一种跨物种编码多肽sorf的预测方法
技术领域
1.本发明具体涉及一种跨物种编码多肽sorf的预测方法,属于生物信息领域。
背景技术:
2.小开放阅读框(smallopenreadingframes,sorfs)是长度小于100个氨基酸的dna序列,许多生物的基因组中均存在sorfs。在过去的十多年里,由于sorfs序列长度短,表达水平低,且相关资料较少,研究人员往往在基因组注释中忽略了sorf。随着测序技术的发展,人们发现许多sorfs也能够编码蛋白质,且普遍存在于基因组各个区域。2016年,德克萨斯大学eric教授等人发现,由sorf编码的小蛋白质dworf对心肌收缩功能具有不可忽视的作用,这引起了研究人员对sorfs的重新思考与认识。
3.近年来,在细菌、酵母、人类中均能检测到由sorf编码的小蛋白质,这些小蛋白质在胚胎发育、肌肉功能、细胞凋亡等生命活动中发挥重要作用。因此,肽编码sorfs逐渐成为生物学领域的一个研究热点。然而传统序列分析方法,如基因组测序、转录组测序、蛋白质组测序(质谱分析)等手段在sorfs识别中表现甚微。近几年,核糖体谱成为继质谱分析等传统测序方法的新技术,并被广泛用于分类识别序列能否编码蛋白质,其主要内容是通过核糖体印记技术分析蛋白质的合成情况,但有证据显示许多ncrna也能与核糖体结合,因此仅靠传统的测序方法分类识别肽编码sorf远远不够。目前,已有许多工具可用于区分编码rna和ncrna,例如:cpat、cnci、plek、cpc2、cppred、lgc、mepiped、deepcpp以及cppred-sorf等。这些方法均建立在一定数据集之上,且能较好地区分“普通”长度的编码rna和ncrna,但对于分类识别肽编码sorf的准确性却不高。因此,发展有效的sorf分类识别技术意义重大。
技术实现要素:
4.本发明的目的在于克服现有背景技术中的不足,提供一种跨物种编码多肽sorf的预测方法,解决样本数量不平衡的问题,便于提取稳健、高效的dna序列特征。
5.为达到上述目的,本发明是采用下述技术方案实现的:
6.一种跨物种编码多肽sorf(smallopenreadingframe,小开放阅读框)的预测方法,包括以下步骤:
7.将非编码序列产生策略应用于多个物种的肽编码sorfs的数据集,分别得到与之对应的非编码sorfs数据集;
8.将各物种的肽编码sorfs和非编码sorfs分别去冗余(max-relevanceandmin-redundancy,mrmr),得到各物种相应的正负样本,构建训练集和测试集;
9.提取各数据集中相应的特征参数;结合mrmr策略和增量选择方法选取表现较好的特征,构建相应特征集;
10.构建基于向量机(supportvectormachine,svm)肽编码sorfs的预测模型,将训练集的特征集用于模型训练;利用贪婪的网格搜索方法对训练模型进一步优化,分别得到
指定参数范围内的最佳预测模型;
11.利用预测模型对测试集进行预测,分析各数据集预测结果,比较评估不同特征选取策略的预测效率,得到表现最好的特征集和预测模型作为最佳的特征集和预测模型。
12.进一步的,从sorf数据库中下载人和小鼠的编码序列(coding sequence, cds),从tair数据库中下载拟南芥的cds,从ncbi数据库中下载部分原核生物基因组的cds;根据数据过滤策略滤除“错误”序列,得到多个物种的肽编码sorfs的数据集。
13.进一步的,“错误”序列过滤策略:
14.滤除sorf长度≥100aa;
15.滤除序列长度不能被3整除的sorf;
16.滤除以终止密码子开头的sorf;
17.滤除不以终止密码子结尾的sorf;
18.滤除序列中带有终止密码子的sorf;
19.进一步的,非编码序列产生策略为:
20.固定起始密码子和终止密码子,随机打乱每个正sorf序列;
21.确保在序列末端的终止密码子之前没有任何终止密码子;
22.进一步的,去冗余方法为:
23.通过cdhit程序,将各物种的肽编码sorfs和非编码sorfs分别去冗余,得到各物种相应的正负样本,构建训练集和测试集;
24.去冗余阈值设为0.80,滤除相似度大于80%的dna序列;
25.进一步的,根据9种不同的特征选取策略,提取各数据集中相应的特征参数;所述9种不同的特征选取策略分别为:cppred、2mer、3mer、tn、itn、cylindrical、spherical、codon、amino。
26.进一步的,利用pycharm软件提取各数据集中dna序列的特征参数;利用pycharm软件pymrmr包实现mrmr策略和增量选择方法,完成特征排序,构建相应的特征集。
27.进一步的,网格搜索方法的参数设置:
28.cmin:惩罚参数c的变化范围的最小值;默认为-5;
29.cmax:惩罚参数c的变化范围的最大值;默认为5;
30.gmin:参数g的变化范围的最小值;默认为-5;
31.gmax:参数g的变化范围的最大值;默认为5;
32.v:交叉验证的参数;默认为3;
33.cstep:参数c步进的大小;默认为1;
34.gstep:参数g步进的大小;默认为1;
35.accstep:最后显示准确率图时的步进大小;默认为1.5。
36.进一步的,利用matlab的libsvm包,利用预测模型对测试集进行跨物种预测,根据sn、sp、acc、mcc,4个指标分析预测结果,对预测模型进行比较评估,将表现最好的特征集和预测模型作为跨物种编码多肽sorf的预测方法的特征集和预测模型。
37.进一步的,评估指标计算公式如下:
38.[0039][0040][0041][0042]
其中,sn为灵敏度,sp为特异度,acc为准确率,mcc为马修斯相关系数,tp为真正例,fn为假负例,tn为真负例,fp假正例。
[0043]
有益效果
[0044]
本发明提出的一种跨物种编码多肽sorf的预测方法,基于高通量测序数据,对人、小鼠、拟南芥以及部分原核生物基因组的肽编码sorfs进行整合筛选,结合严格的非编码sorfs产生策略,解决样本数量不平衡的问题,便于提取稳健、高效的dna序列特征;
[0045]
本发明基于支持向量机(svm),利用最大相关最小冗余(mrmr)策略和增量选择方法筛选dna序列特征参数,构建预测的最佳特征集,有助于分类识别肽编码sorfs,发展一种跨物种编码多肽sorf的预测方法,对肽编码sorfs 的研究和基因注释有重要意义。
附图说明
[0046]
图1为本发明实施例一种跨物种编码多肽sorf的预测方法的数据筛选策略;
[0047]
图2是本发明实施例一种跨物种编码多肽sorf的预测方法的流程图。
具体实施方式
[0048]
下面结合附图对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
[0049]
如图1所示,为本发明实施例一种跨物种编码多肽sorf的预测方法的数据筛选策略,包括如下步骤:
[0050]
步骤1)从sorf数据库中下载人和小鼠的cds,从tair数据库中下载拟南芥的cds,从ncbi数据库中下载部分原核生物基因组的cds;根据数据过滤策略滤除“错误”序列,得到各物种的肽编码sorfs的数据集;
[0051]
本文构建了两个训练集:原核混合训练集(tr1)和真核混合训练集(tr2); 8个测试集:拟南芥测试集1(ara1)、拟南芥测试集2(ara2)、人类测试集 1(hum1)、人类测试集2(hum2)、小鼠测试集1(mou1)、小鼠测试集2 (mou2)、原核测试集(pro)、大肠杆菌测试集(bac)。其中,tr2是从ara1、 hum1和mou1三个数据集中分别随机抽取10%的序列组成的,ara1、hum1和 mou1剩下的90%序列分别构成ara2、hum2和mou2。
[0052]
为构建tr1,从ncbi下载nc_009089、nc_003103、nc_012962、 nc_000913、nc_008380的cds序列,保留长度小于等于100aa且具有明确功能的sorfs;为构建ara1,hum1和mou1,从tair数据库下载了2888个拟南芥sorf,从sorf数据库分别下载了10000个人类sorf和10000个小鼠sorf;为构建pro_6318-6318数据集,从56个原核基因组(表1)中筛选具有明确功能的sorf,其中,56个选定原核基因组的基因组gc含量为20%~70%; bac_150-53数
据集是由hemm等人发布的经实验验证的数据集。
[0053]
表1 56个原核基因组
[0054]
[0055][0056]
根据数据过滤策略,滤除下载数据中的“错误”序列,得到各物种的肽编码sorfs的数据集;
[0057]“错误”序列过滤策略:
[0058]
(i)滤除sorf长度≥100aa;
[0059]
(ii)滤除序列长度不能被3整除的sorf;
[0060]
(iii)滤除以终止密码子开头的sorf;
[0061]
(iv)滤除不以终止密码子结尾的sorf;
[0062]
(v)滤除序列中带有终止密码子的sorf;
[0063]
步骤2)将非编码序列产生策略应用于步骤1中各物种肽编码sorfs的数据集,分别得到与之对应的非编码sorfs数据集;
[0064]
非编码序列产生策略:
[0065]
(i)固定起始密码子和终止密码子,随机打乱每个正sorf序列;
[0066]
(ii)确保在序列末端的终止密码子之前没有任何终止密码子;
[0067]
步骤3)通过cdhit程序,将各物种的肽编码sorfs和非编码sorfs分别去冗余,得到各物种相应的正负样本(表2),构建训练集和测试集;
[0068]
去冗余阈值设为0.80,滤除相似度大于80%的dna序列;
[0069]
表2数据集统计
[0070][0071]
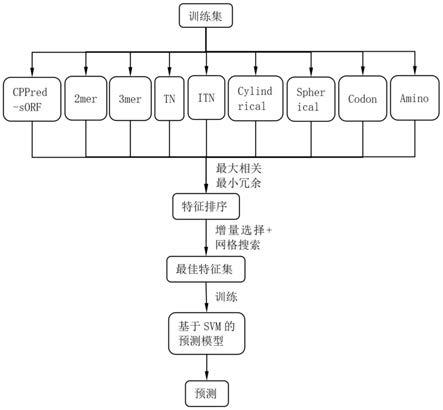
图2是本发明实施例一种跨物种编码多肽sorf的预测方法的流程图,包括如下步骤:
[0072]
步骤4)根据9种特征选取策略(cppred、2mer、3mer、tn、itn、cylindrical、spherical、codon、amino),利用pycharm软件提取各数据集中dna序列的特征参数;利用pycharm软件pymrmr包实现mrmr策略和增量选择方法,完成特征排序,构建相应的特征集;
[0073]
(i)9种特征选取策略:
[0074]
cppred:cppred-sorf为针对cppred进行改进,可用于预测sorf编码潜力。在本项工作中,我们提取cppred-sorf预测工具中涉及到的所有特征,包括序列长度(length)、覆盖率(coverage)、完整性(intergrity)、fickett得分、hexamer得分、预测肽的等电点(pi)、预测肽的亲水性总平均值(gravy)、预测肽的不稳定性(instability)、全局描述符(ctd)、gc含量和11个注意密码子的含量(uac、aac、uau、auc、uuc、gag、aag、gau、gac、 aau和gug),共计40个特征。其中,ctd一共含有30个特征:c表示核苷酸组成,有4个特征,描述了每个核苷酸在转录本序列中所占比例,可用a、t、 c、g表示;t表示核苷酸转换,包含6个特征,描述了四个核苷酸在相邻位置之间转换的百分比频率,可用at、ag、ac、tg、tc和gc表示;d表示核苷酸分布,包含20给特征,描述了每个核苷酸在5个相对位置前所占比比例,分别为0(第一个)、25%、50%、75%和100%(最后一个)。
[0075]
2mer、3mer:k-mer是长度为k的子序列,其中k为整数。对于任意一条 dna序列,指定阅读框长度为k,步长为1,从第一个碱基开始移动至序列末尾,阅读框内截取的长度为k的子序列即为该dna序列的k-mer特征。一般任意一条dna序列含有4种碱基a、t、c、g,所以取
不同的k值时,dna 序列的k-mer数量不同,为4k。考虑到序列长度大小,我们选取了k=2和k=3,并分析不同k值下,各dna子序列所占比例,最终分别获得16和64个特征。
[0076]
tn:tn曲线为一种基于三核苷酸的dna序列三维图形表示方法。在本项工作中,我们提取每条dna序列的tn曲线的6个参数作为序列特征。
[0077]
itn:i-tn曲线为根据tn曲线提出的一种改进的图形表示形式。在本项工作中,我们提取每条dna序列的tn曲线的18个参数作为序列特征。
[0078]
cylindrical:cylindrical为一种蛋白质序列的柱面表示方法。在本项工作中,我们将dna序列转换为蛋白质序列,并将蛋白质序列的柱面表示方法中的参数作为序列特征。
[0079]
spherical:spherical为一种蛋白质序列的球坐标表示方法。本项工作中,我们将dna序列转换为蛋白质序列,并将蛋白质序列的柱球面表示方法中提到的每个氨基酸的物理化学性质作为序列特征。
[0080]
codon:64个密码子的百分含量。
[0081]
amino:20个氨基酸的百分含量。
[0082]
(ii)mrmr和增量选择:
[0083]
利用mrmr(最大相关最小冗余)方法分别对每种特征提取策略中的特征与编码潜力的相关性进行排序,即排名越靠前的特征与序列编码潜力相关性越大,且特征间的冗余度越低。
[0084]
将排序后特征集通过增量选择的方法,即根据排序结果,依序选择前n个特征作为新的特征子集(n=1,2,3...),将每个特征子集做10倍交叉验证,分析平均预测准确率,选取准确率最高的特征子集作为此特征提取策略下的最佳特征集,用于模型训练。
[0085]
步骤5)利用matlab的libsvm包,构建基于svm的肽编码sorfs预测模型;利用训练集tr1的9个特征集构建基于原核训练预测模型,利用训练集tr2 的9个特征集构建基于真核训练预测模型;利用贪婪的网格搜索方法进一步优化预测模型,得到18个指定参数范围内的最佳预测模型;
[0086]
网格搜索方法的参数设置:
[0087]
cmin:惩罚参数c的变化范围的最小值(取以2为底的对数后)。默认为-5。
[0088]
cmax:惩罚参数c的变化范围的最大值(取以2为底的对数后)。默认为5。
[0089]
gmin:参数g的变化范围的最小值(取以2为底的对数后)。默认为-5。
[0090]
gmax:参数g的变化范围的最大值(取以2为底的对数后)。默认为5。
[0091]
v:交叉验证的参数。默认为3。
[0092]
cstep:参数c步进的大小。默认为1。
[0093]
gstep:参数g步进的大小。默认为1。
[0094]
accstep:最后显示准确率图时的步进大小。默认为1.5。
[0095]
步骤6)利用预测模型对测试集进行预测,分析各数据集预测结果的sn、 sp、acc、mcc,比较评估不同特征选取策略的预测效率,得到表现最好的特征集和预测模型作为最佳的特征集和预测模型。
[0096]
根据步骤5)得到的18个预测模型,利用matlab的libsvm包,对测试集进行跨物种预测,其中测试集ara1、hum1、mou1、pro和bac在原核混合训练集训练的预测模型上进行预测,测试集ara2、hum2、mou2、pro和bac在真混合训练集训练的预测模型上进行预测。
[0097]
根据sn、sp、acc、mcc,4个指标评估预测结果(表3,表4),评估指标计算公式如下:
[0098][0099][0100][0101][0102]
其中,sn为灵敏度,sp为特异度,acc为准确率,mcc为马修斯相关系数, tp为真正例,fn为假负例,tn为真负例,fp假正例。表3基于原核混合训练集构建的预测模型的预测结果
[0103][0104][0105]
表4基于原核混合训练集构建的预测模型的预测结果
[0106][0107]
根据预测结果,选取单独基于原核训练集和单独基于真核训练集分别构建原核生物肽编码sorfs预测模型和真核生物肽编码sorfs预测模型,对sorfs 实现同界跨物种预测。针对原核sorfs,选取codon策略提取的最佳特征集训练的模型进行预测效果最好,预测准确率最高可达0.91;针对真核sorfs,选取3mer策略提取的最佳特征集训练的模型进行预测效果最好,预测准确率约为 0.83~0.87。
[0108]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。