基因组、转录组、蛋白质组或同等的基因等的集合)的部分序列信息,基于生物谱系鉴定用序列(例如,16s rdna或标记基因),按照每个同一谱系进行聚类化;(b)根据需要而将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;(c)使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。该方案的示意图示于图1中。草图基因组与本发明的单元生物单元基因组的对应关系示于图2中。
415.(b)为任意工序,可以利用数据库,也可以不利用数据库。如上所述,聚类化的方法包括利用数据库的方法(图3)和不利用数据库的方法(图4)。当利用数据库时,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。当不利用数据库时,新生成聚类。
416.还可从分类后的数据库新确定生物谱系鉴定用序列(标记物)。在该方案中,本发明提供处理单一(single)生物单元(例如,细胞)的序列信息的方法,该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(b)将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。在这种情况下,生物谱系鉴定用序列可用作所谓的生物标记物。该方案的示意图示于图5中。在该方案中,提取所注册的草图基因组所对应的蛋白质家族,并创建草图基因组与蛋白质家族的对应矩阵。计算所对应的蛋白质家族为1的单拷贝基因的比例。其中,可采用存在于全部草图基因组中的蛋白质家族作为标记基因。本发明提供处理单一(single)生物单元的序列信息的方法,其中,该方法包括如下步骤:a)从数据库提取草图中无重复的基因(单拷贝基因)候选;b)计算对应于每个所述基因的草图的个数(或比例);c)按照所述对应草图的个数(或比例)从多至少的顺序排序,选择规定值以上的基因(或排前列的任意数量的基因)作为标记基因候选。
417.在一个方案中,本发明为处理单一生物单元的序列信息的方法,该方法包括如下步骤:(d)基于规定的判断基准(例如,完整率、污染率),将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息(该长度可以为一部分,也可以为全长)的草图;以及(e’)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准(例如,完整率、污染率),对至此所生成的草图进行选择。优选重复(e’)。这是因为优选在改变sag的数量的同时重复进行草图创建。在一部分的实施方式中,作为创建单一生物单元的序列信息草图的步骤,还可进行上述的(d)~(e’)。该方案的示意图示于图7中。
418.在一个优选实施方式中,本发明的处理单一生物单元的序列信息的方法包括如下
步骤:(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;(g)根据需要,重复(g)优选重复进行至该更长的草图达到序列信息的全长为止;以及(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复(d)、(e)以及(e’)。作为更低基准的判断基准,例如,可使用更宽松的参数。该方案的示意图示于图8中。
419.在一个方案中,部分序列信息为sag。在特定的方案中,本发明提供在与判断sag为“同一”聚类(例如,谱系、种)后紧接的阶段相关的方案中使聚类精细化的方法。在该方案中,本发明为处理单一生物单元的序列信息的方法,该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及(i)对于比较结果,基于规定的判断基准(例如,完整率、污染率)来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
420.其中,上述评价可以用标记基因对所提取的部分序列信息(例如,sags)进行循环评价,例如,可通过各sag之间的距离来进行评价。
421.在优选实施方式中,本发明中实施的再次聚类化通过网络分析和社群检测来进行。
422.本发明还提供即使增加部分序列信息(例如,sags)的数量也不再使草图的品质提高的后续阶段的方案中的处理。在该方案中,本发明为处理单一生物单元的序列信息的方法,该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(d)基于规定的判断基准(例如,完整率、污染率),将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度(可以为一部分,也可以为全长)大于该部分序列信息的草图;(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准(例如,完整率、污染率),对至此所生成的草图进行评价;(h)在草图的评价未因序列信息的组的集合数量增大而发生变动(即停留在预定范围内)的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及(j)对于比较结果,基于规定的判断基准(例如,完整率、污染率)来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
423.在本发明中可理解为这些方法的各步骤可以适当组合。在一部分实施方式中,当进行单一(single)生物单元的序列信息的处理以及生物谱系鉴定用序列候选的筛选时,发
出指令使计算机执行这些处理的位置与接收指令并实际实施这些处理等的位置可以不同。在其他实施方式中,本发明的方法的各处理可通过计算机来执行。在另一实施方式中,本发明的数据库可以为通过本发明的聚类或序列分析方法生成的数据库,也可以为独立于本发明的聚类或序列分析方法而生成的数据库。在优选实施方式中,独立于本发明的聚类或序列分析方法而生成的数据库可以为对基于单细胞扩增而扩增的序列进行测序而获得的数据的数据库。在现有技术中,认为追加其他数据库中的序列会导致序列品质降低,但实际上发现:通过将其他数据库的序列追加至聚类中,会使序列品质得到改善。
424.在一部分实施方式中,当从序列数据构建草图基因组时,可除去一定量的包含确认到多个重复读取的序列部位的部分序列信息,并校正(均质化)序列读段的偏差。根据进行了均质化处理的序列数据的聚类,使用由均质化序列数据创建的基因组序列作为参考序列而重复进行均质化处理,由此可预期基因组品质的进一步改善。当供于均质化处理的部分序列信息通过长读段序列来读出时,可预期基因组品质的更进一步的改善。
425.当构建源自单一生物单元的序列的草图基因组时,以数据本身干净且具有预定程度的基因组完整性、多个单细胞数据可汇总获得为前提,这在现有技术中无法实现,而是通过本发明首次实现。并且,无需通过长读段序列来解读源自单一生物单元的序列的草图基因组。并且,在源自单一生物单元的序列中,认识到存在产生嵌合体(原本未相连而分开的基因组序列因扩增时的错误等而产生,并产生被错误解读的序列数据)的问题,因此,并未开发适于具有嵌合体和高扩增偏差的单细胞数据的长读段组装系统。通过有效利用本发明,参考多个单细胞基因组,重复进行映射(mapping)和组装,从而可大幅降低该偏差,由此,可获得极为准确的基因组序列。
426.众所周知,在源自单细胞的基因组序列等的扩增dna的序列中产生偏差。关于这一点,在以往的方法中,均质化处理(降低偏差)采取如下方法,即,钻研酶反应或反应条件而使扩增时不易产生偏差本身(nishikawa et al.plos one);或者积极分解dna以使扩增后产生的偏差消减;等等。然而,在这些方法中存在无法完全消除偏差的问题。在本发明中,即使是已产生偏差的数据,由于以计算机(in silico)执行处理,因而也无需进行如上所述的特别的反应系统的钻研便可将数据均质化。需要说明的是,此时也以数据本身干净且源自多个为前提,因此,只能通过本发明中利用的方法来执行。在以往的方法中,基因组序列的正确性是对近缘种的参考基因组进行映射等,评价偏差或间隙(gap)部等,进行序列校正,但本发明中利用的方法即使在均质化处理时不存在近缘种参考基因组,通过整合分析同一种的多个数据,从而可以参考自身数据来执行均质化处理,因此无参考序列的未知微生物样品的数据也可进行均质化,这一点与现有技术相比,发挥显着的优异效果。并且,在未知微生物的完整基因组解读中非常有效。进而,在无法确定基因组中的基因聚类位置的细胞中,也无需培养便可对基因聚类无间隙地完整解读序列,并能详细获知其功能。另外,还可以进行如下研究开发:将该基因聚类导入至易于处理的其他生物中,制作目标物质。可期待如下所述的应用例和假想实施例。
427.·
抗生物质耐性基因/耐性株的监视
428.·
微生物基因组序列的后处理(finishing)(封闭为环状基因组)(基本上除了培养菌株以外,可封闭的情况较少)
429.·
生合成基因聚类的获得
430.·
基于合成生物学的微生物主体的基因改型评价
431.·
基因组结构变异及各种代谢功能/对主体生物的影响评价。
432.程序和记录介质
433.在一个方案中,本发明提供指示将处理单一(single)生物单元(例如,细胞或细胞结构物)的序列信息的方法安装在计算机中的计算机程序以及储存该程序的记录介质(例如,cd-r、闪存、硬盘、传输介质、云端等)。该程序所安装的该方法包括如下步骤:(a)对多个单一生物单元的序列信息(例如,基因组、转录组、蛋白质组、或同等的基因等的集合)的部分序列信息,基于生物谱系鉴定用序列(例如,16s rdna或标记基因),按照每个同一谱系进行聚类化;(b)根据需要而将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及(c)使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。该方案的示意图示于图1中。草图基因组与本发明的单元生物单元基因组的对应关系示于图2中。
434.(b)为任意工序,可利用数据库,也可以不利用数据库。如上所述,聚类化的方法包括利用数据库的方法(图3)和不利用数据库的方法(图4)。当利用数据库时,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。当不利用数据库时,新生成聚类。
435.还可从分类后的数据库新确定生物谱系鉴定用序列(标记物)。在该方案中,本发明提供指示将处理单一(single)生物单元(例如,细胞)的序列信息的方法安装在计算机中的计算机程序以及储存该程序的记录介质(例如,cd-r、闪存、硬盘、传输介质、云端等)。该程序所安装的该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(b)将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。在这种情况下,生物谱系鉴定用序列可用作所谓的生物标记物。该方案的示意图示于图5中。在该方案中,提取所注册的草图基因组所对应的蛋白质家族,并创建草图基因组与蛋白质家族的对应矩阵。计算所对应的蛋白质家族为1的单拷贝基因的比例。其中,可采用存在于全部草图基因组中的蛋白质家族作为标记基因。本发明提供指示将处理单一(single)生物单元的序列信息的方法安装于计算机中的程序或者储存该程序的记录介质,其中,该方法包括如下步骤:a)从数据库提取草图中无重复的基因(单拷贝基因)候选;b)计算对应于每个所述基因的草图的个数(或比例);c)按照所述对应草图的个数(或比例)从多至少的顺序排序,选择规定值以上的基因(或排前列的任意数量的基因)作为标记基因候选。
436.在一个方案中,本发明提供指示将处理单一生物单元的序列信息的方法安装于计算机中的计算机程序以及储存该程序的记录介质(例如,cd-r、闪存、硬盘、传输介质、云端等)。该程序所安装的该方法包括如下步骤:(d)基于规定的判断基准(例如,完整率、污染率),将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度大于该部分序列信息(该长度可以为一部分,也可以为全长)的草图;以及(e’),选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断
基准(例如,完整率、污染率),对至此所生成的草图进行选择。优选重复(e’)。这是因为优选在改变sag的数量的同时重复进行草图创建。该方案的示意图示于图7中。
437.在一个优选实施方式中,本发明的程序所安装的处理单一生物单元的序列信息的方法包括如下步骤:(f)将所述所选择的草图与所述(e)和(e’)中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;(g)使用(f)中所选择的序列信息和该所选择的草图,生成更长的草图;(g)根据需要,重复(g)优选重复进行至该更长的草图达到序列信息的全长为止;以及(g”)根据需要,在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,重复(d)、(e)以及(e’)。作为更低基准的判断基准,例如,可使用更宽松的参数。该方案的示意图示于图8中。
438.在另一个方案中,本发明的程序编码在与判断sag为“同一”聚类(例如,谱系、种)后紧接的阶段相关的方案中使聚类精细化的方法。在该方案中,本发明提供指示将处理单一生物单元的序列信息的方法安装在计算机中的计算机程序以及储存该程序的记录介质(例如,cd-r、闪存、硬盘、传输介质、云端等)。该程序所安装的该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(h)在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;(i)对于比较结果,基于规定的判断基准(例如,完整率、污染率)来判断(h)中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
439.其中,上述评价可以用标记基因对所提取的部分序列信息(例如,sags)进行循环评价,例如,可通过各sag之间的距离来进行评价。在优选实施方式中,本发明中实施的再次聚类化通过网络分析和社群检测来进行。
440.本发明的程序还提供即使增加部分序列信息(例如,sags)的数量也不再使草图的品质提高的后续阶段的方案中的处理。在该方案中,本发明提供指示将处理单一生物单元的序列信息的方法安装在计算机中的计算机程序以及储存该程序的记录介质(例如,cd-r、闪存、硬盘、传输介质、云端等)。该程序所安装的该方法包括如下步骤:(a)对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(d)基于规定的判断基准(例如,完整率、污染率),将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;(e)基于该排序,从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度(可以为一部分,也可以为全长)大于该部分序列信息的草图;(e”)选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准(例如,完整率、污染率),对至此所生成的草图进行评价;(h)在草图的评价未因序列信息的组的集合数量增大而发生变动(即停留在预定范围内)的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及(j)对于比较结果,基于规定
的判断基准(例如,完整率、污染率)来判断(h)中的再次聚类化是否适当,当适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复进行(d)~(e’)。
441.在另一方案中,本发明提供包括经如下处理的多个单一生物单元的序列信息的部分序列信息的数据结构,所述处理为:对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化。在一个实施方式中,所述数据结构中包含的部分序列信息来源于两个以上的经独立地聚类化而生成的数据库。在一个实施方式中,经独立地聚类化的相关信息与所述部分序列信息关联地储存。在一个实施方式中,当将部分序列信息整合时,构成基因组信息。在一个实施方式中,部分序列信息为按照每个单一生物单元收集的信息。在一个实施方式中,部分序列信息与其来源的单一生物单元的识别信息(id信息)关联地储存。
442.在另一个实施方式中,本发明提供将多个包括经如下处理的多个单一生物单元的序列信息的部分序列信息的数据结构整合而成的单一生物单元的数据结构,所述处理为:对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化。以往不存在整合单细胞等的单一生物单元的高品质数据库,其是本发明首次提供的。
443.系统
444.在一个方案中,本发明提供处理单一(single)生物单元(例如,细胞或细胞结构物)的序列信息的系统。该系统包括:(a)聚类部,其对多个单一生物单元的序列信息(例如,基因组、转录组、蛋白质组、或同等的基因等的集合)的部分序列信息,基于生物谱系鉴定用序列(例如,16s rdna或标记基因),按照每个同一谱系进行聚类化;(b)追加信息追加部(其可与聚类部相同,也可不同),其根据需要而将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中;以及(c)草图创建部,其使用该单一生物单元的序列信息的部分序列信息和该数据库中的该单一生物单元的序列信息,创建该单一生物单元的序列信息草图。该方案的示意图示于图1中。草图基因组与本发明的单元生物单元基因组的对应关系示于图2中。
445.相当于(b)的追加信息追加部是任意的,可以利用数据库,也可以不利用数据库。
446.如上所述,聚类部所实现的聚类化方法包括利用数据库的方法(图3)和不利用数据库的方法(图4)。当利用数据库时,将数据库中的对应于所述聚类的该单一生物单元的部分序列信息追加至所述聚类中。当不利用数据库时,新生成聚类。
447.对于本发明的系统,还可从分类后的数据库新确定生物谱系鉴定用序列(标记物)。在该方案中,本发明提供处理单一(single)生物单元(例如,细胞)的序列信息的系统。该系统包括:(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;以及(b)鉴定部(也称为标记物鉴定部),其将数据库中的对应于所述聚类的部分序列信息与所述聚类的部分序列信息进行比较,计算每个部分序列的相似度,将规定相似度以上的部分序列鉴定为生物谱系鉴定用序列。在这种情况下,生物谱系鉴定用序列可用作所谓的生物标记物。该方案的示意图示于图5中。在该方案中,提取所注册的草图基因组所对应的蛋白质家族,并创建草图基因组与蛋白质家族的对应矩阵。计算所对应的蛋白质家族为1的单拷贝基因的比例。其中,可采用存在于全
部草图基因组中的蛋白质家族作为标记基因。本发明提供用于处理单一(single)生物单元的序列信息的系统,该系统包括:a)提取部,其从数据库提取草图中无重复的基因;b)计算部,其计算对应于每个所述基因的草图的个数或比例;以及c)选择部,其选择所述对应草图的个数或比例为规定值以上的基因作为标记基因候选。
448.在一个方案中,本发明提供处理单一生物单元的序列信息的系统。该系统包括:(d)排序部,其基于规定的判断基准(例如,完整率、污染率)而将多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;以及(e)草图构建部,其基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度(该长度可以为一部分,也可以为全长)大于该部分序列信息的草图,选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准(例如,完整率、污染率),对至此所生成的草图进行选择。优选重复多次草图制作。这是因为优选在改变部分序列信息(例如,sags)的数量的同时重复进行草图创建。该方案的示意图示于图7中。
449.在一个优选实施方式中,本发明的系统包括:(f)选择部(其可以配置为草图构建部的一部分),其将所述所选择的草图与所述(e)草图构建部和(e’)选择部中未选择的单一生物单元的序列信息的部分序列信息进行比较,选择具有未包含在该草图中的部分的序列的、单一生物单元的序列信息的部分序列信息;(g)草图改善部(其也可以配置为草图构建部的一部分),其使用(f)选择部中所选择的序列信息和该所选择的草图,生成更长的草图;(g’)草图构建部,其根据需要而重复(g)草图改善部,优选重复进行至该更长的草图达到序列信息的全长为止;(g”)机构,其根据需要而在全部的构成该草图的部分序列信息中,基于更低基准的判断基准,使(d)排序部、(e)草图构建部以及(e’)选择部重复进行排序、草图构建以及选择。该重复可在草图构建部等中实现。作为更低基准的判断基准,例如,可使用更宽松的参数。该方案的示意图示于图8中。
450.在另一个方案中,本发明的系统编码在与判断sag为“同一”聚类(例如,谱系、种)后紧接的阶段相关的方案中使聚类精细化的方法。在该方案中,本发明提供处理单一生物单元的序列信息的系统。该系统包括:(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(h)再次聚类部(其可以通过聚类部来实现),其在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)比较部(其也可以通过聚类部来实现),其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及(i)注册部,其对于比较结果,基于规定的判断基准(例如,完整率、污染率)来判断(h)再次聚类部中的再次聚类化是否适当,当适当时,作为新群组注册在数据库中。
451.其中,上述评价可以用标记基因对所提取的部分序列信息(例如,sags)进行循环评价,例如,可通过各sag之间的距离来进行评价。
452.在优选实施方式中,本发明中实施的再次聚类化通过网络分析和社群检测来进行。
453.本发明的系统还提供即使增加部分序列信息(例如,sags)的数量也不再使草图的品质提高的后续阶段的方案中的处理。在该方案中,本发明提供处理单一生物单元的序列
信息的系统。该系统包括:(a)聚类部,其对多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列,按照每个同一谱系进行聚类化;(d)排序部,其基于规定的判断基准(例如,完整率、污染率)而将属于该同一谱系聚类的多个单一生物单元的序列信息的部分序列信息按照品质从高至低进行排序;草图构建部,其进行以下两个步骤,即,(e)基于该排序而从高至低地选择该多个单一生物单元的序列信息的部分序列信息中的规定数量的集合,从该部分序列信息构建长度(可以为一部分,也可以为全长)大于该部分序列信息的草图,和选择与该集合不同数量的单一生物单元的序列信息的部分序列信息的组的集合,从该部分序列信息构建长度大于该部分序列信息的草图,基于规定的判断基准(例如,完整率、污染率),对至此所生成的草图进行评价;(h)再次聚类部(其可以通过聚类部来实现),其在草图的评价未因序列信息的组的集合数量增大而发生变动(即停留在预定范围内)的情况下,在该同一谱系聚类中,对构成单一生物单元的序列信息的该多个单一生物单元的序列信息的部分序列信息,基于生物谱系鉴定用序列进行评价,并在同一谱系聚类中进行再次聚类化;(h’)比较部(其也可以通过聚类部来实现),其将从该同一谱系聚类创建的序列信息草图与从该再次聚类化的聚类创建的序列信息草图进行比较;以及判断部(j),其对于比较结果,基于规定的判断基准(例如,完整率、污染率)来判断再次聚类化部(h)中的再次聚类化是否适当,在判断部中,当该判断为适当时,对属于该再次聚类化的聚类的多个单一生物单元的序列信息的部分序列信息,重复由(d)~(e’)实现的步骤。
454.以上,基于实施方式说明了本发明的一个或多个方式涉及的系统、程序、记录介质、方法,但本发明不限定于该实施方式。在不脱离本发明主旨的前提下,本领域技术人员能够想到的对本实施方式进行的各种变形或者组合不同实施方式中的构成要素而构建的方式均可包含在本发明的一个或多个方式的范围内。
455.另外,上述各实施方式中的患病与否判定装置所具备的构成要素的一部分或全部可以由一个系统lsi(large scale integration,大规模集成电路)构成。例如,本发明的处理序列信息的系统可以根据需要与数据库组合,也可以包括或组合用于确定具有生物标记物等的功能的序列的系统(图10)。
456.系统lsi是将多个构成部集成在一个芯片上而制成的超多功能lsi,具体而言,是包括微处理器、rom(read only memory)、ram(random access memory)等而构成的计算机系统。在rom中存储有计算机程序。所述微处理器按照计算机程序运行时,系统lsi实现其功能。此处,虽然称为系统lsi,但根据集成度的不同,有时也称为ic、lsi、超级lsi、超大级lsi。另外,集成电路化的方法不限定于lsi,也可通过专用电路或通用处理器来实现。在制造lsi之后,也可利用能够编程的fpga(field programmable gate array)、或者能够再次构成lsi内部的电路单元的连接和设置的可重组处理器。若由于半导体技术的进步或衍生的其他技术而出现替代lsi的集成电路化的技术,则当然也可以使用该技术来进行功能块的集成化。使生物技术的应用等也成为可能。
457.另外,本发明的一个方案不仅可以是这样的序列信息处理装置或系统,还可以是功能特化的系统(例如,生物标记物筛选装置、药效判定装置、诊断装置等)。另外,本发明的实施方式可以为使计算机执行序列信息处理中包含的表征性的各步骤的计算机程序。另外,本发明的一个方式可以为记录有如上所述的计算机程序的计算机可读取的非暂时性记录介质。
458.需要说明的是,在上述各实施方式中,各构成要素可由专用硬件构成或通过执行适于各构成要素的软件程序来实现。各构成要素可通过cpu或处理器等程序执行部读出并执行记录在硬盘或半导体存储器等记录介质中的软件程序来实现。此处,实现上述各实施方式的疼痛推定装置等的软件可以为本说明书中的上述程序。
459.(使用云端、iot以及ai的实施方式)
460.本发明的序列信息处理技术可以以就一个系统或装置而言所包括的所有形式来提供。或者,也可假设为如下形式:作为序列信息处理装置而主要进行分析和结果的显示,计算或判别模型的计算在服务器或云端来进行。它们的一部分或全部可使用iot(internet of things)和/或人工智能(ai)来实施(图11)。
461.或者,序列信息处理装置还储存各种计算所需的机构并当场进行分析,但分析所需的计算可假设为在服务器或云端中进行的方式即半独立型的方式。在医院等一部分实施场所中,由于未必始终能够发送和接收,因此假设了即使在屏蔽的情况下也可使用的模型。
462.保存部可以是记录介质,例如,cd-r、dvd、blueray、usb、ssd、硬盘等,也可以储存在服务器中,还可以是适当记录在云端上的形式。
463.作为如上所述的云服务,大致相当于“software as service,软件即服务(saas)”。序列信息处理装置被认为搭载有由在实验室环境下的数据创建的判别算法,因此可以提供为具有这些实施方式的两个或三个特征的系统。
464.数据保存也可以根据需要来进行。数据保存通常设置在服务器侧,但在全装备型时,当然可以在终端侧,在云端型时,也可以在终端侧(可选)。当在云端提供服务时,数据保存可提供标准(例如,云端最大10gb)、选项1(例如,云端增加10tb)、选项2(云端设定参数而进行分割存储)、选项3(云端进行分析选项保存)的选项。保存数据,从出售的所有装置抽取数据而制作大数据(例如,序列数据库),实时更新分析模型,或者构建新模型,例如可提供如“疾病判定模型”那样的新的判别模型软件。
465.另外,可具有数据分析选项。此处,可提供服务提供的接收方的要求等。即,可假设为计算方法的选项。
466.在本说明书中,“或”在可以采用文章中所列事项中的“至少一个以上”时使用。“或者”也同样。在本说明书中,记载为“两个值的范围内”时,其范围中也包含两个值本身。
467.在本说明书中所引用的科学文献、专利、专利申请等参考文献的整体以与各自具体的记载相同程度作为参考援引于本说明书中。
468.以上,为了便于理解,示出优选实施方式来说明了本发明。以下,基于实施例来说明本发明,但上述的说明以及以下的实施例仅以例示的目的来提供,并不以限定本发明的目的来提供。因此,本发明的范围仅由权利要求书来进行限定,而不限定于本说明书中具体记载的实施方式和实施例。
469.实施例
470.以下记载实施例。
471.试剂类使用在实施例中具体说明的产品,但也可替代使用其他制造商的同等产品。
472.(实施例1)
473.(草图基因组创建法)
474.(方法)
475.(细胞株样品制备)
476.对由hosokawa等人报道的大肠杆菌e.coli k12(atcc 10798)和枯草芽孢杆菌b.subtilis(atcc 6633),各自分别获得12个sag数据。在hosokawa等人的论文中,这些细胞从atcc获得。e.coli k12在luria-bertani(lb)培养基(1.0%细菌培养用胰蛋白胨、0.5%酵母提取物、1.0%nacl、ph7.0)中进行了培养。b.subtilis在脑心浸液肉汤(atcc培养基44、thermo fisher scientific,san jose,ca,usa)中进行了培养。将回收的细胞用经紫外线(uv)处理的磷酸缓冲生理盐水(-)(pbs,thermo fisher scientific)洗涤3次,供于单一液滴mda以及测序。
477.(小鼠肠道微生物菌群的制备)
478.从7周龄雄性icr小鼠(tokyo laboratory animals science co.,ltd.,tokyo,japan)收集粪便,并在pbs中进行均质化。通过以2000
×
g离心分离2秒钟来回收上清液,并以15000
×
g离心分离3分钟。将所获得的细胞颗粒用pbs洗涤2次,最后再次悬浮于pbs中。
479.(单一液滴mda)
480.制造微流体液滴产生器和mda反应装置,并按照hosokawa等人的报告,用于单一小滴mda中。在分析之前,将细胞悬浮液调整到0.1细胞/液滴的浓度,防止在单一的液滴中封入多个细胞。使用液滴产生器,将单一的微生物细胞封入裂解缓冲液d2(qiagen、hilden、germany)中,然后在65℃下溶解了10分钟。接着,将细胞裂解物注入液滴融合装置,与加入了tween-20和evagreen的mda反应混合液(repli-gsingle cell kit、qiagen)的液滴进行混合。回收到pcr管中后,将液滴在30℃下培养2小时,并在65℃下培养3分钟。关于单细胞测序,逐一选取经荧光标记的液滴,在开放式洁净工作台(koach 500-f,koken ltd,东京,日本)下采用微量移液管转移到新鲜的mda反应混合物中。在30℃下培养2小时后,在65℃下使酶失活3分钟。
481.(16s rdna序列的确定)
482.为了确认源自单一细胞的扩增,将16s rrna基因片段v3-v4扩增,通过对由单一液滴mda获得的sag的sanger测序来进行测序。为了比较谱系学的分布,从肠道菌群的宏基因组样品扩增16s rrna片段(v3-v4),并通过miseq(illumina、san diego、ca、usa)进行了测序。使两端(paird end)读段连结并进行修剪,然后,通过uparse以97%的一致性聚类化为分类单元。分类由rdp分类器来确定。
483.(文库构建和全基因组测序)
484.使用nextera xt dna样品制备试剂盒(illumina)和nextera xt index kit,从单一液滴mda的产物制备单一细胞测序用的illumina文库。接着,将文库以2
×
300的两端读段在illumina miseq系统上进行测序。
485.(sag读取的品质管理以及交叉参考重叠群的构建(ccsag的步骤1))
486.将sag首先基于99%以上的16s rrna相似性和95%以上的ani进行分类。通过全长的原始sag重叠群之间的成对blast推定核苷酸一致性,并计算500bp以上。接着,使用fastx工具包(http://hannonlab.cshl.edu/fastx_toolkit/)以及prinseq,预先对群组化的sag读段进行过滤处理,去除低品质的读段(50%以上的碱基的品质得分<25),修剪低品质(品质得分<20)读段的3个末端,去除短读取(<20bp)以及1%的碱基未被识别的读段,丢弃预
先过滤处理后未配对的读段。之后,以options-careful-disable-rr-sc使用spades-3.9.0,从原始sag读段新组装各个重叠群。最后,为了交叉参考映射而收集500bp以上的原始sag重叠群。
487.(通过交叉参考映射除去嵌合体读段(ccsag的工序2))
488.通过bwa将源自一个sag的品质管理后的读段映射到由相同群组内的其他sag构建的多个原始重叠群。当对参考重叠群的完全比对为与部分比对(soft clipping)同等或其以上的频率时,读段视为干净的,但部分比对比完全比对频繁时,视为潜在嵌合体。接着,将潜在嵌合体分割为比对的片段和未比对的片段,接着,将它们再次映射至多个原始重叠群,然后,如记载那样进行再分类。最后,将完全未比对的读段以及短于20bp的片段化的嵌合体视为未被映射而丢弃。重复交叉参考映射和嵌合体分割的循环,直到检测不到部分比对的潜在嵌合读段为止。
489.(干净的sag与重叠群延伸的共同组装(ccsag的步骤3))
490.使用spade将源自各sag的干净的读段重新组装至干净的复合sag重叠群。同样地,将原始sag读段重新组装至原始复合sag重叠群。通过对原始复合体重叠群的blast映射来填埋干净的复合体重叠群之间的间隙。简而言之,为了使复合体重叠群干净,作为能够潜在使用的原始复合体重叠群,识别到99%以上的一致性。接着,将这种原始复合体重叠群回收到数据库中,通过blast来映射干净的复合体重叠群,基于所获得的比对来填埋间隙,由此生成实质上包括复合体的单一细胞基因组的、填埋了间隙的复合体sag重叠群。
491.sag组装的分析:用quast评价组装品质(gurevich a et al.,bioinformatics.2013apr 15;29(8):1072-5.)。针对细胞株的分析,将伴有f质粒和λ噬菌体序列的nc_00913(e.coli substrain mg1655)的基因组或nc_014479(bacillus subtilis subsp.spizizenii str.w23)的基因组的所有序列数据映射到ncbi参考基因组。为了对通过该实施例获得的未培养的细胞基因组进行分析,将交联的复合sag重叠群用作参考,鉴定潜在的错误组装,并确定各个sag的基因组部分。用checkm评价完整性(complete)和污染(contamination)(parks dh et al.,genome res.2015jul;25(7):1043-55.)。分类是通过amphora2或rnammer中的16s rdna序列的blast检索来进行(lagesen k et al.,nucleic acids res.2007;35(9):3100-8.)。利用kaas(moriya y et al.,nucleic acids res.2007jul;35(web server issue):w182-5.)和maple(takami h et al.,dna res.2016jul 3.pii:dsw030.)进行基因路径分析。另一方面,组装图由bandage(wick rr et al.,bioinformatics.2015oct 15;31(20):3350-2.)生成。关于snp的分析,对交联复合sag重叠群的编码序列映射各个单细胞扩增的基因组。接着,对核苷酸进行至少5个读段的覆盖深度的筛选。其中,99.9%的读段与参照不匹配,显示出均质的碱基(核酸序列)。之后,将相同谱系中包含多个匹配的sags和不匹配的sags双方的核苷酸部位鉴定为snps。
492.(实施例2)微生物基因组数据库的构建
493.图2为用于在本发明中使用的微生物基因组数据库结构。其中,示出由关系数据库系统构建微生物基因组数据库时的实施例。主要在由中央处理装置(cpu)、主存储装置、辅助存储装置、输入/输出装置、其他外围设备组成的计算机系统上安装操作系统和关系数据库管理系统。如图2所示,在关系数据库上创建用于保存草图基因组信息及与其对应的组装
碱基序列、基因信息、基因碱基序列、单一生物单元基因组信息及与其对应的组装碱基序列、基因信息、基因碱基序列、以及部分碱基序列信息的表格。关于组装碱基序列、基因碱基序列、部分碱基序列,还可采取实际数据保存在关系数据库之外并将对实际数据的参照保存在表格中的方式。在草图基因组信息表中创建用于保存草图基因组中附带的信息的列。例如,可以列举暂定谱系分类、完整率、污染率、品质类别、基因组大小、重叠群数量、n50值、gc含量等。暂定谱系分类是用专用分析工具(例如checkm等)分析组装碱基序列而获得的生物谱系分类的信息。由此,可鉴定草图基因组的大致谱系。品质类别用于表示草图基因组的状态。优选为符合国际标准等的类别。在草图基因组的基因信息表中创建用于保存草图基因组的基因中附带的信息的列。例如,可以列举基因名称、基因长度、蛋白质家族、gc含量、标记物类型、单拷贝等。蛋白质家族是通过与蛋白质数据库之间的同源性分析等而获得的信息。标记物类型用于记载是否为用于在谱系分类等中使用的标记物。单拷贝用于表示基因是否为单拷贝基因。可利用图5的方法来鉴定单拷贝基因。单一生物单元基因组信息表中创建用于保存单一生物单元基因组中附带的信息的列。例如,可列举暂定谱系分类、完整率、污染率、基因组大小、重叠群数量、n50值、gc含量等。单一生物单元基因组的基因信息表中创建用于保存单一生物单元基因组的基因中附带的信息。例如,可列举基因名称、基因长度、蛋白质家族、gc含量、单拷贝等。除此之外的各碱基序列表中创建用于保存碱基序列(当作为外部文件时是其参照)的列。优选在各个表格中预先准备用于唯一识别数据的id列。草图基因组信息和单一生物单元基因组信息具有一对多的关系。基因组信息和组装碱基序列具有一对多的对应关系。基因组信息和基因信息具有一对多的关系。基因信息和基因碱基序列具有一对一的对应关系。单一生物单元基因组信息和部分碱基序列具有一对多的对应关系。
494.(实施例3)基于微生物基因组数据库的单一生物单元基因组的聚类化
495.图3示出在获得多个单一生物单元的部分碱基序列时将它们汇总为同一谱系的聚类的方法。例如,用illumina等的dna测序仪对单一生物单元的基因组dna进行分析,能够获得fastq等碱基序列文件。fastq文件中记载有部分碱基序列。将所获得的fastq文件保存至分析用计算机中。在fastq文件内的部分碱基序列中存在混存有接头序列的部分碱基序列或品质低的部分碱基序列。这些低品质的部分碱基序列预先使用fastqc等品质管理工具来删除。将该fastq文件用spades等组装工具进行组装,获得组装碱基序列。对于组装碱基序列,使用quast或checkm等评价工具来计算重叠群数量或基因组大小、完整率、污染率等评价值。接着,从组装碱基序列鉴定基因。基因的鉴定中使用prokka或dfast等基因注释工具。所鉴定的基因碱基序列可通过检索pfam等蛋白质数据库来获得蛋白质家族等功能信息。最后,使用checkm等可进行谱系分类的工具,预先计算暂定谱系分类信息。至此为止是对单一生物单元的部分碱基序列进行的前处理。
496.单一生物单元基因组分析结束之后,参考微生物基因组数据库的草图基因组信息表的暂定谱系分类,提取对应的草图基因组信息和基因信息。参考基因信息的标记物类型,获得生物谱系鉴定用序列。从单一生物单元基因组数据的基因信息提取具有与生物谱系鉴定用序列的蛋白质家族相同的蛋白质家族的基因。若没有对应的基因信息,则结束该处理,并移动至下一个处理。若存在对应的基因信息,则通过blast等的同源性分析工具对单元基因组数据的基因碱基序列和生物谱系鉴定用序列循环进行同源性检索。由于仅以具有某预
定阈值以上的同源性的配对作为对象,因此预定阈值以下(例如同源性70%以下)的配对被排除在外。检测在各生物谱系鉴定用序列中同源性最高的单元基因组数据的基因碱基序列,求出匹配的碱基序列长度和同源性的加权平均,作为两个基因组之间的相似度(距离)。万一检测出多个具有同一相似度的草图基因组时,不对生物谱系鉴定用序列而是对组装碱基序列之间循环进行同源性检索,进行与生物谱系鉴定用序列同样的处理,计算相似度。将相似度最高的草图基因组作为聚类化的基准。
497.(实施例4)单一生物单元基因组的聚类化
498.图4示出当在新测定的单一生物单元基因组数据中没有与微生物基因组数据库对应的草图基因组时用于创建同一谱系的聚类的方法。此处,设定已经通过实施例2中示出的前处理获得各单一生物单元基因组数据(部分碱基序列、基因组信息、组装碱基序列、暂定谱系分类、基因信息、基因碱基破裂)之后,推行实施例。根据暂定谱系分类,对同一谱系的单一生物单元基因组数据进行聚类化。通过使用checkm等工具来处理组装碱基序列,从而确定暂定谱系分类,但大多数情况下单一生物单元的组装碱基序列未覆盖全部基因组,因此谱系分类大多粗略,形成混存有各种谱系的生物的聚类的可能性较高。因此,对于使用注册在微生物基因组数据库中的生物谱系鉴定用序列是否不能将聚类中的单一生物单元基因组数据细分进行评价。图6示出对作为同一谱系的聚类的单一生物单元基因组数据进行细分化的方法。参考注册在微生物基因组数据库中的基因信息表格的标记物类型,获得生物谱系鉴定用序列和与其对应的蛋白质家族。通过与实施例2中示出的求出基因组之间的相似度的方法同样的方法,使用生物谱系鉴定用序列,对聚类中的单一生物单元基因组数据以循环方式计算相似度。通过作为统计处理软件的r等来读入此处求出的相似度矩阵并进行网络分析。在r的情况下,igraph函数可用作网络分析函数。接着,根据网络分析的输出来检测社群。在r中,社群检测安装有基于边介数中心性(edge betweenness centrality)的方法、基于随机移动(random walk)的方法、基于贪心算法(greedy algorithm)的方法、基于固有向量的方法、基于多阶段优化的方法、基于自旋玻璃法(spin-glass)的方法、基于标签传播法的方法、基于infomap法的方法等。基于所检测的社群,对单一生物单元基因组数据进行细分化。此处,示出细分化使用网络分析和社群检测的示例,但也可以考虑使用分层(非分层)聚类来进行细分化的方法。但是,在这种情况下,当在两个单一生物单元基因组数据之间没有用于计算相似度的共用的生物谱系鉴定用序列时,相似度成为缺失值而导致不能进行聚类分析。因此,总而言之,认为优选使用即使在存在缺失值的情况下也能够应对的网络分析和社群检测。
499.(实施例5)新生物谱系鉴定用序列的鉴定
500.图5示出根据注册在微生物基因组数据库中的草图基因组的基因信息来鉴定用于进行谱系分类的新生物谱系鉴定用序列的方法。使用注册在微生物基因组数据库中的草图基因组信息和草图基因组基因信息,创建与如图5所示的草图基因组对应的基因家族的频率表。频率表的表示1的单元表示在该草图基因组中为单拷贝基因。由于生物谱系鉴定用序列应为单拷贝基因,因此忽略除了单拷贝基因以外的单元。在各蛋白质家族中,计算存在于全部草图基因组中的单拷贝基因的比例。单拷贝基因覆盖越多的草图基因组,则作为生物谱系鉴定用序列越优异,因此,将蛋白质家族按照单拷贝基因的比例降序排序,将满足某基准值以上(例如90%以上的草图基因组中存在单拷贝基因)的蛋白质家族再次鉴定为生物
谱系鉴定用序列。或者,也可以将蛋白质家族按照单拷贝基因的比例降序排序,并将从排前列的任意数量的蛋白质家族作为生物谱系鉴定用序列。另外,在迄今为止的研究中,细菌等中已知的生物谱系鉴定用序列等也可与上述鉴定的生物谱系鉴定用序列组合使用。
501.另外,作为与上述不同的生物谱系鉴定用序列的创建方法,认为还可适用在d.h.parks,et.al.,2015中所提倡的方法。这是创建草图基因组的系统树并在各节点定义生物谱系鉴定用序列的方法,其作为checkm的输入数据来使用。
502.(实施例6)最佳草图基因组的构建
503.图7示出构建最佳草图基因组的方法。通过实施例2和实施例3,单一生物单元基因组数据被聚类化为同一谱系。在实施例2的聚类化中,基于微生物基因组数据库的草图基因组进行聚类化,因此从微生物基因组数据库获得与草图基因组对应的单一生物单元基因组的部分碱基序列并追加至聚类中。聚类中的单一生物单元基因组数据根据某基准值来排序。此处,按照污染率由低至高的顺序排序。但是,低于某预定水平的单一生物单元基因组数据被排除在外。此处,排除了完整率低于10%的单一生物单元基因组数据。接着,按照等级从高至低提取两个单一生物单元基因组数据,创建草图基因组构建用的暂定组。接着,按照等级从高至低提取3个单一生物单元基因组数据,同样地创建草图基因组构建用的暂定组。这样按照等级从高到低不断地追加单一生物单元基因组数据,并仅以比聚类中的单一生物单元基因组数据数量少1的数量来创建暂定草图基因组构建用组。对于这些暂定草图基因组构建用组,应用在m.kogawa,et.al.,2018中提出的cleaning and co-assembly of a single-cell amplified genome(ccsag)法,创建暂定草图基因组。暂定草图基因组能够以下述函数的形式来表示,所述函数以用于进行构建的单一生物单元基因组数据数量作为说明变量、并且以完整率或污染率等草图基因组评价值作为目标变量。由此,可以判断:草图基因组处于收敛状态,即使追加更多单一生物单元基因组数据,也不能预期品质提高等。不能预期品质古城的草图基因组由于不进行最佳草图基因组的构建等,因而可期待处理的高速化。选择暂定草图基因组和现有草图基因组中最高品质的草图基因组,当其不是现有草图基因组时,更新微生物基因组数据库的草图基因组。新鉴定为聚类的单一生物单元基因组数据注册在微生物基因组数据库的单一生物单元基因组数据中。
504.(实施例7)草图基因组的后处理
505.图8示出注册在微生物基因组数据库中的草图基因组的后处理方法。通过在实施例5中创建的对草图基因组的收敛状态进行评价的函数,可以从微生物基因组数据库提取处于收敛状态的草图基因组。将该收敛状态的草图基因组作为后处理对象。后处理方法存在如下两种模式。第一种为使用与所提取的草图基因组对应的单一生物单元基因组数据的方法。将所提取的单一生物单元基因组数据根据某基准值进行排序。此处,按照污染率由低到高的顺序排序。但是,低于某预定水平的单一生物单元基因组数据被排除在外。此处,排除了完整率低于10%的单一生物单元基因组数据。使用满足基准的全部单一生物单元基因组数据来实施改变了参数的ccsag。关于参数,设定能够检测稍长的组装碱基序列的参数。将此处所创建的草图基因组作为第一种后处理草图基因组。第二种为从满足基准的全部单一生物单元基因组数据的组装碱基序列中检测结合草图基因组的组装碱基序列的桥接组装碱基序列。通过blast等同源性分析工具,对草图基因组的组装碱基序列和满足基准的全部单一生物单元基因组数据的组装碱基序列以循环方式进行同源性分析。当获得草图基因
组的两个组装碱基序列的一端与单一生物单元基因组数据的一个组装碱基序列的两端匹配的结果时,单一生物单元基因组数据的组装碱基序列发挥结合草图基因组组装碱基序列的作用。将这样地通过成为桥接的组装碱基序列连接的草图基因组作为第二种后处理草图基因组。根据某基准值,将第一种后处理草图基因组和第二种后处理草图基因组与已注册的草图基因组进行比较,选择品质最高的草图基因组。当所选择的草图基因组不是现有草图基因组时,更新微生物基因组数据库的草图基因组。
506.(实施例8)草图基因组的细分化
507.图9示出将注册在微生物基因组数据库中的草图基因组系统地进行细分化的方法。通过在实施例5中创建的对草图基因组的收敛状态进行评价的函数,可以从微生物基因组数据库提取处于收敛状态的草图基因组。将该收敛状态的草图基因组作为细分化对象。将与所提取的草图基因组对应的单一生物单元基因组数据根据某基准值进行排序。此处,按照污染率由低到高的顺序排序。但是,低于某预定水平的单一生物单元基因组数据被排除在外。此处,排除了完整率低于10%的单一生物单元基因组数据。对于满足基准的全部单一生物单元基因组数据进行实施例3中所进行的图6的细分化。针对细分化的各聚类进行图7的最佳草图基因组构建,并获得各聚类的最佳草图基因组。根据某基准值,将该多个最佳草图基因组和已注册的草图基因组进行比较,选择在两者之中品质高的一方的草图基因组。当所选择的草图基因组不是现有草图基因组时,从微生物基因组数据库删除现有草图基因组,并新注册细分化后的草图基因组。
508.(实施例9)外部数据库的序列追加
509.通过利用外部的独立数据库,可如图12所示那样构建更高品质的基因组。例如,若1~4的sag为该项目的数据,则菌株2的基因组仅由一个sag构建。此处,只要能够追加如5及6那样的外部项目数据,菌株2就可以从3个sag构建草图基因组,因此可构建更高品质的基因组。
510.(实施例10)偏差均质化处理
511.目的和方法
512.(扩增)为了改善通过内含偏差的序列数据的组装而获得的基因组序列的品质,进行了偏差均质化处理。具体而言,基于针对参考基因组序列的序列读段的映射结果,除去一定量的确认到多个重复的序列部位的序列读段一定量,进行序列读段的偏差校正,从而进行均质化(图13)。
513.参考基因组序列中可利用通过已知的近缘生物种基因组或进行偏差均质化处理的序列数据本身的组装来创建的dna序列。通过进行均质化处理的序列数据的组装,可改善所取得的草图基因组互补率或序列片段数量。根据情况,使用从均质化序列数据创建的基因组序列作为参考序列,重复进行均质化处理,从而可预期基因组品质的进一步改善。
514.具体如下。
515.使用大肠杆菌k12株单细胞扩增基因组(sag)的nanopore序列数据(gridion)进行基因组组装。直接使用每个基因组区域的读取深度差异很大的序列数据(图14上部线图)进行了组装,结果未获得显示相对较小的读取深度的区域的基因组序列(图14下部带的间隙部分)。接着,将所组装的基因组序列作为参考序列来映射序列数据,以各区域的最大读取深度成为100
×
的方式选择性地进行读段除去,从而进行序列数据的均质化(图15上部线
图)。对均质化后的序列数据进行了再次组装,结果确认到在初始组装中未能获得的基因组区域的序列构建(图15下部带)。进而,将新组装的基因组序列作为参考序列,重复进行均质化―组装,由此,还确认到在基因组互补率提高的同时基因组序列片段数减少(表1)。
516.[表1]
[0517]
表1:均质化―组装循环次数和获得基因组序列的评价
[0518]
均质化―组装循环循环0循环1循环2循环3循环4序列片段数52511187大肠杆菌基因组互补率(%)70.53997.22498.90998.84598.916
[0519]
(注释)
[0520]
如上所述,使用本发明的优选实施方式来举例说明本发明,但可理解为本发明的范围应当仅由权利要求书来解释。可理解为在本说明书中引用的专利、专利申请及其他文献的内容本身应与其内容具体记载在本说明书中同样地作为对本说明书的参考而援用于此。本技术要求在2019年4月26日向日本专利局提出申请的日本特愿2019-85839的优先权,可理解为该申请的内容本身应与其内容具体记载在本说明书中同样地作为对本说明书的参考而援用于此。
[0521]
产业上的可利用性
[0522]
可实现微生物等单细胞数据处理的自动化。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。