新型fxr小分子激动剂制备及其用途
技术领域
1.本发明属于医药领域,涉及一类作为fxr激动剂的非甾体化合物的制备和用途。具体而言,涉及一类可作为fxr激动剂的有机小分子化合物及其对映异构体、非对映异构体、互变异构体、外消旋体、水合物、溶剂合物、前药或其药学上可接受的盐的制备方法以及其在制备治疗fxr相关疾病药物中的应用。
背景技术:
2.类法尼醇x受体(farnesoid x receptor)是核受体超家族的一员,属于配体依赖的核转录因子,其主要表达于肝脏、肠道、肾脏、胆管等系统;fxr因其可被内源性配体胆汁酸激活,参与胆汁酸代谢与胆固醇代谢等重要环节,故又称胆汁酸受体。fxr可直接参与调节包括脂代谢、糖代谢、炎症、纤维化、肝再生、细胞分化和增殖等生理过程在内的300多个基因的表达。在自然环境中其配体包括初级胆汁酸鹅脱氧胆酸、次级胆酸石胆酸、脱氧胆酸等。如,被内源性配体胆汁酸激活后的fxr在甘油三酯(triglyceride,tg)代谢过程中起着重要作用,fxr可通过调控与tg代谢的关键酶、脂蛋白和相应受体,从而使肝脏及循环血液中tg含量达到稳态平衡。所以,截至目前,已有多个fxr合成型配体分子在肝脏等代谢性疾病应用领域。
3.fxr激动剂分子在治疗肝脏疾病,如原发性胆汁性肝硬化(primary biliary cirrhosis,pbc),原发性硬化性胆管炎(primary sclerosing cholangitis,psc)和非酒精性脂肪肝(nonalcoholic steatohepatitis,nash)等方面,已呈现出优异临床效果。至目前,即将最先被批准上市的fxr激动剂分子奥贝胆酸(obeticholic acid,oca)已被证实可显著改善多种代谢性症状,如降低肝脂肪含量,减少炎症反应和抑制肝纤维化等。但,oca也日益凸显出诸多临床短板,如引起瘙痒,高密度脂蛋白(high-density lipoprotein cholesterol,hdlc)降低,低密度脂蛋白(low-density lipoprotein cholesterol,ldlc)升高等。因此,临床需求方面,急需出现新的临床效果好,毒副作用低的fxr激动剂分子。
4.此外,有研究证实,fxr与肿瘤的发生发展密切相关。在多种肿瘤中,fxr扮演着抑癌基因的角色。例如,在肝细胞癌和直肠癌中,fxr呈低表达状态,fxr活化后,通过抑制β-catenin的活性,显著抑制肝癌或直肠癌的进展。新近研究表明,在胆管癌中,fxr的激动剂oca能够显著抑制肝内胆管细胞的增殖、迁移及克隆形成等。
5.再者,fxr激动剂作可为一种新的抗病毒候选药物,有研究证实,fxr配体能够作为一种新的乙型肝炎病毒(hepatitis b virus,hbv)复制的抑制治疗策略。fxr激动剂可抑制hbv表面抗原合成,抑制hbv dna和rna的复制,最重要的是,可抑制hbv cccdna的产生。在丙型肝炎病毒(hepatitis c virus,hcv)方面,fxr激动剂gw4064可通过间接方式抑制hcv入侵肝组织细胞。所以,fxr的激动剂分子亦具有作为开发抗病毒药物的巨大前景。
6.综上所述,本领域尚缺乏制备方法简单,抑制效果好的新型fxr激动剂分子。
技术实现要素:
7.本发明的目的是提供一种制备方法简单,抑制效果好的新型fxr激动剂分子。
8.本发明的第一方面,提供了一种通式i所示的化合物,或其对映异构体、非对映异构体、互变异构体、外消旋体、水合物、溶剂合物、前药,或其药学上可接受的盐。
[0009][0010]
其中,
[0011]
ar选自下组:取代或未取代的c
6-c
10
芳基、取代或未取代的5-9元的杂芳环(包括单环或稠合环,含有1-3个选自氧、硫和氮中的杂原子);
[0012]
a选自下组:取代或未取代的c
6-c
10
芳基、取代或未取代的5-9元的杂芳环(包括单环或稠合环,含有1-3个选自氧、硫和氮中的杂原子);
[0013]
r1选自下组:取代或未取代的c
1-c6烷基、取代或未取代的c
3-c6环烷基、取代或未取代的5-9元的杂环(含有1-3个选自氧、硫和氮中的杂原子);
[0014]
x选自下组:氢或氘;
[0015]
其中,所述的取代指基团上的一个或多个氢原子各自独立地被选自下组的取代基所替代:卤素、卤代c
1-c6烷基、卤代c
1-c6烷氧基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基、c
3-c6环烷氧基、氰基或硝基。
[0016]
在另一优选例中,所述的r1选自下组:取代或未取代的c
1-c6烷基、取代或未取代的c
3-c6环烷基;其中,所述的取代指基团上的一个或多个氢原子各自独立地被选自下组的取代基所替代:卤素、卤代c
1-c6烷基、卤代c
1-c6烷氧基、c
1-c6烷基、c
1-c6烷氧基、c
3-c6环烷基、c
3-c6环烷氧基、氰基或硝基。
[0017]
在另一优选例中,所述的ar选自下组:取代或未取代的c
6-c
10
芳基、取代或未取代的5-9元的杂芳环,且所述的取代基选自下组:氢、氟、氯、溴、甲基、乙基、正丙基、异丙基、正丁基、异丁基、三氟甲基、或三氟甲氧基。
[0018]
在另一优选例中,所述的a选自下组:取代或未取代的c
6-c
10
芳基、取代或未取代的5-9元的杂芳环,其中,所述的芳基或杂芳基的取代基选自下组:氢、氟、氯、溴、甲基、乙基、正丙基、异丙基、正丁基、异丁基、三氟甲基、或三氟甲氧基。
[0019]
在另一优选例中,ar选自下组:取代或未取代的苯基、取代或未取代的5-7元的杂芳环(包括单环或稠合环,含有1-3个选自氧、硫和氮中的杂原子)。
[0020]
在另一优选例中,a选自下组:取代或未取代的苯基、取代或未取代的5-7元的杂芳环(包括单环或稠合环,含有1-3个选自氧、硫和氮中的杂原子)。
[0021]
在另一优选例中,a为取代或未取代的苯并噻唑。
[0022]
在另一优选例中,所述的ar或a各自独立地选自取代或未取代的选自下组的基团:苯环、吡啶环、嘧啶环、哒嗪环、嘧啶环、哒嗪环、呋喃环、噻吩环、吡咯环、噻唑环,或咪唑环。
[0023]
在另一优选例中,所述的r1选自下组:取代或未取代的c
1-c4烷基、取代或未取代的环丙基。
[0024]
在另一优选例中,所述的ar为取代或未取代的苯环。
[0025]
在另一优选例中,所述的ar选自下组:2,5-二氯苯基、2-甲基苯基、2-三氟甲基苯基、2-三氟甲氧基苯基。
[0026]
在另一优选例中,所述的r1选自下组:甲基、乙基、正丙基、异丙基、正丁基、异丁基、环丙基、环丁基或环戊基。
[0027]
在另一优选例中,所述的式(i)化合物具有如下式所示的结构:
[0028][0029]
在另一优选例中,所述的式(i)化合物具有如下式所示的结构:
[0030][0031]
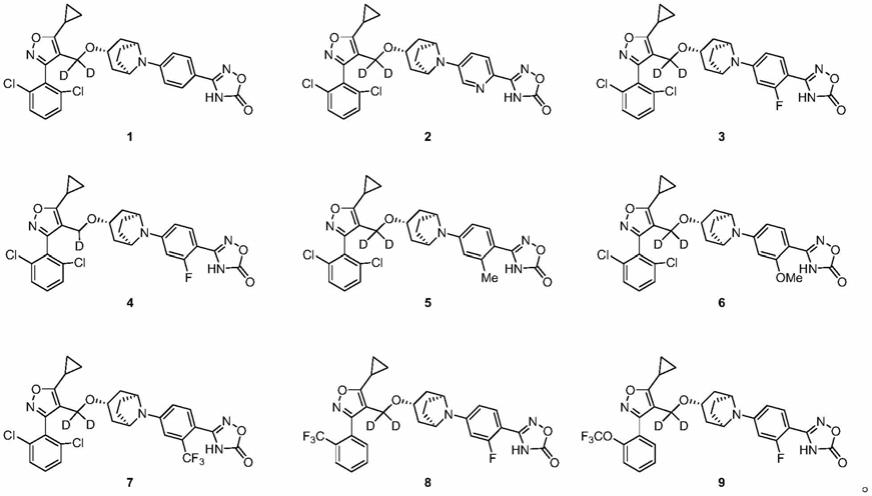
在另一优选例中,所述的化合物选自下组:
[0032][0033]
本发明的第二方面,提供了一种如本发明第一方面所述的化合物的制备方法,所述的方法包括:通过选自下组路线一或路线二所述的方法制备式i化合物:
[0034]
路线一:
[0035][0036]
(a)以取代苯甲醛通式ii所示的化合物为起始原料在碱的作用下与盐酸羟胺反应得到中间体后用n-氯代丁二酰亚胺(ncs)氯代后成通式iii所示的化合物;
[0037]
(b)然后将通式iii所示的化合物在碱的作用下与相应的3-氧代丙酸酯反应得到通式iv所示的化合物;
[0038]
(c)将通式iv所示的化合物中的酯在氘代还原剂的作用下还原成相应的醇即通式v所示的化合物;
[0039]
(d)将通式iv所示的化合物用溴试剂进行溴代后生成vi所示的化合物;
[0040]
(e)通式vi所示的化合物与vii所示的化合物在碱的作用下反应成通式viii所示的化合物;
[0041]
(f)通式viii所示的化合物在碱的作用下与盐酸羟胺反应生成通式ix所示的化合物;
[0042]
(g)通式ix所示的化合物在光气、三光气或者羰基二咪唑的作用下反应生成通式i所示的化合物,
[0043]
其中,x为氘;r1、ar、a的定义如本发明第一方面所述。
[0044]
路线二:
[0045][0046]
(a)通式iv所示的化合物中的酯在还原剂的作用下还原成相应的醇即通式x所示的化合物;
[0047]
(b)通式x所示的化合物在氧化剂的作用下氧化成相应的醛即通式xi所示的化合物;
[0048]
(c)通式xi所示的化合物中的酯在氘代还原剂的作用下还原成通式v所示的化合物;
[0049]
(d)将通式v所示的化合物用溴试剂进行溴代后生成vi所示的化合物;
[0050]
(e)通式vi所示的化合物与vii所示的化合物在碱的作用下反应成通式viii所示的化合物;
[0051]
(f)通式viii所示的化合物在碱的作用下与盐酸羟胺反应生成通式ix所示的化合物;
[0052]
(g)通式ix所示的化合物在光气、三光气或者羰基二咪唑的作用下反应生成通式i所示的化合物,
[0053]
各式中,x为氢;r1、ar、a的定义如本发明第一方面所述。
[0054]
在另一优选例中,通式vii所示的化合物通过以下步骤制备:
[0055][0056]
(k)通式xii所示的化合物与通式xiii所示的化合物在碱的作用下生成通式vii所示的化合物;
[0057]
各式中,a的定义如本发明第一方面所述。
[0058]
在另一优选例中,当产物存在光学异构体时,采用对应的光学构型的原料进行制备。
[0059]
本发明的第三方面,提供了一种药物组合物,其包含如本发明第一方面所述的通式i所示的化合物,或其对映异构体、非对映异构体、互变异构体、外消旋体、水合物、溶剂合
物、前药、药学上可接受的盐;和药学上可接受的载体。
[0060]
本发明的第四方面,提供了一种如本发明第一方面所述的通式i所示的化合物,或其对映异构体、非对映异构体、互变异构体、外消旋体、水合物、溶剂合物、前药或其药学上可接受的盐的用途,其用于制备治疗与fxr活性或表达量相关的疾病或病症的药物组合物。
[0061]
在另一优选例中,所述的fxr相关疾病选自下组:胆汁酸代谢、糖代谢、脂代谢、炎症、和/或肝脏纤维化过程相关疾病。
[0062]
在另一优选例中,所述fxr相关疾病为非酒精性脂肪肝(nash)、原发性胆汁性肝硬化(pbc)、原发性硬化性胆管炎(psc)、胆结石、非酒精性肝硬化、乙型肝炎(hbv)、丙型肝炎(hcv)、肝纤维化、胆汁淤积性肝病、高血脂症、高胆固醇血症或糖尿病。
[0063]
在另一优选例中,所述的药物组合物用作fxr激动剂。
[0064]
在另一优选例中,所述的药物组合物用于降低血清中alp、alt、ast、tba的水平。
[0065]
在另一优选例中,所述的药物组合物用于降低肝脏组织中羟脯氨酸的含量。
[0066]
在另一优选例中,所述的药物组合物用于下调肝脏组织中α-sma及col1α1mrna表达。
[0067]
在另一优选例中,所述的药物组合物用于抑制hbv表面抗原合成,抑制hbv dna和rna的复制,抑制hbv cccdna的产生。
[0068]
在另一优选例中,所述的药物组合物用于减少肝脏中胶原含量。
[0069]
在另一优选例中,所述的药物组合物是通过以下方法制备的:用式i化合物与可药用的辅料(例如赋形剂、稀释剂等)混合,配制成口服给药的片剂、胶囊剂、颗粒剂或糖浆剂等。
[0070]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
[0071]
本技术的发明人经过广泛而深入地研究,研发出一类可作为fxr激动剂的非甾体化合物,在分子水平和细胞水平对fxr均有激动能力,研究表明本技术的化合物能够降低血清中alp、alt、ast、tba的水平,降低肝脏组织中羟脯氨酸的含量,下调肝脏组织中
á-
sma及col1
á
1mrna表达,减少肝脏中胶原含量,抑制hbv表面抗原合成,抑制hbv dna和rna的复制,抑制hbv cccdna的产生。本发明的化合物具有fxr激动活性高、合成简单、原料易得等优点,能够用于制备用于治疗fxr相关疾病的药物。在此基础上,完成了本发明。
[0072]
术语
[0073]
在本发明中,除非特别指出,所用术语具有本领域技术人员公知的一般含义。
[0074]
在本发明中,所述卤素为f、cl、br或i。
[0075]
在本发明中,术语“c1-c6”是指具有1、2、3、4、5或6个碳原子,“c3-c6”是指具有3、4、5或6个碳原子,依此类推。
[0076]
在本发明中,术语“烷基”表示饱和的线性或支链烃部分,例如术语“c1-c6烷基”是指具有1至6个碳原子的直链或支链烷基,非限制性地包括甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基和已基等;优选乙基、丙基、异丙基、丁基、异丁基、仲丁基和叔
丁基。
[0077]
在本发明中,术语“烷氧基”表示-o-(c1-c6烷基)基团。例如术语“c1-c6烷氧基”是指具有1至6个碳原子的直链或支链烷氧基,非限制性地包括甲氧基、乙氧基、丙氧基、异丙氧基和丁氧基等。
[0078]
在本发明中,术语“环烷基”表示饱和的环状烃基部分,例如术语“c3-c6环烷基”是指在环上具有3至6个碳原子的环状烷基,非限制性地包括环丙基、环丁基、环戊基和环己基等。
[0079]
在本发明中,术语“环烷氧基”表示环烷基-o-,环烷基如上所述。
[0080]
在本发明中,术语“芳基”表示包含一个或多个芳环的烃基部分。芳基的例子包括但不限于苯基(ph)、萘基、芘基、芴基、蒽基和菲基。
[0081]
在本发明中,术语“杂芳基”表示包含一个或多个具有至少一个杂原子(例如n,o或s)的芳环的部分。杂芳基的例子包括呋喃基、吡咯基、噻吩基、噁唑基、咪唑基、噻唑基、吡啶基、嘧啶基、喹唑啉基、喹啉基、异喹啉基和吲哚基等。
[0082]
除非另外说明,本文所述的烷基、烷氧基、环烷基、环烷氧基、芳基和杂芳基为取代的和未取代的基团。烷基、烷氧基、环烷基、环烷氧基、芳基和杂芳基上可能的取代基包括,但不限于:羟基、氨基、硝基、腈基、卤素、c
1-c6烷基、c
2-c
10
烯基、c
2-c
10
炔基、c
3-c
20
环烷基、c
3-c
20
环烯基、c
1-c
20
杂环烷基、c
1-c
20
杂环烯基、c
1-c6烷氧基、芳基、杂芳基、杂芳氧基、c
1-c
10
烷基氨基、c
1-c
20
二烷基氨基、芳基氨基、二芳基氨基、c
1-c
10
烷基氨磺酰基、芳基氨磺酰基、c
1-c
10
烷基亚氨基、c
1-c
10
烷基磺基亚氨基、芳基磺基亚氨基、巯基、c
1-c
10
烷硫基、c
1-c
10
烷基磺酰基、芳基磺酰基、酰基氨基、氨酰基、氨基硫代酰基、胍基、脲基、氰基、酰基、硫代酰基、酰氧基、羧基和羧酸酯基。另一方面,环烷基、杂环烷基、杂环烯基、芳基和杂芳基也可互相稠合。
[0083]
本发明中,所述取代为单取代或多取代,所述多取代为二取代、三取代、四取代、或五取代。所述二取代就是指具有两个取代基,依此类推。
[0084]
本发明所述药学上可接受的盐可以是阴离子与式i化合物上带正电荷的基团形成的盐。合适的阴离子为氯离子、溴离子、碘离子、硫酸根、硝酸根、磷酸根、柠檬酸根、甲基磺酸根、三氟乙酸根、乙酸根、苹果酸根、甲苯磺酸根、酒石酸根、富马酸根、谷氨酸根、葡糖醛酸根、乳酸根、戊二酸根或马来酸根。类似地,可以由阳离子与式i化合物上的带负电荷的基团形成盐。合适的阳离子包括钠离子、钾离子、镁离子、钙离子和铵离子,例如四甲基铵离子。
[0085]
在另一优选例中,“药学上可接受的盐”是指式i化合物同选自下组的酸形成的盐类:氢氟酸、盐酸、氢溴酸、磷酸、乙酸、草酸、硫酸、硝酸、甲磺酸、胺基磺酸、水杨酸、三氟甲磺酸、萘磺酸、马来酸、柠檬酸、醋酸、乳酸、酒石酸、琥珀酸、酢浆草酸、丙酮酸、苹果酸、谷氨酸、对甲苯磺酸、萘磺酸、乙磺酸、萘二磺酸、丙二酸、富马酸、丙酸、草酸、三氟乙酸、硬酯酸、扑酸、羟基马来酸、苯乙酸、苯甲酸、谷氨酸、抗坏血酸、对胺基苯磺酸、2-乙酰氧基苯甲酸和羟乙磺酸等;或者式i化合物与无机碱形成的钠盐、钾盐、钙盐、铝盐或铵盐;或者通式i化合物与有机碱形成的甲胺盐、乙胺盐或乙醇胺盐。
[0086]
在另一优选例中,所述的化合物中,a环、ar、x和r1中任一个分别为实施例中所述具体化合物中所对应的基团。
[0087]
本发明的化合物具有不对称中心、手性轴和手性平面,并且可以以外消旋体、r-异构体或s-异构体的形式存在。本领域技术人员能够采用常规技术手段由外消旋体拆分获得r-异构体和/或s-异构体。
[0088]
制备方法
[0089]
本发明的通式i所示的化合物的制备方法,合成路线如下:
[0090]
路线一:
[0091][0092]
(a)以取代苯甲醛通式ii所示的化合物为起始原料在碱的作用下与盐酸羟胺反应得到中间体后用n-氯代丁二酰亚胺(ncs)氯代后成通式iii所示的化合物;
[0093]
(b)然后将通式iii所示的化合物在碱的作用下与相应的3-氧代丙酸酯反应得到通式iv所示的化合物;
[0094]
(c)将通式iv所示的化合物中的酯在氘代还原剂的作用下还原成相应的醇即通式v所示的化合物;
[0095]
(d)将通式v所示的化合物用溴试剂进行溴代后生成vi所示的化合物;
[0096]
(e)通式vi所示的化合物与vii所示的化合物在碱的作用下反应成通式viii所示的化合物;
[0097]
(f)通式viii所示的化合物在碱的作用下与盐酸羟胺反应生成通式ix所示的化合物;
[0098]
(g)通式ix所示的化合物在光气、三光气或者羰基二咪唑的作用下反应生成通式i所示的化合物,
[0099]
其中,x为氘,r1、ar、a的定义如权利要求1所述。
[0100]
路线二:
[0101][0102]
(h)通式iv所示的化合物中的酯在还原剂的作用下还原成相应的醇即通式x所示的化合物;
[0103]
(i)通式x所示的化合物在氧化剂的作用下氧化成相应的醛即通式xi所示的化合物;
[0104]
(j)通式xi所示的化合物中的酯在氘代还原剂的作用下还原成通式v所示的化合物;
[0105]
(d)将通式v所示的化合物用溴试剂进行溴代后生成vi所示的化合物;
[0106]
(e)通式vi所示的化合物与vii所示的化合物在碱的作用下反应成通式viii所示的化合物;
[0107]
(f)通式viii所示的化合物在碱的作用下与盐酸羟胺反应生成通式ix所示的化合物;
[0108]
(g)通式ix所示的化合物在光气、三光气或者羰基二咪唑的作用下反应生成通式i所示的化合物,
[0109]
各式中,x为氢,r1、ar、a的定义如本发明第一方面中所述。
[0110]
在另一优选例中,通式vii所示的化合物通过以下步骤制备:
[0111][0112]
(k)通式xii所示的化合物与通式xiii所示的化合物在碱的作用下生成通式vii所示的化合物;
[0113]
各式中,a的定义如本发明第一方面所述。
[0114]
药物组合物及其治疗用途
[0115]
本发明提供的化合物,可以单独使用,或者将其与可药用的辅料(例如赋形剂、稀释剂等)混合,配制成口服给药的片剂、胶囊剂、颗粒剂或糖浆剂等。该药物组合物可以按照制药学上常规方法制得。本发明的药物组合物包含安全有效量范围内的活性成分,以及药
学上可接受的载体。
[0116]
本发明所述的“活性成分”是指本发明所述的式i化合物。
[0117]
本发明所述的“活性成分”和药物组合物用于制备治疗fxr相关疾病的药物。本发明
[0118]
所述的“活性成分”和药物组合物可用作fxr激动剂。在另一优选例中,所述的活性成分可以用于制备预防和/治疗受fxr激动剂调节的疾病的药物。
[0119]“安全有效量”指的是:活性成分的量足以明显改善病情,而不至于产生严重的副作用。通常,药物组合物含有1-2000mg活性成分/剂,更佳地,含有10-200mg活性成分/剂。较佳地,所述的“一剂”为一个药片。
[0120]“药学上可接受的载体”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的活性成分以及它们之间相互掺和,而不明显降低活性成分的药效。药学上可以接受的载体部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
[0121]
本发明的活性成分或药物组合物的施用方式没有特别限制,代表性的施用方式包括5(但并不限于):口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)等。
[0122]
用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。
[0123]
用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性成分外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、矫味剂和香料。
[0124]
除了活性成分外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。
[0125]
用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。
[0126]
本发明化合物可以单独给药,或者与其他治疗药物(如降血脂药)联合给药。
[0127]
使用药物组合物时,是将安全有效量的本发明化合物适用于需要治疗的哺乳动物(如人),其中施用时剂量为药学上认为的有效给药剂量,对于60kg体重的人而言,日给药剂量通常为1~2000mg,优选20~500mg。当然,具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。
[0128]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件(如sambrook等人,分子克隆:实验室手册(new york:cold spring harbor laboratory press,1989)中所述的条件)或按照制造厂商所建议的条件。除非另外说明,否则百分比和
份数是重量百分比和重量份数。
[0129]
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
[0130]
所用仪器及主要实验材料如下:
[0131]
所用试剂和无水溶剂从中国商业公司购买,除特别说明,均直接使用;1h和
13
c nmr用brukeram-400型和varian mercury plus-400型核磁共振仪,质谱采用agilent6230型质谱仪,及200-300目柱层析硅胶(青岛海洋化工厂),hsgf254 tlc板(烟台市化工研究院)。
[0132]
本发明的化合物按照选自以下路线一、路线二的任一方法,选用合适的起始原料进行制备:
[0133]
路线一:
[0134][0135]
路线二:
[0136][0137]
实施例中间体vii-1合成:
[0138][0139]
将内向-8-氮杂双环[3.2.1]辛烷-3-醇xii(10g,78.7mmol)和对氟苯腈xiii-1(78.7mmol)溶于n,n-二甲基甲酰胺(150ml)中,室温条件下分批加入碳酸钾(197mmol),反应在80℃下过夜。加入乙酸乙酯(500ml)稀释反应液,用水洗涤,并用乙酸乙酯(每次300ml,共3次)萃取水相。混合有机相,用饱和食盐水洗涤,浓缩。经柱层析得到中间体vii-1(11g,收率61%)。1h nmr(400mhz,dmso-d6)δ7.51
–
7.48(m,2h),6.82
–
6.79(m,2h),4.62(s,1h),4.27(d,j=5.2hz,2h),3.78(s,1h),2.28(d,j=6.8hz,2h),1.91
–
1.85(m,4h),1.57(d,j=14.0hz,2h).ms(esi,m/z):229[m h]
。
[0140]
实施例化合物1的合成:
[0141][0142]
0℃下将碳酸钾水溶液(3n,182mmol)逐滴加入搅拌中的盐酸羟胺(182mmol)的乙醇(100ml)溶液中,2,6-二氯苯甲醛ii-1(20g,114mmol)溶于100ml乙醇中,然后加入到上述反应溶液中,将温度升高到90℃,反应两小时。等待反应液冷却到室温然后浓缩至固体。加入水/乙醇(1000ml/100ml)溶液搅拌打碎固体,过滤,50℃下真空干燥过夜,得到化合物中间体(18.4g)。将此中间体溶于n,n-二甲基甲酰胺(50ml),在0℃下逐滴加入n-氯代丁二酰亚胺(97mmol)的n,n-二甲基甲酰胺(100ml)溶液中,搅拌过夜。将反应液倒入0℃的冰水中,然后用甲基叔丁基醚(每次200ml,共3次)萃取,用饱和食盐水洗涤有机相,浓缩得到粗品。往装有粗品的烧瓶中加入正己烷(600ml),利用磁子搅拌,过滤,将固体在真空下(30℃)干燥得到中间体iii-1(18.3g,收率73%)。1h nmr(400mhz,cdcl3)δ7.43
–
7.39(m,2h),7.39
–
7.33(m,1h)。
[0143]
将三乙胺(8.2g)加入到3-环丙基-3-氧代丙酸甲酯(82mmol)中,搅拌30分钟。然后冷却到10℃,再将iii-1(18.3g,82mmol)的无水乙醇(80ml)溶液逐滴加入其中(内温不超过30℃),反应在室温下过夜。加入乙酸乙酯(100ml)稀释反应液,用水洗涤,并用乙酸乙酯(每次100ml,共3次)萃取水相。混合有机相,用饱和食盐水洗涤,浓缩。向浓缩物中加入100ml乙醚搅拌,真空下除去溶剂可得到固体产物iv-1(21.6g,收率84%)。1h nmr(400mhz,cdcl3)δ7.43
–
7.39(m,2h),7.39
–
7.33(m,1h),3.72(s,3h),2.21
–
2.09(m,1h),1.35
–
1.28(m,2h),1.25
–
1.18(m,2h);ms(esi,m/z):312[m h]
。
[0144]
将氘代四氢锂铝(7.3g)加入到四氢呋喃(200ml)中,冷却到0℃,再将iv-1(21.6g,69.2mmol)的四氢呋喃(50ml)逐滴加入其中(内温不超过5℃),反应液在室温下搅拌2h。0℃下加入冰水(9ml)淬灭反应,然后分别逐滴加入15%氢氧化钠水溶液(9ml)以及冰水(27ml),之后再加入无水硫酸镁(100g),将上述混合物在室温下搅拌0.5h,过滤,浓缩,经柱层析得到中间体v-1(19g,收率96%)。1h nmr(400mhz,dmso-d6)δ7.72
–
7.59(m,2h),7.58
–
7.50(m,1h),4.90(s,1h),2.38
–
2.26(m,1h),1.16
–
1.03(m,4h).ms(esi,m/z):286[m h]
。
[0145]
将v-1(19g,66.4mmol)溶于二氯甲烷(200ml)中,冷却到0℃,向溶液中缓慢滴加三溴化磷(66.4mmol),反应液在室温下搅拌2h。将反应液除去溶剂得到油状物,加入乙酸乙酯(100ml)稀释,用饱和碳酸氢钠水溶液调节反应溶液ph值为中性,用水洗涤,并用乙酸乙酯(每次100ml,共3次)萃取水相。混合有机相,用饱和食盐水洗涤之后浓缩。经柱层析得到中间体vi-1(20.2g,收率87%)。1h nmr(400mhz,dmso-d6)δ7.72
–
7.65(m,2h),7.64
–
7.56(m,1h),2.48
–
2.39(m,1h),1.26
–
1.10(m,4h).ms(esi,m/z):348[m h]
。
[0146]
在0℃下,向vii-1(1.96g,8.6mmol)的无水四氢呋喃(150ml)溶液中加入叔丁醇钾(21.4mmol),搅拌15分钟,然后逐滴加入vi-1(8.6mmol)的无水四氢呋喃(50ml)溶液,反应液在室温下搅拌4h。向反应液中加入水(200ml),用乙酸乙酯(每次200ml,共3次)萃取,有机相用饱和食盐水洗涤,浓缩,柱层析得到中间体viii-1(2.1g,收率49%)。1h nmr(400mhz,dmso-d6)δ7.73-7.63(m,2h),7.64
–
7.55(m,3h),6.83(d,j=8.4hz,2h),4.16(s,2h),3.37(s,1h),2.38
–
2.31(m,1h),1.83
–
1.74(m,6h),1.56
–
1.49(m,2h),1.16
–
1.06(m,4h).ms(esi,m/z):496[m h]
。
[0147]
将viii-1(2.1g,4.2mmol),盐酸羟胺(8.4mmol),无水乙醇(80ml)加入到圆底烧瓶中搅拌,缓慢滴加三乙胺(8.4mmol),加热至80℃反应过夜。冷却至室温,除去溶剂,用二氯甲烷(150ml)溶解,再用水,饱和食盐水洗涤,将有机相浓缩,经硅胶柱层析得到中间体ix-1(1.1g,收率50%)。1hnmr(400mhz,dmso-d6)δ7.73
–
7.63(m,2h),7.64
–
7.55(m,3h),6.83(d,j=8.4hz,2h),4.67(s,2h),3.43
–
3.35(m,1h),2.39
–
2.32(m,1h),1.89
–
1.78(m,6h),1.57
–
1.48(m,2h),1.16
–
1.06(m,4h).ms(esi,m/z):529[m h]
。
[0148]
将ix-1(1.1g,2.1mmol),n,n'-羰基二咪唑(3.1mmol),1,4-二氧六环(100ml)加入圆底烧瓶,然后加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(3.1mmol),加热至100℃反应8小时。将反应液冷却至室温,加水(100ml)稀释,用1m盐酸水溶液调节ph约等于3,然后用乙酸乙酯萃取(每次100ml,共3次)。合并有机相,用饱和食盐水洗涤,浓缩所得粗品再经硅胶柱层析得到终产物1(158mg,收率13%)。1hnmr(400mhz,dmso-d6)δ7.65
–
7.63(m,2h),7.59
–
7.55(m,3h),6.83(d,j=8.4hz,2h),4.17(s,2h),3.38(s,1h),2.37
–
2.30(m,1h),1.80
–
1.72(m,6h),1.51(d,j=14.4hz,2h),1.16
–
1.06(m,4h).ms(esi,m/z):555[m h]
。
[0149]
实施例2:
[0150][0151]
实施例2的制备从中间体vii-2出发通过路线1制备得到,合成路线如下:
[0152][0153]
从原料xiii-2出发按照合成中间体vii-1的合成方法合成化合物vii-2,然后通过路线1制备得到2,其中:
[0154]
白色固体viii-2收率77%,1hnmr(400mhz,dmso-d6)δ8.21(s,1h),7.70
–
7.62(m,3h),7.60
–
7.54(m,1h),7.16(d,j=8.4hz,1h),4.29(s,2h),3.43
–
3.35(m,1h),2.37
–
2.29(m,1h),1.85-1.65(m,6h),1.63
–
1.54(m,2h),1.17
–
1.05(m,4h).ms(esi,m/z):497[m h]
。
[0155]
白色固体2收率64%,1hnmr(400mhz,dmso-d6)δ8.20
–
8.16(m,1h),7.70(d,j=8.4hz,1h),7.67
–
7.62(m,2h),7.60
–
7.53(m,1h),7.28
–
7.21(m,1h),4.28(s,br,2h),3.42
–
3.40(m,1h),2.38
–
2.29(m,1h),1.81
–
1.69(m,6h),1.60
–
1.52(m,2h),1.16
–
1.05(m,2h).ms(esi,m/z):556[m h]
。
[0156]
实施例3:
[0157][0158]
实施例3的制备从中间体vii-3出发通过路线1制备得到,合成路线如下:
[0159][0160]
从原料xiii-3出发按照合成中间体vii-1的合成方法合成化合物vii-3,然后通过路线1制备得到3,其中:
[0161]
白色固体viii-3收率67%,hnmr(400mhz,dmso-d6)δ7.68
–
7.45(m,4h),6.69(t,j=12.0hz,2h),4.18(s,2h),3.46
–
3.36(m,1h),2.38
–
2.27(m,1h),1.80
–
1.66(m,6h),1.60
–
1.51(m,2h),1.20
–
1.03(m,4h).ms(esi,m/z):497[m h]
。
[0162]
白色固体3收率38%,1h nmr(400mhz,dmso-d6)δ7.69
–
7.42(m,4h),6.74(d,j=13.6hz,1h),6.63(d,j=8.8hz,1h),4.21(s,2h),3.47
–
3.43(m,1h),2.35
–
2.30(m,1h),1.76
–
1.70(m,6h),1.58
–
1.50(m,2h),1.20
–
1.06(m,4h).ms(esi,m/z):573[m h]
。
[0163]
实施例4:
[0164][0165]
实施例4的制备参考实施例3的操作,从中间体iv-1出发通过路线2制备得到4,合成路线如下:
[0166][0167]
将四氢锂铝(420mg,10mmol)加入到四氢呋喃(8ml)中,冷却到0℃,再将iv-1(2mmol)的四氢呋喃(2ml)逐滴加入其中(内温不超过5℃),反应液在室温下搅拌2h。0℃下加入冰水(0.4ml)淬灭反应,然后分别逐滴加入15%氢氧化钠水溶液(0.4ml)以及冰水(1.2ml),之后再加入无水硫酸镁(8g),将上述混合物在室温下搅拌0.5h,过滤,浓缩,经柱层析得到中间体x-1(1g,收率84%)。ms(esi,m/z):284[m h]
。
[0168]
将x-1(1g,3.53mmol)加入到二氯甲烷(20ml)中,室温下加入氯铬酸吡啶盐(14.14mmol),反应液在室温下搅拌1h。过滤,浓缩,经柱层析得到中间体xi-1(870mg,收率88%)。ms(esi,m/z):282[m h]
。
[0169]
将四氢锂铝(260mg,6.2mmol)加入到四氢呋喃(4ml)中,冷却到0℃,再将xi-1(3.1mmol)的四氢呋喃(1ml)逐滴加入其中(内温不超过5℃),反应液在室温下搅拌2h。0℃下加入冰水(0.2ml)淬灭反应,然后分别逐滴加入15%氢氧化钠水溶液(0.2ml)以及冰水(0.6ml),之后再加入无水硫酸镁(10g),将上述混合物在室温下搅拌0.5h,过滤,浓缩,经柱层析得到中间体v-2(730mg,收率83%)。1h nmr(400mhz,dmso-d6)δ7.62-7.60(m,2h),7.56-7.52(m,1h),4.92(d,j=5.2hz,1h),4.18(d,j=5.2hz,1h),2.35
–
2.28(m,1h),1.14
–
1.04(m,4h).ms(esi,m/z):285[m h]
。
[0170]
将v-2(730mg,2.57mmol)溶于二氯甲烷(10ml)中,冷却到0℃,向溶液中缓慢滴加三溴化磷(3.08mmol),反应液在室温下搅拌2h。将反应液除去溶剂得到油状物,加入乙酸乙酯(20ml)稀释,用饱和碳酸氢钠水溶液调节反应溶液ph值为中性,用水洗涤,并用乙酸乙酯(每次100ml,共3次)萃取水相。混合有机相,用饱和食盐水洗涤之后浓缩。经柱层析得到中间体vi-2(720mg,收率81%)。ms(esi,m/z):347[m h]
。
[0171]
在0℃下,向vii-2(512mg,2.08mmol)的无水四氢呋喃(10ml)溶液中加入叔丁醇钾(4.16mmol),搅拌15分钟,然后逐滴加入vi-2(2.08mmol)的无水四氢呋喃(5ml)溶液,反应液在室温下搅拌4h。向反应液中加入水(20ml),用乙酸乙酯(每次20ml,共3次)萃取,有机相
用饱和食盐水洗涤,浓缩,柱层析得到中间体viii-4(420mg,收率39%)。ms(esi,m/z):513[m h]
。
[0172]
将viii-4(420m g,0.82mmol),盐酸羟胺(1.64mmol),无水乙醇(5ml)加入到圆底烧瓶中搅拌,缓慢滴加三乙胺(1.64mmol),加热至80℃反应过夜。冷却至室温,除去溶剂,用二氯甲烷(20ml)溶解,再用水,饱和食盐水洗涤,将有机相浓缩,经硅胶柱层析得到中间体ix-4(360m g,收率81%)。ms(esi,m/z):546[m h]
。
[0173]
将ix-4(360mg,0.66mmol),n,n'-羰基二咪唑(0.99mmol),1,4-二氧六环(5ml)加入圆底烧瓶,然后加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(0.99mmol),加热至100℃反应8小时。将反应液冷却至室温,加水(10ml)稀释,用1m盐酸水溶液调节ph约等于3,然后用乙酸乙酯萃取(每次10ml,共3次)。合并有机相,用饱和食盐水洗涤,浓缩所得粗品再经硅胶柱层析得到终产物4(76.7mg,收率20%)。1hnmr(400mhz,dmso-d6)δ7.65-7.62(m,2h),7.58
–
7.55(m,1h),7.47(t,j=8.6hz,1h),6.72
–
6.65(m,2h),4.22(s,1h),4.17(s,2h),3.39(s,1h),2.36
–
2.30(m,1h),1.76-1.73(m,6h),1.52(d,j=14.8hz,2h),1.14
–
1.08(m,4h).ms(esi,m/z):572[m h]
。
[0174]
实施例5:
[0175][0176]
实施例5的制备从中间体vii-4出发通过路线1制备得到,合成路线如下:
[0177][0178]
从原料xiii-4出发按照合成中间体vii-1的合成方法合成化合物vii-4,然后通过路线1制备得到5,其中:
[0179]
白色固体viii-5收率47%,1hnmr(400mhz,dmso-d6)δ7.68
–
7.61(m,2h),7.60
–
7.55(m,1h),7.42(d,j=8.4hz,1h),6.71(s,br,1h),6.62(d,j=8.8hz,1h),4.17(s,2h),3.41
–
3.36(m,1h),2.39
–
2.26(m,4h),1.84
–
1.66(m,6h),1.58
–
1.46(m,2h),1.19
–
1.02(m,4h).ms
(esi,m/z):510[m h]
。
[0180]
白色固体3收率11%,1h nmr(400mhz,dmso-d6)δ7.69
–
7.62(m,2h),7.60
–
7.35(m,2h),6.75
–
6.545(m,2h),4.19(s,2h),3.43
–
3.33(m,1h),2.41
–
2.29(m,4h),1.88
–
1.66(m,6h),1.60
–
1.46(m,2h),1.21
–
1.03(m,4h).ms(esi,m/z):569[m h]
。
[0181]
实施例6:
[0182][0183]
实施例6的制备从中间体vii-5出发通过路线1制备得到,合成路线如下:
[0184][0185]
从原料xiii-5出发按照合成中间体vii-1的合成方法合成化合物vii-5,然后通过路线1制备得到6,其中:
[0186]
白色固体viii-6收率32%,1hnmr(400mhz,dmso-d6)δ7.66
–
7.61(m,2h),7.60
–
7.52(m,1h),7.32(d,j=8.4hz,1h),6.39
–
6.29(m,2h),4.22(s,2h),3.83(s,3h),3.41
–
3.32(m,1h),2.36
–
2.28(m,1h),1.84
–
1.66(m,6h),1.60
–
1.49(m,2h),1.20
–
1.02(m,4h).ms(esi,m/z):526[m h]
。
[0187]
白色固体3收率9%,1h nmr(400mhz,dmso-d6)δ7.69
–
7.52(m,3h),7.37
–
7.29(m,1h),6.45
–
6.36(m,2h),4.28(s,2h),3.87(s,3h),3.43
–
3.32(m,1h),2.42
–
2.27(m,1h),1.87
–
1.64(m,6h),1.61
–
1.47(m,2h),1.21
–
1.02(m,4h).ms(esi,m/z):585[m h]
。
[0188]
实施例7:
[0189]
[0190]
实施例7的制备从中间体vii-6出发通过路线1制备得到,合成路线如下:
[0191][0192]
从原料xiii-6出发按照合成中间体vii-1的合成方法合成化合物vii-6,然后通过路线1制备得到7,其中:
[0193]
白色固体viii-7收率56%,1hnmr(400mhz,dmso-d6)δ7.75(d,j=9.2hz,1h),7.67
–
7.52(m,3h),7.08(s,1h),7.02(d,j=8.8hz,1h),4.32(s,2h),3.43
–
3.32(m,1h),2.36
–
2.28(m,1h),1.78
–
1.69(m,3h),1.61
–
1.50(m,2h),1.16
–
1.06(m,4h).ms(esi,m/z):564[m h]
。
[0194]
白色固体7收率49%,1h nmr(400mhz,dmso-d6)δ7.67
–
7.47(m,4h),7.11
–
7.04(m,2h),4.27(s,2h),3.48
–
3.44(m,1h),2.35
–
2.31(m,1h),1.85
–
1.69(m,6h),1.62
–
1.48(m,2h),1.20
–
1.01(m,4h).ms(esi,m/z):623[m h]
。
[0195]
实施例8:
[0196][0197]
实施例8的制备从原料ii-2出发通过路线1制备得到,合成路线如下:
[0198][0199]
其中:
[0200]
胶体v-3收率98%,1hnmr(400mhz,dmso-d6)δ7.88(d,j=7.6hz,1h),7.81
–
7.71(m,2h),7.59(d,j=7.2hz,1h),4.90(s,1h),2.33-2.26(m,1h),1.12-1.05(m,4h).ms(esi,m/z):286[m h]
。
[0201]
白色固体viii-8收率31%,1hnmr(400mhz,dmso-d6)δ7.90(d,j=7.6hz,1h),7.81-7.72(m,2h),7.58(d,j=7.6hz,1h),7.50(t,j=8.4hz,1h),6.74(dd,j=13.8,2.2hz,1h),6.63(dd,j=8.6,2.2hz,1h),4.20(s,1h),3.61
–
3.57(m,1h),3.40
–
3.32(m,1h),2.33
–
2.29(m,1h),1.76-1.69(m,6h),1.54(d,j=14.4hz,2h),1.18
–
1.05(m,4h).ms(esi,m/z):514[m h]
。
[0202]
白色固体8收率29%,1hnmr(400mhz,dmso-d6)δ7.90(d,j=8.0hz,1h),7.81
–
7.72(m,2h),7.58(d,j=7.2hz,1h),7.47(t,j=8.6hz,1h),6.72
–
6.66(m,2h),4.17(s,2h),3.38(s,1h),2.34
–
2.28(m,1h),1.77
–
1.73(m,6h),1.52(d,j=14.8hz,2h),1.13
–
1.05(m,4h).ms(esi,m/z):573[m h]
。
[0203]
实施例9:
[0204][0205]
实施例9的制备从原料ii-3出发通过路线1制备得到,合成路线如下:
[0206][0207]
其中:
[0208]
胶体v-4收率76%,1hnmr(400mhz,dmso-d6)δ7.67-7.63(m,2h),7.53(d,j=6.8hz,2h),4.96(s,1h),2.32-2.27(m,1h),1.13-1.04(m,4h).ms(esi,m/z):302[m h]
。
[0209]
胶体viii-9收率47%,1hnmr(400mhz,dmso-d6)δ7.69
–
7.61(m,2h),7.56
–
7.49(m,3h),6.75(d,j=14.0hz,1h),6.64(d,j=8.8hz,1h),4.21(s,2h),3.45
–
3.43(m,1h),2.36
–
2.29(m,1h),1.77
–
1.69(m,6h),1.56(d,j=14.8hz,2h),1.23
–
1.06(m,4h).ms(esi,m/z):530[m h]
。
[0210]
白色固体8收率21%,1hnmr(400mhz,dmso-d6)δ7.69
–
7.62(m,2h),7.56
–
7.46(m,3h),6.72
–
6.66(m,2h),4.18(s,2h),3.43(s,1h),2.35
–
2.31(m,1h),1.80
–
1.75(m,6h),1.54(d,j=14.4hz,2h),1.13
–
1.06(m,4h).ms(esi,m/z):589[m h]
。
[0211]
药理实验实施例:
[0212]
基于报告基因活性检测的方法检测化合物的fxr激动活性的方法:
[0213]
1.1质粒pgal4-fxr-lbd和pg5-luc的构建及制备
[0214]
报告基因检测系统所用pgal4-fxr-lbd和pg5-luc质粒按常规分子克隆方法进行构建。主要步骤为:利用pcr技术将fxr-lbd(212-476aa)氨基酸序列对应的fxr(nm_001206979.2)cdna序列,插入pgal4载体bamhi和noti酶切位点,获得pgal4-fxr-lbd;pg5-luc和phrl-tk质粒获赠于中国科学院上海药物研究所;利用cacl2法将质粒转化dh5α大肠杆菌,进一步培养扩增后用质粒抽提试剂盒(tiangen,#d107)纯化获得相应质粒dna。
[0215]
1.2质粒共转染hek293t细胞及化合物处理
[0216]
质粒转染前一天将hek293t细胞以1
×
104/well的密度接种于96孔板。按照转染试剂hd(promega,#e2311)的说明书进行细胞转染。主要步骤为:以一个孔为例,将
质粒pgal4-fxr-lbd、pg5-luc和phrl-tk按20ng、50ng和5ng的比例加入10ul的opti-mem
tm
i培养基(gibco,#11058021)中混匀;再加入0.25ul的hd,混匀后室温静置5min;再将此10ul混合物加入至含100ul培养液的细胞孔中。细胞共转染后6h,将化合物以1um为最高浓度,以3倍梯度进行稀释,共分10个浓度加入细胞培养液中进行处理24h,共分2个复孔,以ljn452化合物为阳性对照。
[0217]
1.3dual-glo luciferase检测
[0218]
细胞经化合物处理24h后,按luciferase assay system(promega,#e2940)说明书进行检测。主要步骤为:每孔吸弃50ul培养液,再加入50ule2940)说明书进行检测。主要步骤为:每孔吸弃50ul培养液,再加入50ulluciferase试剂,室温振荡10min;取80ul裂解反应液至白色不透光optiplate-96孔板,用md i3x多功能酶标仪检测萤火虫萤光素酶(firefly luciferase)的发光信号值(firefly-luc);再加入40ulstop&试剂,室温振荡10min;再用md i3x多功能酶标仪检测海肾萤光素酶(renilla luciferase)的发光信号值(renilla-luc)。以firefly-luc/renilla-luc的比值作为化合物对fxr的激活活性,并以溶剂dmso组的比值进行归一化处理,使用graphpad prism6.0软件以四参数拟合剂量-反应曲线,计算ec
50
值。
[0219]
2.结果
[0220]
实验数据表明化合物均有一定的fxr激动活性,其中实施例1,2,3,4的ec
50
均小于5nm,具有非常强的fxr激动活性。其他实施例fxr激动活性数据见表1。
[0221]
表1化合物的fxr激动活性
[0222][0223][0224]
****:ec
50
(nm)《5;***:5《ec
50
(nm)《10;**:10《ec
50
(nm)《50;*:50《ec
50
(nm)。
[0225][0226]
结果显示,本发明的化合物体现出优于现有fxr激动剂化合物ljn452以及无氘代的对照品1的细胞水平活性。特别是本技术实施例3化合物是在无氘代对照品1的结构基础上进行氘代,活性即出现了显著提高,提示该位置为此类化合物的关键氘代位点。
[0227]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
