res.2015,3(2):161-72)中挖掘的基因表达数据证实了ido表达的这种层次结构。在大多数研究中,ido在肿瘤或引流淋巴结中的高表达是不利的预后因素。该类别中的肿瘤包括黑色素瘤、结肠癌、脑肿瘤、卵巢癌、急性髓细胞性白血病、子宫内膜癌、高级别骨肉瘤等等(munn和mellor;trends in immunol.2016,37(3):193-207)。在少数肿瘤类型中,ido表达似乎被诱导或“反应性”——这与t细胞浸润和炎症增加有关。在这种情况下,ido的上调可能代表更强的自发抗肿瘤免疫反应,因此与更有利的预后相关。然而,即使在这些免疫反应患者中,ido本身也没有益处,如果ido被阻断,患者可能会做得更好。
8.由于使用不同抗体在患者样本中观察到的ido表达水平存在差异,因此通过测定血清中kyn和trp的浓度来测量ido活性可能更有意义。实际上,在癌症患者的血清中检测到与正常志愿者相比增加的kyn/trp比率(liu等人;blood.2010,115(17):3520-30)。kyn/trp比率最近被验证为宫颈癌患者的预后工具,由此低trp水平表明肿瘤大小大于4cm并转移到淋巴结(ferns等人;oncoimmunology.2015,4(2):e981457)。因此,患者血清中的高kyn/trp比率与淋巴结转移、figo分期、肿瘤大小、宫旁侵袭和疾病特异性存活率低有关,进一步表明基于trp分解代谢特征的ido靶向的相关性。此外,血清kyn/trp比率是具有成人t细胞白血病/淋巴瘤的患者的一个显著独立的不利预后因素(zhai等人;clin cancer res.2015,21(24):5427-33)。
9.在临床前模型中,用重组ido转染免疫原性肿瘤细胞阻止了它们在小鼠中的排斥反应(uyttenhove等人;nat med.2003,9(10):1269-74)。虽然ido表达的消融导致7,12-二甲基苯并[a]蒽诱导的癌前皮肤乳头状瘤的发生率和生长减少(muller等人;proc natl acad sci usa.2008,105(44):17073-8)。
[0010]
在过表达ido和4t1乳腺癌的b16黑色素瘤的临床前模型中,由肿瘤细胞表达的ido通过募集和激活骨髓衍生性抑制细胞(mdsc)以及使用抗ctla-4和抗pd-1对检查点阻断的抵抗力来促进肿瘤生长。在同一研究中,还注意到人类黑色素瘤肿瘤中的ido表达与mdsc浸润密切相关(holmgaard等人;cell rep.2015,13(2):412-24)。
[0011]
伊马替尼作为用于治疗胃肠道间质瘤(gist)的一种靶向kit(cd117)的小分子受体酪氨酸激酶抑制剂,已显示可调节kyn途径。在gist的小鼠模型中,伊马替尼疗法通过降低ido的肿瘤细胞表达产生了许多免疫反应。为了验证伊马替尼的免疫作用部分由其ido表达降低介导的假设,gist小鼠用kyn途径代谢物-kyn、3-羟基邻氨基苯甲酸(3-haa)和3-羟基犬尿氨酸(3-hk)的混合物治疗,该混合物设计成模拟具有有效ido活性的系统。伊马替尼的抗肿瘤作用因trp代谢物混合物的共同施用而减弱。然而,伊马替尼的抗肿瘤作用并未因ido抑制剂1-甲基-色氨酸(1-mt)的共同施用而增加,这与两种药剂影响相同途径的假设一致balachandran等人;nat med.2011,17(9):1094
–
100)。
[0012]
已经显示,肿瘤的tdo表达阻止了免疫小鼠对其的排斥反应,并且用tdo抑制剂进行的全身治疗恢复了小鼠排斥表达tdo的肿瘤的能力(pilotte等人;proc natl acad sci.2012,109(7):2497-502)。在可移植的胶质瘤模型中,肿瘤细胞中的tdo表达促进了肿瘤生长,而tdo敲低降低了肿瘤发生率(opitz等人;nature 2011,478(7368):197-203)。
[0013]
已发现ido抑制剂可抑制肿瘤和血液中的体内trp代谢,这伴随着结直肠癌实验模型中肿瘤生长的减缓(lin等人;j med chem.2016,59(1):419-30;koblish等人;mol cancer ther.2010,9(2):489-98;kraus等人;aacr年会(4月16-20日,路易斯安那州新奥尔
良)2016:摘要4863;wise等人;aacr年会(4月16-20日,路易斯安那州新奥尔良)2016:摘要5115;liu等人;aacr年会(4月16-20日,路易斯安那州新奥尔良)2016:摘要4877),胰腺癌(koblish等人;mol cancer ther.2010,9(2):489-98),黑色素瘤(yue等人;j med chem.2009,52(23):7364-7),肺(yang等人;j med chem.2013,56(21):8321-31),乳腺癌(holmgaard等人;cell rep.2015,13(2):412-24),神经胶质瘤(hanihara等人;j neurosurg.2016,124(6):1594-601)。
[0014]
1-甲基-色氨酸(1-mt)增强了化学疗法在可移植黑色素瘤(b16)和可移植和本土乳腺癌(4t1)小鼠模型中的作用(hou等人;cancer res.2007,67(2):792-801)。此外,1-mt增强了化学发射疗法以延长患有颅内胶质母细胞瘤(gl-261)的小鼠的生存期。在这种情况下,ido的抑制允许化学放射在肿瘤生长部位触发广泛的补体沉积。ido阻断导致肿瘤微环境中血管内皮上的vcam-1上调。补体成分c3基因缺陷的小鼠失去了ido阻断对化学放射诱导的存活的所有协同作用(li等人;journal immunother cancer.2014,2:21)。ido表达在大量胰腺癌患者接种gvax(放射、gm-csf-分泌、同种异体pdac)疫苗后在肿瘤上皮中被诱导。gvax疫苗接种与ido抑制组合提高胰腺癌临床前模型的存活率,以及与环磷酰胺、gvax疫苗、ido抑制和pd-l1的组合阻断存活的所有小鼠[zheng,约翰霍普金斯医学院(john hopkins school of medicine);itoc3会议(3月21-23日,德国慕尼黑)2016]。在这种情况下,疫苗接种与增加剂量的抗ox40组合在tc1肿瘤模型中也已显示出诱导ido并且1-mt对ido的抑制在同一模型中显示出与抗ox40和疫苗接种的协同作用[khleif,乔治亚癌症中心(georgia cancer center);itoc3会议(3月21-23日,德国慕尼黑)2016]。此外,ido抑制剂epacadostat已被显示可增强抗ox40和抗gitr在临床前模型中的作用(koblish等人;aacr年会(4月1-5日,华盛顿特区)2017:摘要#2618)。
[0015]
在b16f10肿瘤模型中,ido/tdo双重抑制剂nlg919增强了原初静息过继转移的pmel-1细胞对用同源人gp100肽进行的疫苗接种的抗肿瘤反应。这种效果与化学疗法加成,并且一旦化学疗法与吲哚莫德/抗-pd-1组合则效果更加明显(mautino等人;aacr年会(4月5-9日,加利福尼亚州圣地亚哥)2014:摘要5023)。沿着这些思路,当nlg-919与emt-6小鼠模型中的抗pd-l1组合时,检测到肿瘤生长抑制的改善深度和持续时间(spahn等人;journal for immunotherapy of cancer 2015,3(suppl 2):p303)。
[0016]
ido选择性抑制剂在肿瘤小鼠模型中已显示出可增强化学疗法:来自iomet pharma的ido选择性抑制剂增强pan02模型中的化学疗法(吉西他滨和abraxane)(wise等人;aacr年会(4月16-20日,路易斯安那州新奥尔良)2016:摘要5115)。
[0017]
在血浆和肿瘤组织中,抗pd-l1和抗ctla4检查点阻断诱导ido活性,而ido选择性抑制剂(pf-06840003)和抗pd-l1治疗的组合导致ct-26同源小鼠结肠肿瘤模型中的显著肿瘤生长抑制(kraus等人;aacr年会(4月16-20日,路易斯安那州新奥尔良)2016:摘要4863)。在另一项研究中,使用抗ctla-4、抗pd-l1和/或ido抑制剂的双重疗法在b16(siy)黑色素瘤小鼠模型中显示出对肿瘤生长的协同抑制(spranger等人;j immunother cancer.2014,2:3)。与这种改善的功效相关的主要生物学因素恢复了肿瘤浸润cd8 t细胞的il-2产生和增殖。功能恢复不需要新的t细胞迁移到肿瘤。在另一项研究中,1-mt对ido的抑制与靶向免疫检查点(如ctl-4、pd-1/pd-l1和gitr)的疗法的组合协同控制肿瘤生长并提高b16-f10和4t1肿瘤小鼠模型的总生存期(holmgaard等人;j exp med.2013,210(7):1389-402)。在原
位神经胶质瘤模型中,用抗ctla-4、抗pd-l1和1-mt以及epacadostat和抗pd-1的组合进行三重治疗产生了高效的持久生存优势(wainwright等人;clin cancer res.2014,20(20):5290-301;reardon等人;aacr年会(4月1-5日,华盛顿特区)2017:摘要572)。多项临床试验(nct02752074,nct02658890,nct02327078,nct02318277,nct02178722,nct02471846,nct02298153)研究了靶向ido与检查点阻断组合的概念。
[0018]
用tlr9激动剂进行肿瘤内治疗显示出在治疗和远处肿瘤中诱导ido表达,并且ido抑制剂与相同tlr9激动剂的组合在ct-26同源小鼠结肠肿瘤模型中显示出附加的抗肿瘤作用(wang等人;aacr年会(4月16-20日,路易斯安那州新奥尔良)2016:摘要3847)。
[0019]
在几种小鼠模型中,高ido表达诱导免疫抑制性mdsc募集到肿瘤。发现csf-1r在mdsc和csf-1r阻断上表达以抑制瘤内mdsc。因此,用d-1-mt抑制ido在过表达ido的b16模型中显示出与csf-1r阻断协同(holmgaard等人;ebiomedicine 2016,6:50-8)。
[0020]
有实验证据表明,ido抑制还改善对b细胞淋巴瘤中嵌合抗原受体(car)t细胞疗法的治疗反应。在b细胞淋巴瘤的小鼠模型中,肿瘤细胞中的ido表达通过trp代谢物的作用抑制cd19 car t细胞疗法。在该模型中,用ido抑制剂1-mt治疗恢复了car t细胞对肿瘤的控制(ninomiya等人;blood,2015,125(25):3905-16)。
[0021]
dna纳米粒子可以经由依赖于细胞溶质dna的干扰素基因刺激剂(sting)传感器的途径来诱导ido。因此,sting激动剂可以诱导ido并促进致耐受反应。已经在使用低和高抗原性肿瘤的临床前模型中研究了这种情况。在表现出低抗原性的肿瘤中,sting激活ido占主导地位并克服sting/ifn免疫原性反应,而在具有高抗原性的肿瘤中,sting/ifn信号传导反而会增强免疫原性反应并且不能诱导ido。总体而言,这些数据表明ido抑制可以增强对sting激动剂的抗肿瘤反应,尤其是在低抗原性肿瘤中(lemos等人;cancer res.2016,76(8):2076-81)。
[0022]
鉴于jak-stat(信号换能器和转录激活剂)信号传导系统在介导干扰素-γ-诱导的ido表达中的作用,将ido抑制剂与jak/stat抑制剂组合是显而易见的。已经报道了关于这种治疗概念的临床试验(nct02559492)。
[0023]
在中枢神经系统中,作为kyn前体的trp和血清素的宿命都是令人感兴趣的重要途径。由kyn途径产生的代谢物被认为在神经炎症和神经退行性紊乱(如亨廷顿病)的病理机制中发挥作用。来自kyn途径的第一个稳定中间体是kyn。随后,生成了几种神经活性中间体。它们包括犬尿氨酸(kyna)、3-羟基犬尿氨酸(3-hk)和喹啉酸(quin)。3-hk和quin具有不同机制的神经毒性;3-hk是一种有效的自由基发生器(thevandavakkam等人;cns neurol disord.drug targets.2010,9(6):791
–
800;ishii等人;arch biochem biophys.1992,294(2):616
–
622;hiraku等人;carcinogenesis.1995,16(2):349
–
56),而quin是兴奋性毒性n-甲基-d-天冬氨酸(nmda)受体激动剂(stone和perkins;eur j pharmacol.1981,72(4):411
–
2;schwarcz等人;science.1983,219(4582):316
–
8)。另一方面,kyna通过其抗氧化特性以及拮抗α7烟碱型乙酰胆碱受体和nmda受体的甘氨酸协同激动剂位点两者而具有神经保护作用(vecsei和beal;brain res bull.1990,25(4):623-7;foster等人;neurosci lett.1984,48(3):273
–
8;carpenedo等人;eur j neurosci.2001,13(11):2141
–
7;goda等人;adv.exp.med.biol.1999,467:397
–
402)。trp分解代谢物浓度水平的变化可以将平衡转移到病理条件。影响朝向kyn途径神经保护分支(即朝向kyna合成)的代谢的能力可用于预
防神经退行性疾病。
[0024]
在cns中,kyn途径在大多数细胞类型中不同程度地存在,浸润巨噬细胞、激活的小胶质细胞和神经元具有kyn途径酶的完整库。另一方面,神经保护性星形胶质细胞和少突胶质细胞分别缺乏酶kyn 3-单加氧酶(kmo)和ido-1,并且无法合成兴奋性毒素quin(guillemin等人;redox rep 2000,5(2-3):108-11;lim等人;international congress series.2007,1304:213-7)。tdo在脑中以低量表达,并由trp或皮质类固醇诱导(salter和pogson;biochem j.1985,229(2):499-504;miller等人;neurobiol dis.2004,15(3):618-29)。鉴于tdo和ido在多种cns紊乱(如精神分裂症)的发病机制中的作用以及tdo在控制全身trp水平方面的作用,ido和/或tdo抑制剂可用于改善患有多种cns疾病和神经变性的患者的结果。
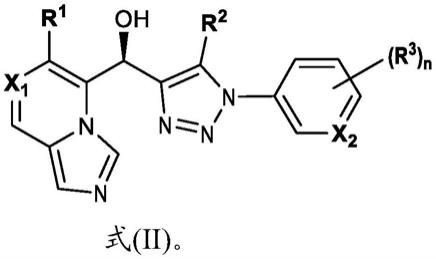
[0025]
ido和/或tdo抑制剂可另外用于治疗肌萎缩侧索硬化(als)(或lou gehrig氏病)。als导致运动皮层、脑干和脊髓中运动神经元的选择性攻击和破坏。尽管多种机制可能导致als,但在神经炎症期间激活的kyn途径正在成为一个促成因素。初始炎症可能对具有易感遗传体质的个体的运动神经元造成非致命性损伤,进而引发渐进性炎症过程,其激活小胶质细胞以产生进一步破坏运动神经元的神经毒性kyn代谢物。在als患者的脑和脊髓中,已经观察到大量激活的小胶质细胞、反应性星形胶质细胞、t细胞和浸润性巨噬细胞(graves等人;amyotroph lateral scler other motor neuron disord.2004,5(4):213
–
9;henkel等人;ann neurol.2004,55(2):221
–
35)。这些细胞释放炎症和神经毒性介质,其中包括ido最有效的诱导剂ifn-γ(mcgeer和mcgeer;muscle nerve.2002;26(4):459
–
70)。ido的神经元和小胶质细胞在als运动皮层和脊髓中的表达增加(chen等人;neurotox res.2010,18(2):132
–
42)。已经提出,免疫激活剂的释放激活kyn途径的限速酶ido,其生成代谢物,如神经毒素quin。因此,抑制ido可减少已明确与als的发病机制有关的神经毒性quin的合成。
[0026]
ido和/或tdo抑制剂可另外用于治疗亨廷顿病(hd)。hd是由亨廷顿(htt)基因中cag重复扩展引起的一种遗传性常染色体显性神经退行性紊乱。受hd影响的患者表现出进行性运动功能障碍,其特征是随意和不随意运动(舞蹈手足徐动症)的异常以及精神和认知障碍。kyn途径内代谢物的生命监测提供了与cag重复次数相关的少数生物标志物之一,因此与疾病的严重程度相关(forrest等人;j neurochem2010,112(1):112-22)。事实上,在hd和hd模型小鼠的患者中,新纹状体和皮质中的3-hk和quin水平增加。此外,hd患者的纹状体中的kyna水平降低。在啮齿动物中纹状体内注射quin再现了hd的行为和病理特征(sapko等人;exp neurol.2006 197(1):31-40)。重要的是,hd果蝇模型中的tdo消融改善了神经变性(campesan等人;curr biol.2011;21(11):961-6)。
[0027]
ido和/或tdo抑制剂可另外用于治疗阿尔茨海默病(ad)。ad是一种与年龄相关的神经退行性紊乱,其特征是神经元丧失和痴呆。该疾病的组织病理学表现为细胞内β-淀粉样蛋白(αβ)的积累和随后的神经炎斑块的形成以及与学习和记忆相关的特定脑区中神经原纤维缠结的存在。这种疾病下的病理机制仍然存在争议,然而,越来越多的证据表明kyn途径代谢物与ad的发展和进展有关。已经显示,αβ(1-42)可以激活原代培养的小胶质细胞并诱导ido表达(guillemin等人;redox rep.2002,7(4):199-206;walker等人;j leukoc biol.2006,79:596
–
610)。此外,在与ad患者脑中的淀粉样斑块相关的小胶质细胞中观察到ido过度表达和quin的增加生产(guillemin等人;neuropathol appl neurobiol.2005,31
(4):395-404)。quin已显示导致人类皮层神经元中的tau过度磷酸化(rahman等人;plos one.2009,4(7):e6344)。因此,小胶质细胞中ido的过度表达和kyn途径的过度激活与ad的发病机制有关。也有证据表明tdo与阿尔茨海默病有关。tdo在患者和ad小鼠模型的脑中上调。此外,tdo与ad患者海马体中的喹啉酸、神经原纤维缠结-tau和淀粉样蛋白沉积物共定位(wu等人;plos one.2013,8(4):e59749)。临床前证据支持使用kmo、tdo、ido和3hao抑制剂来抵消ad中神经炎症的影响。此外,其他观察结果表明ad患者血清中kyn/trp的比率增加(widner等人;j neural transm(vienna).2000,107(3):343-53)。在ad的果蝇模型中,tdo的遗传和药理学抑制都提供了强大的神经保护作用(breda等人;proc natl acad sci.2016,113(19):5435-40)。因此,kyn途径在ad中被tdo和ido两者过度激活,并且可能参与神经原纤维缠结形成并与老年斑形成有关。
[0028]
ido和/或tdo抑制剂可另外用于治疗帕金森病(pd)。pd是一种常见的神经退行性紊乱,其特征是多巴胺能神经元的丧失和局部神经炎症。帕金森病与小胶质细胞的慢性激活有关(gao和hong;trends immunol.2008,29(8):357
–
65)。小胶质细胞激活释放神经毒性物质包括活性氧物质(ros)和促炎细胞因子,如inf-γ(block等人;nat rev neurosci.2007;8(1):57
–
69),经由诱导ido表达对kyn途径的有效激活。激活的小胶质细胞中的kyn途径导致3hk和quin的上调。3hk是有毒的,主要是由于转化为ros(okuda等人;j neurochem.1998;70(1):299
–
307)。quin对ros和nmda受体介导的兴奋性毒性的综合作用导致神经元功能障碍及其死亡(stone和perkins;eur j pharmacol.1981,72(4):411
–
2;braidy等人;neurotox res.2009,16(1):77-86)。然而,通过神经元中kyn途径激活产生的吡啶甲酸(pic)具有保护神经元免受quin诱导的神经毒性影响的能力,是一种nmda激动剂(jhamandas等人;brain res.1990,529(1-2):185-91)。小胶质细胞可以被促炎介质和垂死神经元的刺激过度激活,并导致进一步的小胶质细胞激活小胶质细胞增生的持续循环。过度的小胶质细胞增生会对邻近的神经元造成神经毒性并导致神经元死亡,从而导致帕金森病的进展。因此,pd与脑内kyn途径的两个主要分支之间的不平衡有关。星形胶质细胞的kyna合成减少,同时小胶质细胞产生的quin增加。重要的是,tdo的遗传和药理学抑制都在pd的果蝇模型中提供了强大的神经保护(breda等人;proc natl acad sci.2016,113(19):5435-40)。
[0029]
ido和/或tdo抑制剂可另外用于治疗多发性硬化症(ms)。ms是一种自身免疫性疾病,其特征在于神经系统白质中的炎性病变,包括对髓鞘质片的特异性免疫反应,导致炎症和轴突丧失(trapp等人;curr opin neurol.1999,12:295
–
302;owens;curr opin neurol.2003,16:259
–
265)。由免疫系统的激活引起的神经毒性kyn代谢物的积累与ms的发病机制有关。发现quin在ms的自身免疫动物模型eae的大鼠脊髓中选择性升高(flanagan等人;j neurochem.1995,64:1192
–
6)。eae中增加的quin的起源被认为是巨噬细胞。quin是脂质过氧化的引发剂,髓鞘质附近高水平的quin可导致eae中的脱髓鞘作用并可能导致ms。干扰素-βlb(ifn-pib)在巨噬细胞中诱导kyn途径代谢,其浓度与ifn-β治疗患者的血清中发现的浓度相当,这可能是其治疗ms疗效的限制因素(guillemin等人;j interferon cytokine res.2001,21:1097
–
1101)。在施用ifn-β后,在接受ifn-β注射的ms患者的血浆中发现与健康受试者相比增加的kyn水平和kyn/trp比率,这表明ifn-β诱导了ido(amirkhani等人;eur.j.neurol.2005,12,625
–
31)。ifn-pib导致产生quin,其浓度足以干扰神经元树
突整合传入信号和杀死少突胶质细胞的能力(cammer等人;brain res.2001,896:157
–
160)。在ifn-pib治疗的患者中,用ido/tdo抑制剂同时阻断kyn途径可提高ifn-pib的疗效。
[0030]
已确定ido(ido2)的同源物与ido具有44%的氨基酸同源性,但其功能在很大程度上与ido的功能不同(ball等人,gene 2007,396(1):203
–
13;yuasa等人,j mol evol 2007,65(6):705
–
14。ido抑制剂可以调节ido1和/或ido2。目前的证据表明ido2是在b细胞中起作用以调节自身免疫疾病的一种免疫调节酶。虽然其酶促功能特征不佳,ido2的免疫调节机制与其研究得较好的同源物ido1不同。ido2在自身免疫性炎症疾病(包括类风湿性关节炎、接触性超敏反应和系统性红斑狼疮)的多种模型中充当促炎介质(merlo和mandik-nayak,clinical medicine insights:pathology 2016,9(s1):21
–
28)。因为ido2在促进炎症上起作用,它可能是用于治疗这些疾病的靶向治疗的候选者,特别是在联合治疗设置中如此。
[0031]
大多数trp是通过kyn途径处理的。一小部分trp被加工成5-ht并因而加工成褪黑激素,这两者也是ido的底物。早就已知的是,除了其他影响外,急性trp耗竭可引发抑郁发作,甚至在健康个体中产生情绪的深刻变化。这些观察与血清素能药物在增强情绪和刺激神经发生方面的临床益处密切相关。
[0032]
近年来,精神分裂症病理生理学的一般观点(即多巴胺[da]传输障碍)已扩展到还涉及脑的谷氨酸能功能障碍。因此,临床观察显示,全身施用n-甲基-d-天冬氨酸(nmda)受体拮抗剂(例如,苯环利定[pcp]和氯胺酮)在健康个体中引发精神分裂症样症状,并引发精神分裂症患者的症状(holtze等人;j psychiatry neurosci.2012,37(1):53-7)。此外,谷氨酸缺乏理论已经得到遗传学发现的一些支持。脑的低谷氨酸能状态也可以通过内源性nmda受体拮抗剂kyna的升高来实现。事实上,在精神分裂症患者的死后脑中发现了kyna和产生kyna的酶的脑水平改变(barry等人;j psychopharmacol.2009,23(3):287-94)。特别是,在额叶皮质中发现升高的kyn和kyna水平,并且在精神分裂症患者的前扣带回皮质中观察到kyn途径的第一步的上调(miller等人;brain res.2006,1073-1074:25-37)。然而,其他研究人员已经发现精神分裂症中kyna减少而3-haa增加(miller等人;neurochem int.2008,52(6):1297-303)。精神分裂症中kyn代谢物升高的机制尚未完全阐明。机制包括kmo多态性和tdo上调(miller等人;neurobiol dis.2004,15(3):618-29)。因此,ido和/或tdo抑制剂可用于治疗精神分裂症。
[0033]
ido和/或tdo抑制剂可另外用于治疗疼痛和抑郁症。疼痛和抑郁症通常是共病紊乱。已经显示,ido在这种共病症中起着关键作用。最近的研究已经显示,ido活性与(a)降低的血清素含量和抑郁(dantzer等人;nat rev neurosci.2008,9(1):46-56;sullivan等人;pain.1992,50(1):5-13)以及(b)通过其衍生物(例如喹啉酸)对谷氨酸受体的影响而增加的kyn含量和神经可塑性变化(heyes等人;brain.1992,115(pt5):1249-73)有关。
[0034]
在大鼠中,慢性疼痛引起抑郁行为和双侧海马体中的ido上调。ido的上调导致双侧海马体中kyn/trp比率增加以及血清素/trp比率降低。此外,海马体ido活性的ido基因敲除或药理学抑制减弱了伤害感受性和抑郁行为(kim等人;j clin invest.2012,122(8):2940-54)。
[0035]
由于促炎细胞因子与疼痛和抑郁症的病理生理有关,促炎细胞因子对脑ido的调节通过调节trp代谢作为疼痛与抑郁症之间的共病关系的关键机制环节。
[0036]
此外,kyn途径与创伤性脑损伤(tbi)相关。tbi已被显示会随着quin的持续增加而
诱导kyn途径的显著激活(yan等人;journal of neuroinflammation 2015,12(110):1-17)。quin的过量产生以及脑损伤区域中ido1激活和mrna表达的增加表明tbi选择性地诱导了对kyn途径的神经毒性分支的强烈刺激。quin的不利作用得到了其与不良结果的关联的支持,从而可能成为tbi后的早期预后因素。因此,ido和/或tdo抑制剂可另外用于预防/治疗tbi。
[0037]
受细菌、寄生虫或病毒感染诱导强烈的ifn-y依赖性炎症反应。ido可以抑制保护性宿主免疫,从而间接导致病原体负担增加。例如,在感染了鼠白血病病毒(mulv)的小鼠中,发现ido被高度表达,并且ido的消融增强了对病毒复制的控制并增加了存活率(hoshi等人;j immunol.2010,185(6):3305-3312)。在流感感染的模型中,ido的免疫抑制作用可能使肺部容易发生继发性细菌感染(van der sluijs等人;j infect dis.2006,193(2):214-22)。因此,社区获得性肺炎(cap)中的ido活性增加,并且该活性与该疾病的严重程度和结果相关。这些结果表明ido活性可以预测cap的预后(suzuki等人;j infect.2011sep;63(3):215-22)。
[0038]
在由克氏锥虫(trypanosoma cruzi)寄生虫引起的恰加斯病中,kyn在患者体内增加并与疾病的严重程度相关(maranon等人;parasite immunol.2013,35(5-6):180-7)。沙眼衣原体感染诱导大量ifn-γ的产生,进而导致ido诱导。一项研究显示,ido介导的trp池消耗导致衣原体转化为高度适应在恶劣环境中生存的持久形式(barth和raghuraman;crit rev microbiol.2014,40(4):360-8)。在慢性皮肤利什曼病患者中,高水平的ido mrna表达已经在感染性病变中检测到,并且与病变内treg细胞的积累有关。小鼠中的硕大利什曼原虫感染诱导局部皮肤病变和引流淋巴结中的ido表达。ido的遗传和药理学消融导致对硕大利什曼原虫的控制得到改善。脑型疟疾可能是人类恶性疟原虫感染的致命表现。脑型疟疾期间小鼠脑中的ido活性增加,并且在疟疾小鼠模型中抑制ido增强了抗疟疾t细胞的功能并略微降低了寄生虫负荷(barth和raghuraman;crit rev microbiol.2014,40(4):360-8)。
[0039]
测量肺结核患者体内的kyn和trp血清浓度以及评估ido活性显示,与对照受试者相比,kyn浓度和ido活性显著增加而trp浓度显著降低。有趣的是,在肺结核患者中,非幸存者具有显著更高的kyn浓度和显著更低的trp浓度,从而导致ido活性与幸存者体内的相比显著增加。最重要的是,多变量分析显示了,ido活性是肺结核死亡的重要独立预测因子(suzuki等人;clin vaccine immunol.2012,19(3):436
–
442)。
[0040]
因此,ido抑制剂可用于改善患有多种传染性疾病和炎症病症的患者的结果。鉴于tdo在控制全身trp水平方面的作用,tdo抑制剂还可用于改善患有多种传染性疾病和炎症病症的患者的结果。
[0041]
感染hiv的患者具有长期降低的血浆trp水平和升高的kyn水平,以及增加的ido表达(murray;lancet infect dis.2003,3(10):644-52)。在hiv患者中,ido的上调起作用以抑制对hiv抗原的免疫反应,从而导致病毒的免疫逃避。晚期hiv感染期间的一个特有的特征是优先消耗胃肠道和血液两者中的th17细胞。有趣的是,hiv感染中th17细胞的丢失伴随着诱导性treg细胞频率的同时升高,并与ido活性直接相关。treg细胞可抑制有效的hiv特异性细胞免疫反应,而th17细胞的逐渐消耗可增加对粘膜感染的易感性。因此,持续的ido激活可为hiv持续存在建立一个有利的环境,并导致具有进行性疾病的hiv感染者中出现免
疫缺陷(barth和raghuraman;crit rev microbiol.2014,40(4):360-8)。hiv患者,尤其是hiv相关痴呆患者(kandanearatchi&brew;febs j.2012,279(8):1366-74)的csf中通常具有显著升高的kyn水平。这些水平与神经认知衰退[hiv相关神经认知紊乱(hand)]的发展并常常与严重精神病症状的存在直接相关(stone&darlington;trends pharmacol sci.2013,34(2):136-43)。因此,ido和/或tdo抑制剂可另外用于治疗hiv(aids,包括其表现,例如恶病质、痴呆和腹泻)。
[0042]
与hiv感染一样,与已解决hcv感染的患者和健康个体相比,慢性感染hcv的患者血液中kyn与trp的比率增加(larrea等人;j virol.2007,81(7):3662-6)。此外,已经提出,ido的表达与该疾病的发病机制相关,并且ido在hcv感染患者的进行性肝硬化中的高表达可能有助于肝细胞癌的发展(asghar等人;exp ther med.2015,9(3):901-4)。因此,ido和/或tdo抑制剂可用于治疗慢性感染hcv的患者。
[0043]
ido在调节对肠道微生物群的粘膜免疫方面发挥作用。ido已被显示可调节肠道内共生诱导的抗体产生;ido缺陷小鼠血清中的免疫球蛋白a(iga)和免疫球蛋白g(igg)的基线水平升高以及肠道分泌物中的iga增加。由于抗体产生增加,ido缺陷小鼠比wt小鼠更能抵抗革兰氏阴性肠道细菌病原体啮齿柠檬酸杆菌(citrobacter rodentium)的肠道定植。ido缺陷小鼠也表现出对由啮齿柠檬酸杆菌(c.rodentium)感染引起的结肠炎增强的抵抗力(harrington等人;infect immunol.2008,76(7):3045-53)。
[0044]
因此,ido/tdo活性的药理学靶向可能代表一种操纵肠道免疫和控制由肠道病原体引起的病理(包括结肠炎)的新方法(harrington等人;infect immunol.2008,76(7):3045-53)。
[0045]
最近的文献强调了ido在代谢紊乱中的作用(laurans等人;nature medicine https://doi.org/10.1038/s41591-018-0060-4(2018);natividad等人;cell metabolism 2018,28:1-13)。发现,喂食高脂肪饮食的ido1敲除小鼠与野生型小鼠相比获得较少的体重,具有较低的脂肪量,较好的葡萄糖和胰岛素耐受性,并且巨噬细胞浸润到脂肪组织中较少。用ido抑制剂l-1-mt治疗,同时高脂肪饮食对胰岛素和葡萄糖耐量的影响与敲除中的情况相似。抗生素治疗阻止了ido1敲除小鼠因高脂肪饮食而增重,并且ido1敲除小鼠和wt小鼠的共同饲养具有与ido1敲除小鼠相似的代谢测量结果,这些事实表明来自ido1敲除小鼠的微生物群具有保护作用。与这些假设一致的是,ido-1敲除小鼠具有不同的肠道微生物群组成。trp可以通过ido代谢以产生kyn,或者通过肠道微生物群代谢以产生吲哚衍生物,例如吲哚-3-乙酸,ahr的配体。ido的消耗增加了粪便中吲哚-3-乙酸的水平。吲哚-3-乙酸诱导肠道免疫细胞中ahr的激活,增加了il-17和il-22的产生。il-22水平降低伴随着肠道屏障功能障碍。这些数据支持ido在控制kyn和吲哚-3-乙酸激活ahr平衡方面的重要性。与小鼠的观察结果一致的是,肥胖或2型糖尿病患者的血浆和粪便中kyn水平较高,其粪便中吲哚-3-乙酸的水平较低(laurans等人;nature medicine https://doi.org/10.1038/s41591-018-0060-4(2018)。在另一项研究中,代谢综合征患者的粪便样本中也发现与健康受试者相比升高的kyn水平(natividad等人;cell metabolism 2018,28:1-13)。目前尚不清楚肠道微生物群对ahr激动剂产生的改变是否是代谢综合征发病机制中的主要事件。然而,通过应用ahr激动剂纠正这一缺陷的治疗效果显示其参与发病机制((natividad等人;cell metabolism 2018,28:1-13)。因此,ido抑制剂通过改变trp衍生性ahr激动剂平衡的
平衡可用于调节代谢紊乱,例如肥胖、2型糖尿病和/或脂肪酸性肝病。
[0046]
白内障是导致视力下降的眼内晶状体混浊。最近的研究表明,kyn可化学改变人类晶状体中的蛋白质结构,从而导致白内障形成。在人晶状体中,ido活性主要存在于前上皮中(takikawa等人;adv exp med biol.1999,467:241-5)。在晶状体中已经检测到几种kyn,如kyn、3-hk和3-羟基犬尿氨酸葡萄糖苷(3-hk-g);它们被认为通过吸收紫外线来保护视网膜,因此通常被称为紫外线过滤器。然而,最近的几项研究显示,kyn易于脱氨基和氧化以形成α,β-不饱和酮,其发生化学反应并改变晶状体蛋白(taylor等人;exp eye res.2002;75(2):165-75)。kyn介导的修饰可有助于衰老和白内障发生期间的晶状体蛋白修饰。它们还可降低保持晶状体透明度所必需的a-晶状体蛋白的伴侣功能。
[0047]
在晶状体中过度表达人类ido的转基因小鼠系在出生后3个月内发展为双侧白内障。已证明ido介导的kyn产生导致纤维细胞分化及其凋亡缺陷(mailankot等人;lab invest.2009;89(5):498-512)。因此,抑制ido/tdo可减缓白内障形成的进展。
[0048]
子宫内膜异位症是子宫腔外存在子宫内膜,是一种常见的引起腹痛、性交困难和不孕症的妇科失调。通过微阵列分析发现患有子宫内膜异位症的女性的在位子宫内膜中ido表达更高(burney等人;endocrinology.2007;148(8):3814-26;aghajanova等人;reprod sci.2011,18(3):229-251)。此外,ido被显示可提高子宫内膜基质细胞的存活率和侵袭性(mei等人;int j clin exp pathol.2013;6(3):431-44)。因此,ido/tdo抑制剂可用于治疗子宫内膜异位症。
[0049]
胚胎植入过程需要防止同种异体移植物排斥的机制;并且对胎儿同种异体移植物的耐受性代表用于维持妊娠的重要机制。在胎儿-母体界面中表达ido的细胞保护同种异体胎儿免受母体免疫反应的致命排斥。通过将怀孕小鼠暴露于1-甲基色氨酸对ido的抑制诱导了t细胞介导的同种异体孕体排斥,而同基因孕体不受影响;这表明胎儿-母体界面的ido表达对于防止胎儿同种异体移植物的排斥是必要的(munn等人;science 1998,281(5380):1191-3:)。越来越多的证据表明,胎儿-母体界面的ido产生和正常功能可在妊娠耐受性中发挥重要作用(d
ü
rr和kindler;j leukoc biol.2013,93(5):681-700)。因此,ido/tdo抑制剂可用作避孕或流产剂。
[0050]
在实验性慢性肾功能衰竭中,ido的激活导致kyn的血液水平升高(tankiewicz等人;adv exp med biol.2003,527:409-14),并且在尿毒症患者的尿液中存在kyn修饰的蛋白质(sala等人;j biol chem.2004,279(49):51033-41)。此外,肾脏ido表达在炎症期间可能是有害的,因为它增强了肾小管细胞损伤。
[0051]
在冠心病中,炎症和免疫激活可能经由干扰素-y介导的ido激活与kyn血液水平升高有关(wirleitner等人;eur j clin invest.2003,33(7):550-4)。
[0052]
涉及体外循环的心脏手术可导致认知功能障碍。由于此类手术与炎症迹象相关,并且促炎介质激活色氨酸氧化成神经活性犬尿氨酸,其调节nmda受体功能和氧化应激。麻醉后认知功能障碍通常与这些后遗症有关。最近,这些缺陷已被显示与心脏手术后和康复的中风患者体内kyn途径标志物的变化有关,但不与细胞因子的变化无关(forrest等人;j.neurochem.201,119(1):136-52)。
[0053]
一般而言,据报道,trp分解代谢在中风中改变。中风急性期kyn途径的激活可通过直接机制参与缺血性损伤,该直接机制包括兴奋性毒性和氧化应激等,因为抑制kyn途径减
少中风动物模型中的脑损伤。可能的是,中风后可存在免疫系统和kyn途径之间的相互作用,但不同的炎症独立机制也可在该途径的调节中发挥作用,从而调节trp分解代谢的限速酶。有趣的是,脑缺血后的kyn途径也可在这种病理的慢性阶段期间发挥作用,在该阶段,中风幸存者呈现残疾高发率,如痴呆和抑郁症,甚至是中风结果和死亡的危险因素。总之,kyn和trp分解代谢可在脑缺血后发挥重要作用,并且ido/tdo抑制剂可为中风的急性和慢性阶段提供新的药理学工具(cuartero等人;curr pharm des.2016;22(8):1060
–
1073)。
[0054]
本发明提供抑制ido和/或tdo酶的活性的新型式(i)化合物。
[0055]
1)本发明的第一实施方案涉及式(i)化合物其中x1表示氮或碳(尤其是碳);x2表示氮或碳(尤其是碳);r1表示
·c1-4-烷基(尤其是甲基或乙基);
·c3-5-环烷基(尤其是环丙基);或者
·
卤素(尤其是氯);r2表示
·
氢;
·c1-3-烷基(尤其是甲基或乙基);或者
·
卤素(尤其是氯);每个r3独立地表示
·c1-4-烷基(尤其是甲基);
·c1-3-烷氧基-c
1-4-烷基(尤其是甲氧基甲基);
·
卤素(尤其是氟,氯或者溴);
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);
·-nr
n1rn2
,其中
·rn1
表示氢以及r
n2
表示-(c=o)-r
co
,其中r
co
表示c
1-3-烷氧基(尤其是甲氧基);
·rn1
以及r
n2
独立地表示氢或者c
1-3-烷基(尤其是甲基);
·rn1
以及r
n2
,与它们所连接的氮原子一起,形成4至6元饱和杂环,其包含一个氮
环原子(尤其是氮杂环丁烷基,吡咯烷基或者哌啶基;尤其是吡咯烷基);或者
·rn1
表示c
1-3-烷基(尤其是甲基)以及r
n2
表示1,2-乙烷二基使得式(i)的片段表示1-(c
1-3-烷基)-2,3-二氢-吲哚-5-基(尤其是1-甲基-2,3-二氢-吲哚-5-基);
·
2-氧杂-6-氮杂-螺旋[3.3]庚-6-基或者6-氧杂-1-氮杂-螺旋[3.3]庚-1-基;以及n表示0,1,2,3,4或者5(尤其是0,1,2或者3)(即(r
3)n
表示0,或者1至5个取代基r3,其中应理解当n=0时,r3是不存在的)。
[0056]
[在实施方案1的子实施方案中),一个取代基r3(尤其是-or4或者-nr
n1rn2
)连接在关于与分子其余部分的连接点的对位,以及不存在另外的r3,或者剩余的r3,如果存在,尤其是选自卤素(尤其是氟,氯或者溴)]
[0057]
2)本发明的另一个实施方案涉及根据实施方案1)的化合物,其中x1表示氮或碳(尤其是碳);x2表示氮或碳(尤其是碳);r1表示
·c1-4-烷基(尤其是甲基或乙基);
·c3-5-环烷基(尤其是环丙基);或者
·
卤素(尤其是氯);r2表示
·
氢;或者
·c1-3-烷基(尤其是甲基);每个r3独立地表示
·c1-4-烷基(尤其是甲基);
·c1-3-烷氧基-c
1-4-烷基(尤其是甲氧基甲基);
·
卤素(尤其是氟,氯或者溴);
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);或者
·-nr
n1rn2
,其中r
n1
表示氢以及r
n2
表示-(c=o)-r
co
,其中r
co
表示c
1-3-烷氧基(尤其是甲氧基);以及n表示0,1,2,3,4或者5(尤其是0,1,2或者3)(即(r
3)n
表示0,或者1至5个取代基r3,其中应理解当n=0时,r3是不存在的)。
[0058]
[在实施方案2的子实施方案中),一个取代基r3(尤其是-or4或者-nr
n1rn2
)连接在关于与分子其余部分的连接点的对位,以及不存在另外的r3,或者剩余的r3,如果存在,尤其是选自卤素(尤其是氟,氯或者溴)]
[0059]
下文提供的定义旨在统一适用于如实施方案1)至20)中任一项所定义的式(i)/(ii)化合物,并且经必要修改后,贯穿整个说明书和权利要求,除非另有明确规定的定义提
烷基的优选示例是甲氧基甲基。
[0068]
术语“氧杂环丁烷-3-基-c
1-3-烷基”是指如前所定义的烷基基团,其中其氢原子之一已被氧杂环丁烷环取代,其中所述氧杂环丁烷环在环3位中连接至所述烷基基团。代表性示例包括氧杂环丁烷-3-基-甲基,1-(氧杂环丁烷-3-基)-乙基,2-(氧杂环丁烷-3-基)-乙基以及1-(氧杂环丁烷-3-基)-丙基;尤其是氧杂环丁烷-3-基-甲基。
[0069]
术语“(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基”是指如前所定义的氧杂环丁烷-3-基-c
1-3-烷基基团,其中氧杂环丁烷环的3位中的氢原子已被氟取代。代表性示例包括(3-氟-氧杂环丁烷-3-基)-甲基,2-(3-氟-氧杂环丁烷-3-基)-乙基以及3-(3-氟-氧杂环丁烷-3-基)-丙基;尤其是(3-氟-氧杂环丁烷-3-基)-甲基。
[0070]
3)另一个实施方案涉及根据实施方案1)或2)中任一项的化合物,其中x1表示碳。
[0071]
4)另一个实施方案涉及根据实施方案1)或2)中任一项的化合物,其中x1表示氮。
[0072]
5)另一个实施方案涉及根据实施方案1)至4)中任一项的化合物,其中x2表示碳。
[0073]
6)另一个实施方案涉及根据实施方案1)至4)中任一项的化合物,其中x2表示氮。
[0074]
7)另一个实施方案涉及根据实施方案1)至6)中任一项的化合物,其中r1表示c
3-5-环烷基(尤其是环丙基)或者卤素(尤其是氯);尤其是r1表示环丙基。
[0075]
8)另一个实施方案涉及根据实施方案1)至6)中任一项的化合物,其中r1表示c
1-4-烷基;尤其是r1表示乙基。
[0076]
9)另一个实施方案涉及根据实施方案1)至8)中任一项的化合物,其中r2表示氢。
[0077]
10)另一个实施方案涉及根据实施方案1)至8)中任一项的化合物,其中r2表示氢或者c
1-3-烷基(尤其是甲基或乙基)。
[0078]
11)另一个实施方案涉及根据实施方案1)至10)中任一项的化合物,其中r3独立地表示
·c1-3-烷氧基-c
1-4-烷基(尤其是甲氧基甲基);
·
卤素(尤其是氟,氯或者溴);
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);
·-nr
n1rn2
,其中r
n1
表示氢以及r
n2
表示-(c=o)-r
co
,其中r
co
表示c
1-3-烷氧基(尤其是甲氧基)。
[0079]
12)另一个实施方案涉及根据实施方案1)至10)中任一项的化合物,其中r3独立地表示
·c1-3-烷氧基-c
1-4-烷基(尤其是甲氧基甲基);
·
卤素(尤其是氟,氯或者溴);
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基)。
[0080]
13)另一个实施方案涉及根据实施方案1)至12)中任一项的化合物,其中n表示1,2
或者3(尤其是2或者3)。
[0081]
14)另一个实施方案涉及根据实施方案1)和3)至10)中任一项的化合物,其中n表示1,2或者3;一个取代基r3表示
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);或者
·-nr
n1rn2
,其中
·rn1
表示氢以及r
n2
表示-(c=o)-r
co
,其中r
co
表示c
1-3-烷氧基(尤其是甲氧基);
·rn1
以及r
n2
独立地表示氢或者c
1-3-烷基(尤其是甲基);
·rn1
以及r
n2
,与它们所连接的氮原子一起,形成4至6元饱和杂环,其包含一个氮环原子(尤其是氮杂环丁烷基,吡咯烷基或者哌啶基;尤其是吡咯烷基);或者
·
2-氧杂-6-氮杂-螺旋[3.3]庚-6-基或者6-氧杂-1-氮杂-螺旋[3.3]庚-1-基;其中所述一个取代基在关于与分子其余部分的连接点的对位连接以及剩余的r3,如果存在,选自卤素(尤其是氟或者氯)。
[0082]
15)另一个实施方案涉及根据实施方案1)至10)中任一项的化合物,其中n表示1,2或者3;一个取代基r3表示
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);或者
·-nr
n1rn2
,其中r
n1
表示氢以及r
n2
表示-(c=o)-r
co
,其中r
co
表示c
1-3-烷氧基(尤其是甲氧基);其中所述一个取代基在关于与分子其余部分的连接点的对位连接以及剩余的r3,如果存在,选自卤素(尤其是氟或者氯)。
[0083]
16)另一个实施方案涉及根据实施方案1)至10)中任一项的化合物,其中n表示1,2或者3;一个取代基r3表示
·-or4,其中r4表示氢,c
1-4-烷基(尤其是甲基或乙基),羟基-c
2-5-烷基(尤其是2-羟基-2-甲基丙基或者3-羟基-3-甲基丁基),(氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(氧杂环丁烷-3-基)-甲基)或者(3-氟-氧杂环丁烷-3-基)-c
1-3-烷基(尤其是(3-氟-氧杂环丁烷-3-基)-甲基);其中所述一个取代基在关于与分子其余部分的连接点的对位连接以及剩余的r3,如果存在,选自卤素(尤其是氟或者氯)。
[0084]
17)另一个实施方案涉及根据实施方案1)至10)中任一项的化合物,其中式(i)的片段
表示
·
苯基,4-羟基苯基,4-甲氧基苯基,3-溴-4-甲氧基苯基,4-甲基苯基,3-氯-4-羟基苯基,3-氯-4-甲氧基苯基,3-氟-4-羟基苯基,3-氟-4-甲氧基苯基,2-氟-3-氯-4-甲氧基苯基,3-氯-4-甲氧基-5-氟苯基,2-氟-4-甲氧基-5-氯苯基,2,5-二氟-4-甲氧基苯基,4-((氧杂环丁烷-3-基)甲氧基)-苯基,3-氟-4-((氧杂环丁烷-3-基)甲氧基)-苯基,4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,3-氟-4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,4-(甲氧基-甲酰胺基)-苯基,4-(2-羟基-2-甲基丙氧基)-苯基,4-(甲氧基甲基)-苯基;或者4-乙氧基吡啶-3-基;或者,除了上面列出的,还表示3-氟-4-(2-羟基-2-甲基丙氧基)-苯基,或者6-乙氧基吡啶-3-基。
[0085]
18)另一个实施方案涉及根据实施方案1)和3)至10)中任一项的化合物,其中式(i)的片段表示
·
苯基,4-羟基苯基,4-甲氧基苯基,3-溴-4-甲氧基苯基,4-甲基苯基,3-氯-4-羟基苯基,3-氯-4-甲氧基苯基,3-氟-4-羟基苯基,3-氟-4-甲氧基苯基,2-氟-3-氯-4-甲氧基苯基,3-氯-4-甲氧基-5-氟苯基,2-氟-4-甲氧基-5-氯苯基,2,5-二氟-4-甲氧基苯基,4-((氧杂环丁烷-3-基)甲氧基)-苯基,3-氟-4-((氧杂环丁烷-3-基)甲氧基)-苯基,4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,3-氟-4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,4-(甲氧基-甲酰胺基)-苯基,4-(2-羟基-2-甲基丙氧基)-苯基,4-(甲氧基甲基)-苯基;或者4-乙氧基吡啶-3-基;或者,除了上面列出的,还表示3-氟-4-(2-羟基-2-甲基丙氧基)-苯基,或者6-乙氧基吡啶-3-基;或者
·
3-氟-4-(3-羟基-3-甲基丁氧基)-苯基,3-氯-4-(3-羟基-3-甲基丁氧基)-苯基,2,5-二氟-4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,2,5-二氟-4-((氧杂环丁烷-3-基)甲氧基)-苯基,2,5-二氟-4-(2-羟基-2-甲基丙氧基)-苯基,4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基,4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基,1-甲基-2,3-二氢-1h-吲哚-5-基,4-氨基-苯基,4-(甲基氨基)-苯基,4-(吡咯烷-1-基)-苯基,4-二甲基氨基-苯基,2-氟-苯基,或者2,4-二氟-苯基。
[0086]
19)另一个实施方案涉及根据实施方案1)至2)中任一项的化合物,其中x1表示碳;r1表示甲基,乙基,环丙基或者氯;r2表示氢;以及其中式(i)的片段表示4-羟基苯基,4-甲氧基苯基,3-氯-4-羟基苯基,3-氯-4-甲氧基苯基,3-氟-4-羟基苯基,3-氟-4-甲氧基苯基,2-氟-4-甲氧基-5-氯苯基,2,5-二氟-4-甲氧基苯基,3-氟-4-((氧杂环丁烷-3-基)甲氧基)-苯基,4-((3-氟-氧杂环丁烷-3-基)甲氧基)-苯基,4-(甲氧基-甲酰胺基)-苯基,或者4-(2-羟基-2-甲基丙氧基)-苯基;或者4-乙氧基吡
啶-3-基。
[0087]
20)另一实施方案涉及根据实施方案1)至19)中任一项的化合物,其也是式(ii)化合物(即其中带有连接片段[1,2,3]三唑-1,4-二基的oh基团的不对称碳原子具有式(ii)中描述的绝对构型[即所述不对称碳原子为绝对(r)-构型)]。
[0088]
21)另一实施方案涉及根据实施方案1)或2)中任一项的化合物,其选自由以下项组成的组:(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(r)-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-(1-p-tolyl-1h-[1,2,3]三唑-4-基)-甲醇;(4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯基)-氨基甲酸甲酯;2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚;2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚;2-氯-4-{4-[(s)-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚;[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;(r)-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(6-乙氧基-吡啶-3-基)-1h-[1,2,3]三唑-4-基]-甲醇;
基}-2-氟-苯酚;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丙-2-醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-[1-(3-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;(r)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;以及(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。
[0089]
应理解在实施方案21)中列出的所有化合物尤其是富集的(r)-立体异构形式,以及尤其是基本上纯的(r)-立体异构形式。
[0090]
22)另一实施方案涉及根据实施方案1)的化合物,其选自由以下项组成的组:(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-乙基-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-甲基-1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;[1-(5-氯-2-氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-(6-氯-咪唑并[1,5-a]吡啶-5-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-吡咯烷-1-基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-[1-(4-氨基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇;
2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯酚;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-二甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(1-甲基-2,3-二氢-1h-吲哚-5-基)-1h-[1,2,3]三唑-4-基]-甲醇;4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯酚;4-(2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯氧基)-2-甲基-丁-2-醇;4-(4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丁-2-醇;(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-氯-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,4-二氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇;(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇;1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2,5-二氟-苯氧基)-2-甲基-丙-2-醇;以及(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2-氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇。
[0091]
23)另一实施方案涉及根据实施方案1)或2)中任一项的化合物,其选自由以下项组成的组:(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(r)-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇;2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚;
(r)-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;(r)-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(3-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚;(r)-[1-(3-溴-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲氧基甲基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-[1-(5-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-[1-(3-氯-5-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丙-2-醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-[1-(3-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇;以及(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。24)另一实施方案涉及根据实施方案1)的化合物,其选自由以下项组成的组:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-吡咯烷-1-基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;
(r)-[1-(4-氨基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-二甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(1-甲基-2,3-二氢-1h-吲哚-5-基)-1h-[1,2,3]三唑-4-基]-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇;以及1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2,5-二氟-苯氧基)-2-甲基-丙-2-醇。
[0092]
基于如上文公开的不同实施方案1)至20)的从属性,因此以下实施方案是可能的和预期的,并且特此以个体化的形式具体公开如下:2 1,3 1,3 2 1,5 1,5 2 1,5 3 1,5 3 2 1,7 1,7 2 1,7 3 1,7 3 2 1,7 5 1,7 5 2 1,7 5 3 1,7 5 3 2 1,10 1,10 2 1,10 3 1,10 3 2 1,10 5 1,10 5 2 1,10 5 3 1,10 5 3 2 1,10 7 1,10 7 2 1,10 7 3 1,10 7 3 2 1,10 7 5 1,10 7 5 2 1,10 7 5 3 1,10 7 5 3 2 1,12 1,12 2 1,12 3 1,12 3 2 1,12 5 1,12 5 2 1,12 5 3 1,12 5 3 2 1,12 7 1,12 7 2 1,12 7 3 1,12 7 3 2 1,12 7 5 1,12 7 5 2 1,12 7 5 3 1,12 7 5 3 2 1,12 10 1,12 10 2 1,12 10 3 1,12 10 3 2 1,12 10 5 1,12 10 5 2 1,12 10 5 3 1,12 10 5 3 2 1,12 10 7 1,12 10 7 2 1,12 10 7 3 1,12 10 7 3 2 1,12 10 7 5 1,12 10 7 5 2 1,12 10 7 5 3 1,12 10 7 5 3 2 1,13 1,13 2 1,13 3 1,13 3 2 1,13 5 1,13 5 2 1,13 5 3 1,13 5 3 2 1,13 7 1,13 7 2 1,13 7 3 1,13 7 3 2 1,13 7 5 1,13 7 5 2 1,13 7 5 3 1,13 7 5 3 2 1,13 10 1,13 10 2 1,13 10 3 1,13 10 3 2 1,13 10 5 1,13 10 5 2 1,13 10 5 3 1,13 10 5 3 2 1,13 10 7 1,13 10 7 2 1,13 10 7 3 1,13 10 7 3 2 1,13 10 7 5 1,13 10 7 5 2 1,13 10 7 5 3 1,13 10 7 5 3 2 1,13 12 1,13 12 2 1,13 12 3 1,13 12 3 2 1,13 12 5 1,13 12 5 2 1,13 12 5 3 1,13 12 5 3 2 1,13 12 7 1,13 12 7 2 1,13 12 7 3 1,13 12 7 3 2 1,13 12 7 5 1,13 12 7 5 2 1,13 12 7 5 3 1,13 12 7 5 3 2 1,13 12 10 1,13 12 10 2 1,13 12 10 3 1,13 12 10 3 2 1,13 12 10 5 1,13 12 10 5 2 1,13 12 10 5 3 1,13 12 10 5 3 2 1,13 12 10 7 1,13 12 10 7 2 1,13 12 10 7 3 1,13 12 10 7 3 2 1,13 12 10 7 5 1,13 12 10
7 5 2 1,13 12 10 7 5 3 1,13 12 10 7 5 3 2 1,15 1,15 2 1,15 3 1,15 3 2 1,15 5 1,15 5 2 1,15 5 3 1,15 5 3 2 1,15 7 1,15 7 2 1,15 7 3 1,15 7 3 2 1,15 7 5 1,15 7 5 2 1,15 7 5 3 1,15 7 5 3 2 1,15 10 1,15 10 2 1,15 10 3 1,15 10 3 2 1,15 10 5 1,15 10 5 2 1,15 10 5 3 1,15 10 5 3 2 1,15 10 7 1,15 10 7 2 1,15 10 7 3 1,15 10 7 3 2 1,15 10 7 5 1,15 10 7 5 2 1,15 10 7 5 3 1,15 10 7 5 3 2 1,20 1,20 2 1,20 3 1,20 3 2 1,20 5 1,20 5 2 1,20 5 3 1,20 5 3 2 1,20 7 1,20 7 2 1,20 7 3 1,20 7 3 2 1,20 7 5 1,20 7 5 2 1,20 7 5 3 1,20 7 5 3 2 1,20 10 1,20 10 2 1,20 10 3 1,20 10 3 2 1,20 10 5 1,20 10 5 2 1,20 10 5 3 1,20 10 5 3 2 1,20 10 7 1,20 10 7 2 1,20 10 7 3 1,20 10 7 3 2 1,20 10 7 5 1,20 10 7 5 2 1,20 10 7 5 3 1,20 10 7 5 3 2 1,20 13 1,20 13 2 1,20 13 3 1,20 13 3 2 1,20 13 5 1,20 13 5 2 1,20 13 5 3 1,20 13 5 3 2 1,20 13 7 1,20 13 7 2 1,20 13 7 3 1,20 13 7 3 2 1,20 13 7 5 1,20 13 7 5 2 1,20 13 7 5 3 1,20 13 7 5 3 2 1,20 13 10 1,20 13 10 2 1,20 13 10 3 1,20 13 10 3 2 1,20 13 10 5 1,20 13 10 5 2 1,20 13 10 5 3 1,20 13 10 5 3 2 1,20 13 10 7 1,20 13 10 7 2 1,20 13 10 7 3 1,20 13 10 7 3 2 1,20 13 10 7 5 1,20 13 10 7 5 2 1,20 13 10 7 5 3 1,20 13 10 7 5 3 2 1,20 13 12 1,20 13 12 2 1,20 13 12 3 1,20 13 12 3 2 1,20 13 12 5 1,20 13 12 5 2 1,20 13 12 5 3 1,20 13 12 5 3 2 1,20 13 12 7 1,20 13 12 7 2 1,20 13 12 7 3 1,20 13 12 7 3 2 1,20 13 12 7 5 1,20 13 12 7 5 2 1,20 13 12 7 5 3 1,20 13 12 7 5 3 2 1,20 13 12 10 1,20 13 12 10 2 1,20 13 12 10 3 1,20 13 12 10 3 2 1,20 13 12 10 5 1,20 13 12 10 5 2 1,20 13 12 10 5 3 1,20 13 12 10 5 3 2 1,20 13 12 10 7 1,20 13 12 10 7 2 1,20 13 12 10 7 3 1,20 13 12 10 7 3 2 1,20 13 12 10 7 5 1,20 13 12 10 7 5 2 1,20 13 12 10 7 5 3 1,20 13 12 10 7 5 3 2 1,20 15 1,20 15 2 1,20 15 3 1,20 15 3 2 1,20 15 5 1,20 15 5 2 1,20 15 5 3 1,20 15 5 3 2 1,20 15 7 1,20 15 7 2 1,20 15 7 3 1,20 15 7 3 2 1,20 15 7 5 1,20 15 7 5 2 1,20 15 7 5 3 1,20 15 7 5 3 2 1,20 15 10 1,20 15 10 2 1,20 15 10 3 1,20 15 10 3 2 1,20 15 10 5 1,20 15 10 5 2 1,20 15 10 5 3 1,20 15 10 5 3 2 1,20 15 10 7 1,20 15 10 7 2 1,20 15 10 7 3 1,20 15 10 7 3 2 1,20 15 10 7 5 1,20 15 10 7 5 2 1,20 15 10 7 5 3 1,20 15 10 7 5 3 2 1。
[0093]
在上面的列表中,数字是指根据上文提供的其编号的实施方案,而“ ”表示从属于另一实施方案。不同的个体化实施方案用逗号分隔。换句话说,例如“3 2 1”指的是实施方案3)从属于实施方案2),从属于实施方案1),即实施方案“3 2 1”对应于实施方案1),其特征还在于实施方案2)和3)的特征。
[0094]
式(i)化合物包括这样的化合物,其具有至少一个(即片段[1,2,3]三唑-1,4-二基所连接的不对称碳原子)和可能更多个不对称中心,例如一个或更多个不对称碳原子,其允许以(r)-和(s)-构型存在。式(i)化合物可进一步包括具有允许以z-和e-构型存在的一个或更多个双键的化合物;和/或在环系统中具有允许相对于彼此以顺式-和反式-构型存在的取代基的化合物。式(i)化合物因此可以作为立体异构体的混合物或优选以立体异构体富集的形式存在,尤其是作为基本上纯的立体异构体存在。在式(ii)中,除了片段[1,2,3]三唑-1,4-二基所连接并具有式(ii)所示的定义的绝对构型的不对称碳原子外,所述式的
化合物可以包含允许以(r)-和(s)-构型存在的另外的不对称碳原子。式(ii)化合物因此可以作为立体异构体的混合物或优选作为纯立体异构体存在。立体异构体的混合物可以以本领域技术人员已知的方式分离。
[0095]
在本专利申请中,虚线(例如)显示所绘游离基的连接点。
[0096]
在特定化合物(或通用结构)被指定为(r)-或(s)-对映体的情况下,则此类指定应理解为指的是富集的,尤其是基本纯的对映体异构体形式的相应化合物(或通用结构)。同样,在化合物中的特定不对称中心被指定为处于(r)-或(s)-构型或处于某种相对构型的情况下,则此类指定应理解为是指相对于所述不对称中心的相应构型是富集的,尤其是基本上纯的形式。类似地,顺式或反式指定应理解为是指富集的,尤其是基本上纯的形式的相应立体异构体。同样,在特定化合物(或通用结构)被指定为z-或e-立体异构体的情况下(或在化合物中的特定双键被指定为z-或e-构型的情况下),则此类指定应理解为是指以富集的,尤其是基本上纯的立体异构形式的相应化合物(或通式结构)(或相对于双键的相应构型是富集的,尤其是基本上纯的形式的化合物)。
[0097]
当在立体异构体的上下文中使用时,术语“富集的”在本发明的上下文中应理解为意指相应立体异构体相对于相应的其他立体异构体/相应的其他立体异构体的整体以至少70:30的比率,尤其是以至少90:10的比率存在(即,纯度为至少70重量%,尤其是至少90重量%)。
[0098]
当在立体异构体的上下文中使用时,术语“基本上纯的”在本发明的上下文中应理解为意指相应立体异构体相对于相应的其他立体异构体/相应的其他立体异构体的整体以至少95重量%,尤其是至少99重量%的纯度存在。
[0099]
本发明还包括同位素标记的尤其是2h(氘)标记的式(i)化合物,该化合物与式(i)化合物相同,不同之处在于一个或多个原子各自被原子数相同但原子质量与自然界中通常发现的原子质量不同的原子取代。同位素标记的尤其是2h(氘)标记的式(i)化合物及其盐在本发明的范围内。用更重的同位素2h(氘)取代氢可导致更高的代谢稳定性,例如引起体内半衰期增加或剂量要求减少,或可导致代谢改变,例如引起安全配置文件改进。在本发明的一个实施方案中,式(i)化合物没有进行同位素标记,或者它们仅用一个或多个氘原子标记。在一个子实施方案中,式(i)的化合物根本没有进行同位素标记。同位素标记的式(i)化合物可以以类似于下文描述的方法来制备,但是使用合适试剂或起始材料的适当同位素变化。
[0100]
在复数形式用于化合物、盐、药物组合物、疾病时,这旨在也意指单一化合物、盐、组合物和疾病。
[0101]
本文通篇使用的术语“调节”或“调节剂”涉及酶或受体活性的增加或减少。术语ido和/或tdo抑制剂是指能够抑制ido和/或tdo酶活性的药剂。
[0102]
上文或下文中对式(i)化合物的任何提及应理解为还指式(i)化合物的盐,尤其是药学上可接受的盐,视情况而定。
[0103]
术语“药学上可接受的盐”是指保留主题化合物的所需生物活性并且表现出最小的不希望的毒理学作用的盐。这种盐包括无机或有机酸和/或碱加成盐,具体取决于主题化
合物中碱性和/或酸性基团的存在。参见例如
‘
handbook of pharmaceutical salts.properties,selection and use.’,p.heinrich stahl,camille g.wermuth(eds.),wiley-vch,2008,以及
‘
pharmaceutical salts and co-crystals’,johan wouters and luc qu
éré
(eds.),rsc publishing,2012。
[0104]
式(i)化合物及其药学上可接受的盐可用作药物,例如以用于肠内(例如尤其是口服)或肠胃外(包括局部应用或吸入)施用的药物组合物的形式。
[0105]
式(i)化合物适于抑制ido和/或tdo酶,以及预防和/或治疗哺乳动物(例如尤其是人类)中与ido和/或tdo酶相关的疾病或病症(例如尤其是癌症)。
[0106]
药物组合物的生产可以以本领域任何技术人员熟悉的方式通过将所述的式(i)化合物或其药学上可接受的盐,任选地与其他有治疗价值的物质组合,与合适的、无毒的、惰性的、药学上可接受的固体或液体载体材料以及如果需要,还有常用的药物佐剂一起制成盖仑制剂施用形式进行(参见例如remington,the science and practice of pharmacy,第21版(2005),第5部分,“pharmaceutical manufacturing”[由lippincott williams&wilkins出版])。
[0107]
在本发明的一个优选实施方案中,包含的施用量介于每天1mg和1000mg之间,特别是介于每天5mg和500mg之间,更特别是介于每天25mg和400mg之间,尤其是介于每天50mg和200mg之间。
[0108]
每当使用“介于
……
之间”一词来描述数字范围时,应理解所指示范围的端点明确包括在该范围内。例如:如果温度范围被描述为介于40℃和80℃之间,这意指端点40℃和80℃包括在该范围内;或者,如果变量被定义为介于1和4之间的整数,这意指该变量是整数1、2、3或4。
[0109]
除非用于温度,否则置于数值“x”之前的术语“约”在本技术中是指从x减去x的10%延伸到x加上x的10%的区间,并且优选地是从x减去x的5%延伸到x加上x的5%的区间。在温度的特殊情况下,放在温度“y”之前的术语“约”在当前申请中是指从温度y减去10℃延伸到y加上10℃的区间,并且优选指从y减去5℃延伸到y加上5℃的区间。
[0110]
为避免任何疑问,如果化合物被描述为可用于预防或治疗某些疾病,则此类化合物同样适用于制备用于预防或治疗所述疾病的药物。
[0111]
本发明还涉及一种用于预防或治疗上文提及的疾病或紊乱的方法,其包括将药学活性量的式(i)化合物单独或与其他药理学活性化合物和/或疗法组合地施用给受试者。
[0112]
术语“预防”的含义也可以理解为“防范”(prophylaxis)。
[0113]
一种或多种式(i)化合物可用于预防和/或治疗与ido和/或tdo酶相关的疾病或紊乱;例如尤其是癌症。
[0114]
癌症可被定义为包括皮肤癌,包括黑色素瘤;转移性黑色素瘤;肺癌,包括非小细胞肺癌;膀胱癌,包括膀胱三角癌(urinary bladder cancer);尿道上皮细胞癌;肾癌,包括肾细胞癌;转移性肾细胞癌;转移性肾透明细胞癌;胃肠癌,包括结直肠癌;转移性结直肠癌;家族性腺瘤性息肉病(fap);食道癌;胃癌;胆囊癌;胆管癌;肝细胞癌;和胰腺癌,例如胰腺腺癌或胰腺导管癌;子宫内膜癌;卵巢癌;宫颈癌;成神经细胞瘤;前列腺癌,包括去势抵抗性前列腺癌;脑肿瘤,包括脑转移瘤、恶性胶质瘤、多形性胶质母细胞瘤、髓母细胞瘤、脑膜瘤、神经母细胞瘤、星形细胞瘤;乳腺癌,包括三阴性乳腺癌;口腔肿瘤;鼻咽肿瘤;胸癌;
头颈癌;间皮瘤;白血病,包括急性髓性白血病、成人t细胞白血病;癌;腺癌;甲状腺癌,包括甲状腺乳头状癌;绒毛膜癌;肉瘤,包括尤文氏肉瘤(ewing’s sarcoma);骨肉瘤;横纹肌肉瘤;卡波西肉瘤;淋巴瘤,包括伯奇氏淋巴瘤(burkitt’s lymphoma)、霍奇金氏淋巴瘤(hodgkin’s lymphoma)、malt淋巴瘤;多发性骨髓瘤;和病毒诱导的肿瘤。
[0115]
此外,癌症可以定义为包括脑癌、皮肤癌、膀胱癌、卵巢癌、乳腺癌、胃癌、胰腺癌、前列腺癌、结肠癌、血癌、肺癌和骨癌。此类癌症类型的示例包括神经母细胞瘤;肠癌,如直肠癌、结肠癌、家族性腺瘤性息肉病和遗传性非息肉病性结直肠癌;食道癌、唇癌、喉癌、下咽癌、舌癌、唾液腺癌、胃癌、腺癌、甲状腺髓样癌、甲状腺乳头状癌、肾癌、肾实质癌、卵巢癌、宫颈癌、子宫体癌、子宫内膜癌、绒毛膜癌、胰腺癌、前列腺癌、睾丸癌、乳腺癌、泌尿系统癌、黑色素瘤;脑肿瘤,如胶质母细胞瘤、星形细胞瘤、脑膜瘤、髓母细胞瘤和外周神经外胚层肿瘤;霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、伯奇氏淋巴瘤、急性淋巴性白血病(all)、慢性淋巴性白血病(cll)、急性髓性白血病(aml)、慢性粒细胞白血病(cml),成人t细胞白血病淋巴瘤、弥漫性大b细胞淋巴瘤(dlbcl)、肝细胞癌、胆囊癌、支气管癌、小细胞肺癌、非小细胞肺癌、多发性骨髓瘤、基底细胞瘤、畸胎瘤、视网膜母细胞瘤、脉络膜黑色素瘤、精原细胞瘤、横纹肌肉瘤、颅咽管瘤、骨肉瘤、软骨肉瘤、肌肉瘤、脂肪肉瘤、纤维肉瘤、尤文氏肉瘤和浆细胞瘤。
[0116]
癌症可以尤其是定义为包括皮肤癌,特别是晚期黑色素瘤和默克尔细胞癌;肺癌,包括非小细胞肺癌;膀胱癌;头颈癌;肾细胞癌;霍奇金氏淋巴瘤;宫颈癌;子宫内膜癌;乳腺癌;结肠癌;胃肠道间质瘤;胰腺癌;前列腺癌;白血病,包括急性髓性白血病;淋巴瘤;胃癌;卵巢癌;食道癌;肝癌;和脑肿瘤,特别是胶质母细胞瘤、间皮瘤、神经母细胞瘤;肉瘤,特别是高级骨肉瘤、星形细胞瘤、骨髓瘤。
[0117]
癌症可以尤其是定义为包括具有特定遗传特征的实体瘤,称为错配修复缺陷和高度微卫星不稳定性(microsatellite instability);皮肤癌,特别是晚期黑色素瘤、默克尔细胞癌和皮肤鳞状细胞癌;肺癌[尤其是非小细胞肺癌(nsclc)];膀胱癌;晚期宫颈癌;晚期胃癌;头颈癌;肾细胞癌;具有错配修复缺陷(dmmr)或高度微卫星不稳定性(msi-h)的转移性结直肠癌;原发性纵隔大b细胞淋巴瘤;晚期肝癌;和霍奇金氏淋巴瘤。
[0118]
一种或多种式(i)化合物可单独或与其他药理学活性化合物和/或疗法组合地用于预防和/或治疗任何癌症,尤其是上文提及的癌症。
[0119]
除了癌症,尤其是如上所列的癌症之外,与ido和/或tdo酶相关的其他疾病或紊乱可定义为包括神经退行性紊乱,例如帕金森氏病、阿尔茨海默氏病、亨廷顿氏病和肌萎缩侧索硬化症;中枢神经系统(cns)紊乱,例如精神紊乱(精神分裂症、抑郁症);疼痛;中风;癫痫;慢性传染病,如hiv(aids,包括其表现,如恶病质、痴呆和腹泻)和hcv;由各种细菌(如衣原体菌株和肠道致病菌株)、寄生虫(如锥虫、利什曼原虫、疟原虫)或病毒(如流感、人乳头瘤病毒、巨细胞病毒、单纯疱疹病毒、爱泼斯坦-巴尔病毒、脊髓灰质炎病毒、水痘带状疱疹病毒和柯萨奇病毒)引起的感染和炎症;以及其他感染(例如皮肤感染、胃肠道感染、尿路感染、生殖泌尿系统感染、全身感染);自身免疫性疾病,包括哮喘、类风湿性关节炎、多发性硬化症、过敏性炎症、炎症性肠炎疾病、银屑病和系统性红斑狼疮、器官移植(例如器官移植排斥);代谢紊乱,如肥胖、2型糖尿病和/或脂肪酸肝病;白内障;子宫内膜异位症;避孕和堕胎。
[0120]
进一步的自身免疫疾病包括胶原病,例如类风湿性关节炎、系统性红斑狼疮、夏普综合征(sharp's syndrome)、crest综合征(钙质沉着症、雷诺氏综合征、食道运动障碍、毛细血管扩张症)、皮肌炎、血管炎[莫布斯韦格纳氏(morbus wegener's)]和修格兰氏综合征(sjogren's syndrome);肾病,例如古德帕斯彻氏综合征(goodpasture's syndrome)快速进展性肾小球肾炎和ii型膜增生性肾小球肾炎;内分泌疾病,如i型糖尿病、自身免疫性多内分泌疾病-念珠菌病外胚层营养不良(apeced)、自身免疫性甲状旁腺病(autoimmune parathyroidism)、恶性贫血、性腺功能不全、特发性莫布斯爱迪生氏甲状腺功能亢进症、桥本甲状腺炎和原发性粘液性水肿;皮肤疾病,例如寻常型天疱疮、大疱性类天疱疮、妊娠疱疹、大疱性表皮松解症和重症多形红斑;肝病,例如原发性胆汁性肝硬化、自身免疫性胆管炎、自身免疫性1型肝炎、自身免疫性2型肝炎、原发性硬化性胆管炎;神经元疾病,例如多发性硬化症、重症肌无力、肌无力兰伯特-伊顿综合征、获得性神经肌切断、格林-巴利综合征(穆勒-费舍尔综合征)、僵人综合征、小脑变性、共济失调、眼阵挛、感觉神经病和失弛缓症;血液病,如自身免疫性溶血性贫血、特发性血小板减少性紫癜[莫布斯韦尔霍夫(morbus werlhof)],以及与自身免疫反应相关的传染病,如艾滋病、疟疾和查加斯病(chagas disease)。
[0121]
术语“放疗”或“放射疗法”或“放射肿瘤学”,是指电离放射在预防(辅助治疗)和/或治疗癌症中的医学用途;包括外部和内部放射疗法。
[0122]
术语“靶向疗法”是指用一种或多种抗肿瘤剂(例如作用于特定类型的癌细胞或基质细胞的小分子或抗体)来预防/防范(辅助治疗)和/或治疗癌症。一些靶向疗法阻断某些酶、蛋白质或其他参与癌细胞生长和扩散的分子的作用。其他类型的靶向疗法帮助免疫系统杀死癌细胞(免疫疗法);或将有毒物质直接递送到癌细胞并杀死它们。特别适合与本发明化合物组合的靶向疗法的一个示例是免疫疗法,尤其是靶向程序性死亡1(pd-1)受体或其配体pd-l1的免疫疗法(feig c等人,pnas 2013)。
[0123]
免疫疗法还指(i)刺激(包括共刺激)受体的激动剂或(ii)t细胞上抑制(包括共抑制)信号的拮抗剂,这两者都会导致扩增抗原特异性t细胞反应(通常称为免疫检查点调节剂)。某些刺激和抑制分子是免疫球蛋白超家族(igsf)的成员。与共刺激或共抑制受体结合的一个重要的膜结合配体家族是b7家族,其包括b7-1,b7-2,b7-hl(pd-ll),b7-dc(pd-l2),b7-h2(icos-l),b7-h3,b7-h4,b7-h5(vista)和b7-h6。与共刺激或共抑制受体结合的另一个膜结合配体家族是与同源tnf受体家族成员结合的tnf分子家族,其包括cd40和cd40l,ox-40,ox-40l,cd70,cd27l,cd30,cd30l,4-lbbl,cd137(4-lbb),trail/apo2-l,trailr1/dr4,trailr2/dr5,trailr3,trailr4,opg,rank,rankl,tweakr/fnl4,tweak,baffr,edar,xedar,taci,april,bcma,ltpr,light,dcr3,hvem,vegi/tlla,tramp/dr3,edar,edal,xedar,eda2,tnfrl,淋巴毒素a/tnfp,tnfr2,tnfa,ltpr,淋巴毒素a 1p2,fas,fasl,relt,dr6,troy,ngfr。
[0124]
当与式(i)化合物组合使用时,术语“靶向治疗”尤其是指药剂,例如:a)表皮生长因子受体(egfr)抑制剂或阻断抗体(例如吉非替尼、厄洛替尼、阿法替尼、埃克替尼、拉帕替尼、帕尼单抗、扎鲁木单抗、尼妥珠单抗、马妥珠单抗和西妥昔单抗)以及曲妥珠单抗(赫赛汀);b)ras/raf/mek途径抑制剂[例如维莫非尼、索拉非尼、达帕菲尼、gdc-0879、plx-4720、lgx818、rg7304、曲美替尼(gsk1120212)、考比替尼(gdc-0973/xl518)、比美替尼(mek162,
arry-162),司美替尼(azd6244)];c)janus激酶(jak)抑制剂[例如鲁索替尼、伊他替尼、莫洛替尼(momelotinib)];d)芳香酶抑制剂[例如依西美坦、来曲唑、阿那曲唑、伏氯唑(vorozole)、福美司坦、法倔唑];e)信号转导抑制剂(sti)。“信号转导抑制剂”是选择性抑制癌细胞正常功能中信号途径中一个或多个重要步骤从而导致细胞凋亡的药剂。合适的stis包括但不限于:(i)bcr/abl激酶抑制剂,例如sti 571达沙替尼;(ii)表皮生长因子(egf)受体抑制剂,例如激酶抑制剂(ssi-774)和抗体(imclone:c225[goldstein等人,clin.cancer res.,1:1311-1318(1995)],以及abgenix:abx-egf);(iii)her-2/neu受体抑制剂,例如法尼基转移酶抑制剂(fti),例如l-744,832(kohl等人,nat.med.,1(8):792-797(1995));(iv)akt家族激酶或akt途径的抑制剂,例如雷帕霉素[参见,例如sekulic等人,cancer res.,60:3504-3513(2000)];(v)细胞周期激酶抑制剂,例如夫拉平度(flavopiridol)和ucn-o1[参见例如sausville,curr.med.chem.anti-cane.agents,3:47-56(2003)];以及(vi)磷脂酰肌醇激酶抑制剂,例如ly294002[参见,例如vlahos等人,j biol.chem.,269:5241-5248(1994)];f)血管生成抑制剂,尤其是vegf信号抑制剂,如贝伐单抗(阿瓦斯汀)、雷莫芦单抗、索拉非尼或阿西替尼;g)免疫检查点抑制剂(例如:抗pd1抗体,例如帕博利珠单抗[兰洛利珠单抗(lambrolizumab),mk-3475]、纳武单抗(nivolumab)、匹地利珠单抗(pidilizumab)(ct-011),amp-514/medi0680、pdr001、shr-1210;regn2810、bgba317、pf-06801591、mga-012、tsr042、js-001、bcd100、ibi-308、bi-754091;靶向pd-1的融合蛋白,例如amp-224;小分子抗pd1剂,例如wo2015/033299、wo2015/044900和wo2015/034820中公开的化合物;抗pd1l抗体,例如bms-936559、阿特珠单抗(mpdl3280a,rg7446)、阿维单抗(msb0010718c)、度伐利尤单抗(medi4736);抗pdl2抗体,例如amp224;抗ctla-4抗体,例如伊匹单抗、曲美木单抗;抗淋巴细胞激活基因3(lag-3)抗体,如瑞拉利单抗(bms-986016),imp701,imp731,mk-4280,immufact imp321;抗t细胞免疫球蛋白粘蛋白-3(tim-3)抗体,例如mbg453,tsr-022;具有ig和itim域(tigit)抗体的抗t细胞免疫受体,例如rg6058(抗tigit,mtig7192a);抗杀伤细胞免疫球蛋白样受体(kir),例如利丽单抗(lirilumab)(iph2102/bms-986015)、半乳糖凝集素(例如半乳糖凝集素-1、半乳糖凝集素-9)、btla的拮抗剂;h)疫苗接种方法[例如树突细胞疫苗接种、dna、肽或蛋白质疫苗接种(例如使用gp100肽或mage-a3肽)以及重组病毒];i)重新引入患者来源或同种异体(非自身)的癌细胞,其经基因改造以分泌免疫调节因子,例如粒细胞单核细胞集落刺激因子(gmcsf)基因转染肿瘤细胞疫苗(gvax)或fms相关酪氨酸激酶3(flt-3)配体基因转染肿瘤细胞疫苗(fvax),或基于toll样受体增强型gm-csf肿瘤的疫苗(tegvax);j)基于t细胞的过继免疫疗法,包括嵌合抗原受体(car)工程化t细胞(例如ctl019);k)基于细胞因子或免疫细胞因子的疗法(例如干扰素α、干扰素β、干扰素γ、白介素2、白介素6、白介素10、白介素15、tgf-β);l)toll样受体(tlr)激动剂[例如瑞喹莫特、咪喹莫特、莫托莫特(motolimod)、吡喃葡萄糖基脂质a、cpg寡脱氧核苷酸];m)沙利度胺类似物(例如来那度胺、泊马度胺);n)t细胞共刺激受体的激活剂(例如抗cd137/4-1bb抗体,如bms-663513(乌瑞芦单抗)、乌托鲁单抗(utomilumab)(pf-05082566);抗ox40/cd134(肿瘤坏死因子受体超家族,成员4)(如rg7888(moxr0916),9b12;medi6469,gsk3174998,medi6383,medi0562),抗ox40-配体/cd252;抗糖皮质激素诱导的tnfr家族相关基因(gitr)(如trx518,medi1873,mk-4166,bms-986156,bms-986153)、抗cd40(tnf受体超家族成员5)
抗体[如达西组单抗(dacetuzumab)(sgn-40),hcd122,cp-870,893,rg7876,adc-1013,apx005m,sea-cd40];抗cd40-配体抗体(例如bg9588);抗cd27抗体,例如伐立鲁单抗(varlilumab);抗cd28抗体;抗icos抗体;o)结合肿瘤特异性抗原以及t细胞表面标志物的分子,例如双特异性抗体或抗体片段、抗体模拟蛋白,例如设计的锚蛋白重复蛋白(darpins)、双特异性t细胞接合剂(bite,例如amg103、amg330);p)靶向集落刺激因子1受体(csf-1r)的抗体或小分子量抑制剂[例如依米妥珠单抗(emactuzumab)(rg7155)、卡比利珠单抗(cabiralizumab)(fpa-008)、plx3397];q)靶向自然杀伤细胞上免疫细胞检查点的药物,例如针对杀伤细胞免疫球蛋白样受体(kir)的抗体,例如利丽单抗(iph2102/bms-986015);r)靶向腺苷受体或将三磷酸腺苷(atp)转化为腺苷的外切核酸酶cd39和cd73的药剂,例如medi9447(抗cd73抗体)、pbf-509;cpi-444(腺苷a2a受体拮抗剂);s)包括ccr2或ccr4的趋化因子受体的拮抗剂;t)补体系统的调节剂;v)消耗或抑制t调节细胞[例如,使用抗cd25单克隆抗体(例如达利珠单抗)或通过离体抗cd25珠消耗]或逆转/防止t细胞无反应性或耗竭的药剂。
[0125]
当与式(i)化合物组合使用时,优选免疫检查点抑制剂,例如f)下所列的那些,尤其是靶向程序性细胞死亡受体1(pd-1受体)或其配体pd-l1的那些。
[0126]
术语“化学疗法”是指用一种或多种细胞毒性抗肿瘤剂(“细胞毒性化学治疗剂”)治疗癌症。化学疗法通常与其他癌症治疗(例如放射疗法或手术)结合使用。该术语尤其是指通过杀死快速分裂的细胞起作用的常规化学治疗剂,这是大多数癌细胞的主要特性之一。化学疗法可一次使用一种药物(单剂化学疗法)或一次使用几种药物(联合化学疗法或多化学疗法)。使用仅在光暴露时转化为细胞毒性活性的药物的化学疗法称为光化学疗法或光动力疗法。
[0127]
如本文所用,术语“细胞毒性化学治疗剂”或“化学治疗剂”是指诱导细胞凋亡或坏死细胞死亡的活性抗肿瘤剂。当与式(i)化合物组合使用时,该术语尤其是指常规细胞毒性化学治疗剂,例如:1)烷化剂(包括但不限于氮芥、乙烯亚胺衍生物、烷基磺酸盐、亚硝基脲和三氮烯),例如尿嘧啶芥、甲氯乙胺、苯丁酸氮芥、环磷酰胺、异环磷酰胺、链脲佐菌素、卡莫司汀、洛莫司汀、美法仑、白消安、丙卡巴肼、达卡巴嗪、替莫唑胺、哌溴甘、三亚乙基三聚氰胺、三亚乙基硫代磷胺、噻替哌或阿维甲胺;特别是替莫唑胺);2)铂类药物(如顺铂、卡铂或奥沙利铂);3)抗代谢药物(例如5-氟尿嘧啶、氟尿苷、喷司他汀、卡培他滨、6-巯基嘌呤、甲氨蝶呤、吉西他滨、阿糖胞苷、氟达拉滨或培美曲塞);4)抗肿瘤抗生素(例如柔红霉素、多柔比星、表柔比星、伊达比星、放线菌素-d、博来霉素、丝裂霉素-c或米托蒽醌);5)有丝分裂抑制剂(例如紫杉醇、多西他赛、伊沙匹隆、长春碱、长春新碱、长春瑞滨、长春地辛或雌莫司汀);或6)拓扑异构酶抑制剂(例如依托泊苷、替尼泊苷、拓扑替康、伊立替康、二氟替康或依洛替康)。细胞毒性剂,例如生物反应调节剂;生长抑制剂;抗激素治疗剂;亚叶酸;替加氟;和造血生长因子也是合适的。
[0128]
当与式(i)化合物组合使用时,优选的细胞毒性化学治疗剂是上述烷化剂(尤其是福替莫司汀、环磷酰胺、异环磷酰胺、卡莫司汀、达卡巴嗪及其前药,例如尤其是替莫唑胺或这些化合物的药学上可接受的盐;特别是替莫唑胺);有丝分裂抑制剂(特别是紫杉醇、多西他赛、伊沙匹隆;或这些化合物的药学上可接受的盐;特别是紫杉醇);铂类药物(尤其是顺铂、奥沙利铂和卡铂);以及依托泊苷和吉西他滨。1)化学疗法可能是出于治愈目的给出,或
者其可旨在延长生命或缓解症状。2)联合模式化学疗法是将药物与其他癌症治疗(例如放射治疗或手术)一起使用。3)诱导化学疗法是化学疗法药物对癌症的一线治疗。这种类型的化学疗法用于治疗目的。4)缓解后给出巩固化学疗法以便延长总体无病时间并提高总体生存率。施用的药物与达到缓解的药物相同。5)强化化学疗法与巩固化学疗法相同,但使用的药物与诱导化学疗法不同。6)联合化学疗法涉及同时使用多种不同药物治疗患者。药物的机制和副作用不相同。最大的优势是最大限度地减少对任何一种药剂产生耐药性的机会。此外,药物通常可以以较低的剂量使用,从而降低毒性。7)新辅助化学疗法是在手术等局部治疗之前给出的,并且旨在缩小原发肿瘤。它还用于具有微转移疾病高风险的癌症。8)局部治疗(放疗或手术)后给出辅助化学疗法。其在几乎没有癌症存在的证据时可以使用,但有复发的风险。它还可用于杀死已扩散到身体其他部位的任何癌细胞。这些微转移可以用辅助化学疗法进行治疗,并可以降低由这些播散细胞引起的复发率。9)维持化学疗法是一种延长缓解期的重复低剂量治疗。10)挽救性化学疗法或姑息性化学疗法并非以治愈为目的,而只是为了减少肿瘤负荷并延长预期寿命。对于这些方案,通常预期有更好的毒性特征。
[0129]
式(i)化合物的制备:
[0130]
式(i)化合物可以通过以下给出的方法、通过实施例中给出的方法或通过类似的方法制备。最佳反应条件可随所用的特定反应物或溶剂而变化,但此类条件可由本领域技术人员通过常规优化程序确定。
[0131]
在以下方案中,通用基团r1,r2,r3,r4,x1,x2以及n如对式(i)化合物所定义的那样。为免生疑问,x是指卤素,或者当包含在杂环(例如x1或者x2)中时,它是指氮或碳。在某些情况下,所述通用基团可能与方案中所示的组装不相容,或者将需要使用保护基团(pg)。保护基团的使用是本领域公知的(参见例如“protective groups in organic synthesis",t.w.greene,p.g.m.wuts,wiley-interscience,1999)。为了讨论的目的,将假设这种保护基团必要时在适当的位置。在某些情况下,最终产物可以被进一步修饰(例如,通过操纵取代基)以得到新的最终产物。这些操纵可以包括但不限于本领域技术人员公知的还原、氧化、烷化、酰化和水解反应。所得化合物也可以以本领域已知的方式转化成盐,尤其是药学上可接受的盐。
[0132]
本发明的式(i)化合物可根据如下概述的一般合成方案制备。
[0133]
式(i)化合物的合成
[0134]
通常,其中r2=h的式(i)化合物是通过使用标准铜催化叠氮化物-炔烃环加成(cuaac)条件,例如在室温(rt)下在极性溶剂(如dmf和水)的混合物中的硫酸铜、抗坏血酸钠盐,进行炔丙醇中间体1与叠氮化物2的反应得到的(方案1)。可以使用标准方法(例如,从卤化物、硼酸或胺)制备叠氮化物。
方案1:制备醇3的cuaac反应
[0135]
对映体纯醇3a和3b可以通过cuaac的所得产物3的手性分离获得(方案1)。
[0136]
替代地,可以通过对映体纯炔丙醇1a和合适的叠氮化物2的cuaac反应获得其中r2=h的式(i)对映体纯化合物(方案2)。对映体纯炔丙醇可通过外消旋炔丙醇1的手性分离获得。方案2:对映体纯炔丙醇1a和叠氮化物2之间的反应
[0137]
替代地,其中r2=me的式(i)化合物可通过溴化镁物质5与醛4的反应获得(方案3)。格氏试剂5可以使用合适的叠氮化物2和丙炔基溴化镁原位制备。方案3:醛4上的格氏加成
[0138]
替代地,式(i)化合物可以通过从中间体6和合适的醛7开始的去质子化/加成序列获得(方案4,例如其中x1=碳)。化合物6可以使用在溶剂(如thf)中的碱(如n-buli)以及
在-78℃至0℃的温度范围内去质子化,所得阴离子可以用在溶剂(如thf)中的醛7以及在-78℃至rt的温度范围内处理。然后可以使用手性制备型hplc分离外消旋化合物以得到醇3a和3b。方案4:去质子化/加成序列
[0139]
替代地,保护/导向基团策略可用于制备式(i)化合物(方案5)。在大约-78℃或更高的温度下,使用在溶剂(如thf)中的碱(如n-buli或lda)对中间体8进行去质子化,随后加入合适的醛7得到醇9。在范围从rt至90℃的温度下使用在溶剂(例如乙醇和水的混合物)中的催化剂(例如雷尼镍)进行硫醚官能团的去除以得到化合物3。然后可以使用手性制备型hplc分离外消旋化合物以得到醇3a和3b。方案5:定向去质子化/加成序列
[0140]
替代地,式(i)化合物可以通过在范围从rt到100℃的温度下在溶剂(例如dmf)中在nai和碱(例如k2co3)的存在下使用标准烷基化条件,例如卤化物(r
4-x)进行10的烷基化(方案6)来制备。然后可以使用手性制备型hplc分离外消旋化合物11,得到醇11a和11b。
方案6:羟基芳基化合物的烷基化
[0141]
醛4和炔丙醇中间体1的合成
[0142]
炔丙醇1可以使用方案7中描述的合成顺序来制备(例如其中x1=碳)。方案7:炔丙醇1的合成
[0143]
从羧酸12开始,使用标准酯化条件,例如亚硫酰氯在溶剂(例如etoh)中并在rt左右的温度下将酸官能团转化为相应的甲酯或乙酯。然后使用在溶剂(如dmf)中的zn(cn)2作为氰化物源、钯催化剂[如pd2(dba)3]和配体(如dppf),以及在范围从rt至110℃的温度下通过金属催化的氰化将2-cl杂芳基13转化为相应的2-cn杂芳基14。在氢气氛下(例如在hcube-pro装置中生成)在溶剂(如etoh)中并在二碳酸二叔丁酯的存在下使用阮内镍将腈官能团还原为相应的胺,以便生成boc-保护的胺15。然后除去保护基团并且在碱(例如dipea)的存在下在范围从rt至50℃的温度下使用甲酰化剂(例如甲酸乙酯)将伯胺转化为相应的甲酰化胺。然后可以使用脱水剂(例如pocl3)在溶剂(例如甲苯或dcm)中并在范围从0℃至110℃的温度下将甲酰化胺16环化为双环系统17。使用还原剂(如nabh4)在溶剂(如thf、meoh或etoh或混合物)中将酯官能团还原为相应的醇。使用氧化剂(如戴斯-马丁高碘烷或mno2,在溶剂(如dcm或ch3cn)中并在范围从0℃至70℃的温度下将醇18氧化为醛4。替代地,醛4可以经由weinreb酰胺22从酯17获得(方案8)。醛4可以在范围从0℃至rt的温度下在溶剂(如thf)中使用乙炔基溴化镁经由格氏反应转化为炔丙醇1。
[0144]
炔丙醇1的替代合成途径在方案8中示出(例如其中x1=氮)。
方案8:炔丙醇1的替代合成
[0145]
市售的(或通过相应羧酸的酯化制备的,或通过fgi以引入r1取代基的)溴化物19被转化为溴甲基21,例如在溶剂(如ccl4)中在aibn的存在下并在范围从rt至80℃的温度下使用标准的自由基溴化反应条件,如nbs经由甲基衍生物20的溴化进行。进而甲基衍生物可以通过在溶剂(如二噁烷)中并在碱(如k2co3)的存在下使用例如三甲基环硼氧烷、钯基催化剂[如pd(dppf)2]以及在范围从rt到110℃的温度下进行交叉偶联反应获得。然后可以使用两步一锅程序将溴化物21转化为甲酰胺16,该程序包括通过在溶剂(如dmf)中并在rt左右的温度下与二甲酰胺钠的反应形成双甲酰胺,然后在碱性条件下使用例如nahco3作为碱水解甲酰基基团之一。类似于方案7中所描述的,然后可以在溶剂(如甲苯或dcm)中使用脱水剂(例如pocl3)并在范围从0℃至110℃的温度下将甲酰化胺16环化为双环系统17。然后通过在范围从0℃至50℃的温度下在溶剂(如thf和水)的混合物中使用碱(如lioh)进行皂化,然后通过在范围从0℃至rt的温度下在溶剂(如dmf)中在碱(如dipea)的存在下使用n,o-二甲基羟胺偶联剂(如hatu)进行标准酰胺偶联,将酯官能团转化为weinreb酰胺22。weinreb酰胺22可以通过在范围从20℃至rt的温度下在溶剂(如thf或甲苯)中使用还原剂(如dibalh)还原为醛4。醛4可以在范围从0℃至rt的温度下在溶剂(如thf)中使用乙炔基溴化镁经由格氏反应转化为炔丙醇1。
[0146]
替代地,r1可以在方案7和8中所示的合成的任何适当阶段相互转化(cl至环丙基或甲基或乙基)。例如,醛4可以使用标准金属催化的偶联反应,例如suzuki交叉偶联反应(方案9)转化为相应的中间体,其中r1是烷基/环烷基基团(例如环丙基、甲基或乙基)。替代地,可以使用金属催化的偶联反应,如negishi交叉偶联反应(方案10)将r1取代基引入到酯17上。方案9:将r1=cl转化为r1=烷基并随后形成炔丙醇1(例如其中x1=碳)方案10:r1=cl转化为r1=烷基/环烷基并随后形成炔丙醇1(例如其中x1=氮)
[0147]
叠氮化物2的合成
[0148]
叠氮化物,如果不是市售的,可以使用标准方法制备,例如从溴化物、硼酸(chan-lam偶联)或胺(sandmeyer反应)开始,或通过适当取代的叠氮化物的fgi制备。
[0149]
替代地,叠氮化物24可以在范围从室温至50℃的温度下在溶剂(例如dmf)中在nai和碱(如k2co3)的存在下通过使用标准烷基化条件,例如卤化物(r4-x)对23的烷基化(方案11)来制备。
方案11:羟基芳基叠氮化物的烷基化
[0150]
中间体6的合成
[0151]
伯胺26(市售或通过从相应的羧酸或酯25(方案12)或从相应的醇的官能团互相转化而制备)被转化为相应的甲酰化胺27,其可在溶剂(如甲苯或dcm)中使用脱水剂(如pocl3)并在范围从0℃至110℃的温度下环化为双环系统6。方案12:中间体6的合成
[0152]
中间体8的合成
[0153]
胺26可以在溶剂(例如meoh)中在碱(例如et3n)的存在下并在范围从0℃至70℃的温度下使用标准条件,例如二硫化碳被内环化成硫醇28(方案13)。硫醇28可以在溶剂(例如丙酮)中在碱(例如k2co3)的存在下并在范围从rt至45℃的温度下使用标准烷基化条件(例如eti)进行烷基化,得到中间体8。方案13:中间体8的合成
[0154]
醛7的合成
[0155]
醛7,如果不是市售的,可以通过标准方法制备,其中两种方法在方案14和方案15中描述。醛7可以通过适当取代的β-酮酯29和叠氮化物2之间的碱催化的环加成反应(方案14)制备。酯30可被还原成醇,该醇随后可被氧化成所述醛。替代地,酯30可以转化为相应的weinreb酰胺,该weinreb酰胺进而可以还原为醛7。方案14:醛7的合成
[0156]
替代地,典型的铜催化的炔烃-叠氮化物偶联反应可用于偶联炔烃31和叠氮化物2(方案15)。可以使用上述方法将所得酯转化为相应的醛。
方案15:醛7的基于cuaac的合成
[0157]
每当以对映体混合物的形式获得式(i)化合物时,可以使用本领域技术人员已知的方法,例如通过非对映体盐的形成和分离或通过手性固定相[例如regis whelk-o1(r,r)(10μm)柱,daicel chiralcel od-h(5-10μm)柱,或daicel chiralpak ia(10μm),ia,ib,ic,ie,或者if(5μm)或者ad-h(5μm)柱]上的hplc分离对映体。手性hplc的典型条件是洗脱液a(etoh,存在或不存在胺,如三乙胺或二乙胺)和洗脱液b(庚烷)的等度混合物,流速为0.8至150ml/min。
[0158]
提供以下实施例来说明本发明。这些实施例仅是说明性的并且不应以任何方式解释为限制本发明。
[0159]
实验部分
[0160]
化学
[0161]
所有温度均以℃表示。
[0162]
制备型hplc条件:
[0163]
根据待纯化化合物的性质,在以下给出的可能性中选择用于制备型hplc纯化的条件。每个问题不止一个选项可以导致成功的结果。设备:hplc泵:gilson 333/334或等效自动进样器:gilson lh215(带gilson 845z注射器)或等效脱气机:dionex srd-3200或等效补充泵:dionex iso-3100a或等效dad检测器:dionex dad-3000或等效ms检测器:单四极杆质量分析器thermo finnigan msq plus或等效mra分流器:mra100-000分流器或等效els检测器:polymer laboratories pl-els1000或等效物。方法:柱:可变waters atlantis t3 30x75mm 10μm(仅限酸性条件);waters xbridge c18,30x75mm 10μm(酸性/碱性条件);waters xbridge c18,50x150mm 10μm(酸性/碱性条件);流速:可变75ml/min(用于尺寸为30x75mm的柱)、150ml/min(用于尺寸为50x150mm的柱)。流动相:梯度模式a:水 0.5%甲酸(酸性条件)a:水 0.5%氢氧化铵溶液(25%)(碱性条件)b:乙腈梯度:可变,例如对于75ml/min:“极极性”:t[min]%a%b流量ml/min:0.000 100 0 75;1.000 100 0 75;3.500 80 20 75;4.000 5 95 75;6.000 5 95 75;6.200 100 0 75;6.600 100 0 75.“非常极性”:t[min]%a%b流量ml/min:0.000 95 5 75;0.100 95 5 75;3.000 50 50 75;4.000 5 95 75;6.000 5 95 75;6.200 95 5 75;6.600 95 5 75;“极性”:t[min]%a%b流量ml/min:0.000 90 10 75;0.010 90 10 75;4.000 5 95 75;6.000 5 95 75;6.200 90 10 75;6.600 90 10 75;“正常”:t[min]%a%b流量ml/min:0.000 80 20 75;0.010 80 20 75;4.000 5 95 75;6.000 5 95 75;6.200 80 20 75;6.600 80 20 75;“亲脂”:t[min]%a%b流量ml/min:0.000 70 30 75;0.010 70 30 75;3.500 5 95 75;6.000 5 95 75;6.200 70 30 75;6.600 70 30 75;“非常亲脂”:t[min]%a%b流量ml/min:0.000 50 50 75;0.010 50 50 75;3.000 5 95 75;6.000 5 95 75;6.200 50 50 75;6.600 50 50 75.注射量:100-2500μl.收集:uv/ms/elsd如果可用,及所有可能的组合;补充流速:0.50ml/min.补充
洗脱液ms:乙腈/水/tfa70:30:0.025(v/v/v);ms电离模式:esi 。
[0164]
lc-ms-条件:
[0165]
碱性条件:柱:waters beh c18,3.0x50mm,2.5μm/01593635616710;温度:40℃;注射量:0.30μl;洗脱液a:水/nh3其中c(nh3)=13mmol/l;洗脱液b:乙腈;电离:esi ;梯度:0.0min=5%b,0.01min=5%b,1.20min=95%b,1.90min=95%b,2.00min=5%b;流量=1.6ml/min。
[0166]
酸性条件:柱:zorbax rrhd sb-aq,2.1x50mm,1.8μm/useaf01579;温度:40℃;注射量:0.15μl;洗脱液a:水0.04%tfa;洗脱液b:乙腈;电离:esi ;梯度:0.0min=5%b,0.01min=5%b,1.20min=95%b,1.90min=95%b,2.10min=5%b;流量=0.8ml/min。
[0167]
qc条件:柱:acquity uplc csh c18 1.7μm 2.1x50mm;温度:60℃;注射量:0.25μl,部分循环2μl;洗脱液a:h2o 0.05%v/v甲酸;洗脱液b:乙腈 0.045%v/v甲酸;梯度:在2.0min内2%b到98%b;流量=1.0ml/min.检测:214nm处uv和ms(xevo三重四极杆检测器仪器);电离:esi 。
[0168]
缩写(如前文或后文所用):ac
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
醋酸乙酯aq.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
水性acn
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙腈aibn
ꢀꢀꢀꢀꢀꢀꢀꢀ
偶氮二异丁腈bpds
ꢀꢀꢀꢀꢀꢀꢀꢀ
红菲咯啉二磺酸二钠盐水合物brp
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
背压调节器boc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
叔丁氧羰基ccl4
ꢀꢀꢀꢀꢀꢀꢀꢀ
四氯化碳cuaac
ꢀꢀꢀꢀꢀꢀꢀ
铜催化的叠氮化物-炔环加成反应dba
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二亚苄基丙酮dcm
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二氯甲烷dea
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二乙胺dibalh
ꢀꢀꢀꢀꢀꢀ
二异丁基氢化铝dipea
ꢀꢀꢀꢀꢀꢀꢀ
二异丙基乙胺(h
ü
nig碱)dmf
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基甲酰胺dmso
ꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基亚砜dppf
ꢀꢀꢀꢀꢀꢀꢀꢀ
1,1'-双(二苯基膦)二茂铁et
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙基etoac
ꢀꢀꢀꢀꢀꢀꢀ
乙酸乙酯et2o
ꢀꢀꢀꢀꢀꢀꢀꢀ
二乙醚etoh
ꢀꢀꢀꢀꢀꢀꢀꢀ
乙醇fc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
快速色谱fgi
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
官能团相互转换h
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
小时hatu
ꢀꢀ
(1-[双(二甲氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶3-氧化
六氟磷酸盐)lc-ms
ꢀꢀꢀꢀꢀꢀꢀꢀ
液相色谱与质谱联用me
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
甲基meoh
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
甲醇mecn
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙腈min.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
分钟ml
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
毫升nabh4ꢀꢀꢀꢀꢀꢀꢀꢀ
硼氢化钠nbs
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
n-溴代琥珀酰亚胺n-bu
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
正丁基org.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
有机的pd(ii)dppf
ꢀꢀꢀ
[1,1'-双(二苯基膦)二茂铁]二氯钯(ii)prephplc
ꢀꢀꢀꢀꢀ
制备型hplcrt
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
室温rflx
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
反流sat.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
饱和sfc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
超临界流体色谱tfa
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
三氟乙酸thf
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
四氢呋喃t
r hplc
ꢀꢀꢀꢀꢀꢀ
保留时间(分钟)
[0169]
中间体合成
[0170]
中间体a:外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0171]
步骤1:制备3,6-二氯吡啶甲酸乙酯
[0172]
在0℃下向3,6-二氯吡啶-2-羧酸(9701mg,48mmol)在etoh(42ml)中的浅黄色溶液中滴加亚硫酰氯(8.84ml,120mmol)。将所得乳状悬浮液回流1小时以完成。减压蒸发溶剂。向混合物中加入饱和nahco3,将ph调节至7并用et2o萃取混合物。合并的有机层用盐水洗涤,在mgso4上干燥,过滤并减压浓缩。将浅绿色油溶于et2o并用活性炭处理10min,然后过滤并在减压下浓缩,得到为无色油的11.4g的3,6-二氯吡啶甲酸乙酯。lcms(酸性):tr=0.86min,[m h]
=220.08。
[0173]
步骤2:制备3-氯-6-氰基吡啶甲酸乙酯
[0174]
向3,6-二氯吡啶甲酸乙酯(2403mg,10.9mmol)在dmf(47ml)中的脱气溶液中加入氰化锌(1374mg,11.5mmol)、三(二亚苄基丙酮)二钯(0)(619mg,0.655mmol)和1,1'-双(二苯基膦)二茂铁(371mg,0.655mmol)。将所得黑色悬浮液在110℃下搅拌2小时30分钟,然后在rt下搅拌过夜。加入更多氰化锌98%(65.4mg,0.546mmol)、三(二亚苄基丙酮)二钯(0)(103mg,0.109mmol)和1,1'-双(二苯基膦基)二茂铁(61.8mg,0.109mmol)并将反应混合物加热至110℃达2小时30分钟以接近完成。将混合物冷却至rt,然后减压浓缩。将残余物重新溶解在et2o中,过滤并减压浓缩。将残余物通过fc(硅胶;etoac/庚烷)纯化,得到为黄色油的1.44g的3-氯-6-氰基吡啶甲酸乙酯。lcms(酸性):tr=0.82min,[m h]
=211.14。
[0175]
步骤3:制备6-(((叔丁氧基羰基)氨基)甲基)-3-氯吡啶甲酸乙酯
[0176]
将3-氯-6-氰基吡啶甲酸乙酯(1440mg,6.63mmol)溶解在etoh(70ml)中并加入二碳酸二叔丁酯(4431mg,19.9mmol)。反应在hcube-pro中使用ra-ni催化剂(7cm长)在以下条件下进行:t=70℃,p=10巴,f=1.0ml/min,100%h2模式(1次通过)。在减压下浓缩混合物。将残余物通过fc(硅胶;etoac/庚烷)纯化,得到为白色固体的3.89g的6-(((叔丁氧基羰基)氨基)甲基)-3-氯吡啶甲酸乙酯。lcms(酸性):tr=0.91min,[m h]
=315.25。
[0177]
步骤4:制备3-氯-6-(甲酰胺甲基)吡啶甲酸乙酯
[0178]
将6-(((叔丁氧基羰基)氨基)甲基)-3-氯吡啶甲酸乙酯(2840mg,9.02mmol)溶解在三氟乙酸(9ml,116mmol)中。将混合物在rt下搅拌30min并在减压下浓缩。将残余物溶解在sat.aq.nahco3中并通过添加固体nahco3将ph调至8。加入dcm(9ml)并剧烈搅拌混合物。加入甲酸(2.4ml,62.4mmol)和乙酸酐(2.4ml,25.2mmol)的预热(50℃下30min)混合物。将所得混合物在rt下搅拌过夜。分离各层并用dcm(3x)萃取水层。将合并的有机萃取物在mgso4上干燥,过滤并减压浓缩。将所得粗制黄色油在dcm/et2o/戊烷中结晶,得到为白色固体的1.87g的3-氯-6-(甲酰氨基甲基)吡啶甲酸乙酯。lcms(酸性):tr=0.64min,[m h]
=243.03。
[0179]
步骤5:制备6-氯咪唑并[1,5-a]吡啶-5-羧酸乙酯
[0180]
将3-氯-6-(甲酰氨基甲基)吡啶甲酸乙酯(1875mg,7.73mmol)溶解在甲苯(40ml)中。在0℃下加入pocl3(1.44ml,15.5mmol)并将混合物加热至110℃达10分钟。在减压下浓缩混合物。将残余物重新溶解在dcm中并加入sat.aq.nahco3。用dcm(3x)萃取水层。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩。将粗制红色油通过fc(硅胶;etoac/庚烷)纯化,得到1.613g的6-氯咪唑并[1,5-a]吡啶-5-羧酸乙酯,其为亮黄色油,在rt下固化。lcms(碱性):tr=0.83min,[m h]
=225.13。
[0181]
步骤6:制备(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇
[0182]
向6-氯咪唑并[1,5-a]吡啶-5-羧酸乙酯(1613mg,7.18mmol)在etoh(92ml)中的冰冷亮黄色溶液中加入nabh4(823mg,21.5mmol)。将所得橙色悬浮液在rt下搅拌20h以完成。减压除去etoh,加入水并用dcm萃取混合物。将合并的org.层干燥(mgso4),过滤并减压浓缩。将粗制残余物在et2o/戊烷中研磨并过滤,得到为灰白色固体的1.0g的(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇。lcms(碱性):tr=0.56min,[m h]
=183.24。
[0183]
步骤7:制备6-氯咪唑并[1,5-a]吡啶-5-甲醛
[0184]
于0℃在n2气氛下向(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇(1000mg,5.48mmol)在dcm(30ml)中的悬浮液中加入戴斯-马丁高碘烷(3667mg,8.21mmol)在dcm(20ml)中的悬浮液。在0℃下搅拌黄色悬浮液,然后升温至rt达2小时。加入水性nahco3和na2s2o3的饱和溶液并用dcm(3x)萃取混合物。将有机萃取物用盐水洗涤,在mgso4上干燥,过滤并减压浓缩,得到为红色固体的869mg的6-氯咪唑并[1,5-a]吡啶-5-甲醛。lcms(酸性):tr=0.65min,[m h]
=181.26。
[0185]
步骤8:制备6-环丙基咪唑并[1,5-a]吡啶-5-甲醛
[0186]
在80℃下将6-氯咪唑并[1,5-a]吡啶-5-甲醛(88.5mg,0.49mmol)、环丙基硼酸(126mg,1.47mmol)、三环己基膦(41.2mg,0.147mmol)、乙酸钯(ii)(11.2mg,0.049mmol)和k2co3(135mg,0.98mmol)在甲苯(8.5ml)和水(3.4ml)中的脱气混合物加热过夜(转化一半)。加入更多的环丙基硼酸(378mg,4.4mmol)、三环己基膦(185mg,0.66mmol)和乙酸钯(ii)
(50.4mg,0.22mmol)并将混合物加热至100℃达3小时以完成反应。将混合物冷却至rt,用etoac稀释并通过硅藻土短垫过滤。分离各层并用etoac将水层再萃取两次。将合并的有机萃取物在mgso4上干燥,过滤并减压浓缩。将棕色残余物溶解在mecn中,依次用庚烷和戊烷洗涤,然后减压浓缩。将棕色油在et2o/戊烷中研磨,并通过过滤收集所得沉淀,得到为米色固体的654mg的6-环丙基咪唑并[1,5-a]吡啶-5-甲醛。lcms(酸性):tr=0.47min,[m h]
=187.33。
[0187]
步骤9:制备外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇(中间体a)
[0188]
将6-环丙基咪唑并[1,5-a]吡啶-5-甲醛(654mg,3.51mmol)在thf(25ml)和et2o(10ml)中的溶液冷却至-10℃,并用在thf(9ml,4.5mmol)中的乙炔基溴化镁溶液0.5m进行处理。将反应混合物在-10℃下搅拌并升温至rt达4h以完成反应。加入冰和sat.aq.nh4cl并用etoac(3x)萃取混合物。将合并的org.层在mgso4上干燥,过滤并减压浓缩,得到为棕色泡沫的701mg的外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇。lcms(酸性):tr=0.52min,[m h]
=213.16。
[0189]
中间体b:外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇
[0190]
步骤1:制备3-氯-6-甲基吡嗪-2-羧酸甲酯
[0191]
6-溴-3-氯吡嗪-2-羧酸甲酯(13.156g,49.7mmol)、三甲基环硼氧烷(7.02ml,49.7mmol)、k2co3(13.738g,99.4mmol)和pd(ii)dppf(2.029g,2.48mmol)悬浮在二噁烷(158ml)中。将混合物用n2脱气10min,然后在100℃下加热36h。将混合物冷却至rt并通过硅藻土垫过滤。减压浓缩滤液。将粗制产物通过fc(硅胶;etoac/庚烷)纯化,得到为黄色油的7.7g的3-氯-6-甲基吡嗪-2-羧酸甲酯。lcms(酸性):tr=0.67min,[m h]
=187.18。
[0192]
步骤2:制备6-(溴甲基)-3-氯吡嗪-2-羧酸甲酯
[0193]
将3-氯-6-甲基吡嗪-2-羧酸甲酯(5.84g,29.7mmol)溶解在ccl4(82ml)中。依次加入nbs(8.018g,44.6mmol)和aibn(249mg,1.49mmol)。将混合物回流24h。加入更多的aibn并将混合物回流搅拌直至反应几乎完成。将混合物冷却至rt并减压浓缩。向残余物中加入水和etoac,分离各层,并用etoac(2x)进一步萃取水相。将合并的有机层干燥(mgso4),过滤并减压浓缩。通过fc(硅胶;etoac/庚烷)将粗制产物纯化,得到为淡黄色油的3.57g的6-(溴甲基)-3-氯吡嗪-2-羧酸甲酯。lcms(酸性):tr=0.79min,[m h]
=无质量。
[0194]
步骤3:制备3-氯-6-(甲酰胺甲基)吡嗪-2-羧酸甲酯
[0195]
向6-(溴甲基)-3-氯吡嗪-2-羧酸甲酯(8790mg,33.1mmol)在dmf(116ml)中的溶液中加入二甲酰胺钠(3568mg,36.4mmol)。将sat.aq.nahco3溶液加入并将反应混合物在rt下搅拌过夜直至完全转化为所需产物。加入etoac,分离各层并用etoac(3x)萃取aq.层。将合并的org.萃取物用盐水洗涤,干燥(mgso4),过滤并减压浓缩,得到为黑色油的7.5g的3-氯-6-(甲酰氨基甲基)吡嗪-2-羧酸甲酯。lc-ms(酸性):tr=0.52min,[m h]
=230.23。
[0196]
步骤4:制备6-氯咪唑并[1,5-a]吡嗪-5-羧酸甲酯
[0197]
将3-氯-6-(甲酰氨基甲基)吡嗪-2-羧酸甲酯(3.077g,13.4mmol)溶解在甲苯(24ml)中。添加pocl3(2.5ml,26.8mmol)并将混合物在70℃下加热1h。向混合物中加入aq.nahco3直至ph=7。将产物用etoac(3x)萃取。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩。将粗制产物通过fc(硅胶;etoac/庚烷)纯化,得到为棕色固体的1.82g的6-氯咪
唑并[1,5-a]吡嗪-5-羧酸甲酯。lc-ms(酸性):tr=0.64min,[m h]
=212.06。
[0198]
步骤5:制备6-环丙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯
[0199]
于70℃在n2下将6-氯咪唑并[1,5-a]吡嗪-5-羧酸甲酯(735mg,3.47mmol)、在thf(10.4ml,5.21mmol)中的环丙基溴化锌溶液0.5m和在thf(7ml)中的四(三苯基膦)钯(0)(40.2mg,0.035mmol)的混合物搅拌1小时15分钟。加入sat.aq.nahco3,将混合物用etoac稀释并过滤。分离各层并用etoac(2x)萃取水相。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩。将粗制产物通过fc(硅胶;庚烷/etoac)纯化,得到为黄色固体的262mg的6-环丙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯。lc-ms(酸性):tr=0.70min,[m h]
=218.15。
[0200]
步骤6:制备6-环丙基-n-甲氧基-n-甲基咪唑并[1,5-a]吡嗪-5-甲酰胺
[0201]
步骤6.1:皂化:将6-环丙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯(524mg,2.41mmol)溶解在thf(7.7ml)和水(3.85ml)中。加入氢氧化锂一水合物(123mg,2.89mmol)并将混合物在rt下搅拌2小时15分钟。将混合物在减压下浓缩。
[0202]
步骤6.2:酰胺偶联:将残留物溶解在dmf(10ml)中。加入dipea(1.24ml,7.24mmol),n,o-二甲基羟胺盐酸盐(288mg,2.89mmol)和hatu(1.101g,2.89mmol)并将混合物在rt下搅拌2小时30分钟。将混合物在减压下浓缩。将粗制产物通过制备型hplc(碱性条件)纯化,得到为黄色固体的374mg的6-环丙基-n-甲氧基-n-甲基咪唑并[1,5-a]吡嗪-5-甲酰胺。lc-ms(酸性):tr=0.60min,[m h]
=247.14。
[0203]
步骤7:制备6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛
[0204]
以逐滴方式向6-环丙基-n-甲氧基-n-甲基咪唑并[1,5-a]吡嗪-5-甲酰胺(323mg,1.31mmol)在thf(8.5ml)中的冰冷溶液中加入二异丁基氢化铝溶液(1.0m在甲苯中,1.31ml,1.31mmol)。将所得溶液在0℃下搅拌30min。在0℃下加入更多的二异丁基氢化铝溶液(1.0m在甲苯中,0.66ml,0.66mmol)并将混合物在该温度下搅拌1h。加入sat.aq.nh4cl并用etoac(3x)萃取混合物。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩,得到为黄色固体的252mg的6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛。lc-ms(碱性):tr=0.63min,[m h]
=188.29.1h nmr(500mhz,dmso)δ:10.68(s,1h),9.49(s,1h),9.28(s,1h),8.08(d,1h),3.00(m,1h),1.24-1.25(m,2h),1.12(dd,j1=8.0hz,j2=2.9hz,2h)。
[0205]
步骤8:制备外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇(中间体b)
[0206]
将6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛(245mg,1.31mmol)溶解在thf(5.8ml)中。将溶液冷却至0℃并逐滴加入在thf(7.86ml,3.93mmol)中的乙炔基溴化镁溶液0.5m。将反应混合物在0℃下搅拌1h。加入aq.nh4cl并用etoac(3x)萃取产物。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩,得到为黄白色固体的285mg的外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇。lc-ms(碱性):tr=0.58min,[m h]
=214.27。
[0207]
中间体ba:(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇
[0208]
在手性固定相上分离外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇:
[0209]
柱:chiralpak as-h,30x250mm,5μm;温度:40℃;bpr:100巴;检测器波长:227nm;流动相:acn/etoh/dea 50:50:0.1;流量:160.00ml/min;注射体积:6ml。
[0210]
通过上述方法分离613mg的外消旋体,得到312mg的s-对映体和350mg的r-对映体。
[0211]
中间体c:外消旋-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0212]
将6-氯咪唑并[1,5-a]吡啶-5-甲醛-中间体a,步骤7-(73.9mg,0.409mmol)在thf(1.6ml)中的冰冷溶液用在thf(2.46ml,17.9mmol)中的乙炔基溴化镁溶液0.5m进行处理。将反应混合物在0-10℃下搅拌2h(直至反应完成),然后加入水和aq.nh4cl。用dcm(3x)萃取混合物,将合并的有机萃取物在mgso4上干燥,过滤并减压浓缩,得到为橙色固体的77.8mg的外消旋-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇。lc-ms(碱性):tr=0.65min,[m h]
=207.19。
[0213]
中间体3a和3b:(s)-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇以及(r)-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0214]
在手性固定相上分离外消旋-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0215]
柱:chiralpak ih,30x250mm,5μm;温度:40℃;bpr:100巴;检测器波长:225nm;流动相:25%etoh以及75%co2;流量:160.00ml/min;注射体积:1ml。
[0216]
通过上述方法分离233mg的外消旋体,得到100mg的s-对映体以及114mg的r-对映体。
[0217]
中间体d:外消旋-1-(6-甲基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0218]
步骤1:制备6-甲基咪唑并[1,5-a]吡啶-5-甲醛
[0219]
于110℃将6-氯咪唑并[1,5-a]吡啶-5-甲醛-中间体a,步骤7-(117mg,0.648mmol)、三甲基环硼氧烷(0.453ml,3.24mmol)、k2co3(181mg,1.3mmol)、[1,1'-双(二苯基膦)二茂铁]二氯钯(ii)与二氯甲烷的络合物(26.5mg,0.0324mmol)在二噁烷(2ml)中的脱气悬浮液加热1h以完成。将混合物冷却至rt,用etoac稀释并通过硅藻土垫过滤。将滤液减压浓缩,将残余的棕色油在et2o/戊烷中研磨并过滤,提供为橙色固体的0.114g的6-甲基咪唑并[1,5-a]吡啶-5-甲醛。lc-ms(碱性):tr=0.57min,[m h]
=161.14。
[0220]
步骤2:外消旋-1-(6-甲基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇(中间体d)
[0221]
将6-甲基咪唑并[1,5-a]吡啶-5-甲醛(60.9mg,0.38mmol)在thf(1.6ml)中的冰冷溶液用在thf(2.28ml,1.14mmol)中的乙炔基溴化镁溶液0.5m进行处理。将反应混合物在0-10℃下搅拌2h(直到反应完成)。加入水和aq.nh4cl,并用dcm(3x)萃取混合物。将合并的有机层在mgso4上干燥,过滤并减压浓缩,得到为棕色固体的70mg的外消旋-1-(6-甲基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇。lc-ms(酸性):tr=0.45min,[m h]
=187.21。
[0222]
中间体e:外消旋-1-(6-乙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇
[0223]
步骤1:制备6-乙基咪唑并[1,5-a]吡啶-5-羧酸乙酯
[0224]
将6-氯咪唑并[1,5-a]吡啶-5-羧酸乙酯-中间体a,步骤5-(679mg,3.02mmol)在thf(7.2ml)中的脱气亮黄色溶液冷却至0℃,然后加入[1,3-双(二苯基膦基)丙烷]二氯镍(ii)(82mg,0.151mmol),接着加入在thf(4.53ml,4.53mmol)中的乙基溴化镁溶液1.0m的脱气溶液。将反应混合物在0℃下搅拌30min,然后加热至70℃1h。加入水,接着加入sat.aq.nahco3。将产物用etoac(3x)萃取。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩。将粗制残余物通过制备型hplc(碱性条件)纯化,得到为黄色油的0.380g的6-乙基咪唑并[1,5-a]吡啶-5-羧酸乙酯。lc-ms(酸性):tr=0.60min,[m h]
=219.30。
[0225]
步骤2:制备(6-乙基咪唑并[1,5-a]吡啶-5-基)甲醇
[0226]
向6-乙基咪唑并[1,5-a]吡啶-5-羧酸乙酯(380mg,1.74mmol)在etoh(12ml)和dcm
(2ml)中的冰冷溶液中分批加入nabh4(200mg,5.22mmol)。将混合物在rt下搅拌过夜。加入更多的nabh4并将混合物在rt下搅拌直至反应完成。减压除去etoh,加入水,并将混合物用dcm萃取3次。将合并的org.萃取物用在mgso4上干燥,过滤并减压浓缩,得到为淡黄色泡沫的0.320g的(6-乙基咪唑并[1,5-a]吡啶-5-基)甲醇。lc-ms(酸性):tr=0.43min,[m h]
=177.40。
[0227]
步骤3:制备6-乙基咪唑并[1,5-a]吡啶-5-甲醛
[0228]
将(6-乙基咪唑并[1,5-a]吡啶-5-基)甲醇(298mg,1.69mmol)在ch3cn/dcm 1:1(6ml)中的溶液在rt下用mno2(817mg,8.46mmol)处理并在微波放射下加热至70℃1h。加入更多的mno2(817mg,8.46mmol)并于70℃在微波放射下将混合物进一步搅拌45min。过滤悬浮液并用dcm冲洗滤饼。减压浓缩滤液,将残余物悬浮在et2o中,过滤,并减压浓缩滤液,得到为棕色油的0.233g的6-乙基咪唑并[1,5-a]吡啶-5-甲醛。lc-ms(碱性):tr=0.65min,[m h]
=175.24。
[0229]
步骤4:制备外消旋-1-(6-乙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇(中间体e)
[0230]
将6-乙基咪唑并[1,5-a]吡啶-5-甲醛(256mg,1.47mmol)在thf(10ml)以及et2o(4ml)中的溶液冷却至0℃,并通过在20min内逐滴加入在thf(3.8ml,1.91mmol)中的乙炔基溴化镁溶液0.5m进行处理。将反应混合物在0℃下搅拌1h并在rt下搅拌过夜。加入在thf中的更多的乙炔基溴化镁溶液0.5m并进一步搅拌混合物直至反应完成。加入饱和水性nh4cl,并将产物用etoac萃取3次。将合并的有机萃取物在mgso4上干燥,过滤并减压浓缩。将粗制残余物通过fc(硅胶;庚烷/etoac)纯化,得到为棕色固体的0.060g的外消旋-1-(6-乙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇。lc-ms(酸性):tr=0.50min,[m h]
=201.28。
[0231]
中间体f:外消旋-1-(6-乙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇
[0232]
步骤1:制备6-乙烯基咪唑并[1,5-a]吡嗪-5-羧酸甲酯
[0233]
在rt下向在etoh(4ml)中的6-氯咪唑并[1,5-a]吡嗪-5-羧酸甲酯-中间体b,步骤4-(200mg,0.93mmol)的溶液中加入乙烯基三氟硼酸钾(144mg,1.02mmol)以及et3n(0.19ml,1.39mmol)并将混合物搅拌5min。然后加入[1,1
’‑
双(二苯基膦)二茂铁]二氯钯(ii)(68mg,0.09mmol),并将混合物用n2脱气5min。将反应混合物于90℃在微波放射下加热2h并减压浓缩。将残余物用水和etoac稀释,过滤,分离各层,并用etoac(2x)萃取水层。将合并的有机萃取物干燥(mgso4),过滤并减压浓缩。将粗制残余物通过fc(硅胶,het/etoac)纯化,得到为黄色固体的132mg的6-乙烯基咪唑并[1,5-a]吡嗪-5-羧酸甲酯。lc-ms(酸性):tr=0.65min,[m h]
=204.26。
[0234]
步骤2:制备6-乙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯
[0235]
在0℃下,向在meoh(12ml)中的6-乙烯基咪唑并[1,5-a]吡嗪-5-羧酸甲酯(132mg,0.65mmol)的脱气(3次真空/n2循环)溶液中加入炭载pd(10%pd,69mg,0.06mmol)。将所得黑色悬浮液在h2气氛下于0℃搅拌10min。将混合物通过whatman 0.45um玻璃微纤维过滤器过滤并用meoh洗涤。减压浓缩滤液,得到为黄色粘性油的106mg的6-乙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯。lc-ms(酸性):tr=0.58min,[m h]
=206.26。
[0236]
步骤3:制备6-乙基-n-甲氧基-n-甲基咪唑并[1,5-a]吡嗪-5-甲酰胺
[0237]
向在thf(1.65ml)和水(0.83ml)中的6-乙基咪唑并[1,5-a]吡嗪-5-羧酸甲酯
4-基)-甲醇
[0249]
将在dmf(1ml)和水(0.2ml)中的中间体c外消旋-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇(38.8mg,0.188mmol)、在叔丁基甲基醚(0.45ml,0.226mmol)中的叠氮苯溶液~0.5m、五水硫酸铜(ii)(4.69mg,0.0188mmol)、l( )-抗坏血酸钠盐(7.52mg,0.0376mmol)的脱气溶液在rt下搅拌过夜。
[0250]
将混合物过滤并通过制备型hplc(碱性)纯化,得到为灰白色固体的36.2mg的外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=0.659min,[m h]
=326.1.1h nmr(500mhz,dmso)δ:9.10(s,1h),8.62(s,1h),7.93(d,j=7.9hz,2h),7.66(d,j=9.5hz,1h),7.59(t,j=7.6hz,2h),7.49(m,2h),7.02(d,j=4.2hz,1h),6.91(d,j=9.5hz,1h),6.82(d,j=4.1hz,1h)。
[0251]
实施例1a:(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0252]
在手性固定相上分离外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇:
[0253]
柱:chiralpak as-h 30x250mm,5μm;检测器波长:uv 226nm;洗脱液:75%co2以及25%etoh 0.1%dea;流量:160.00ml/min;bpr:100巴;温度:40℃.注射体积:2000μl。
[0254]
通过上述方法分离25mg的外消旋体,得到9mg的r-对映体以及9mg的s-对映体。
[0255]
实施例1a:lc-ms(qc):tr=0.659min,[m h]
=326.1。
[0256]
实施例2:外消旋-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0257]
使用中间体d外消旋-1-(6-甲基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和叠氮苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=0.519min,[m h]
=306.2.1h nmr(500mhz,dmso)δ:9.06(d,1h),8.48-8.61(m,1h),7.85-8.02(m,2h),7.57-7.61(m,2h),7.47-7.50(m,2h),7.33(s,1h),6.69-6.81(m,1h),6.54-6.66(m,2h),2.47(m,3h)。
[0258]
实施例2a:(r)-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0259]
在手性固定相上分离外消旋-(6-甲基-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇:
[0260]
柱:chiralpak as-h,30x250mm,5μm;温度:40℃;bpr:100巴;检测器波长:222nm;洗脱液:75%co2以及25%etoh 0.1%dea;流量:160.00ml/min;注射体积:2.5ml。
[0261]
通过上述方法分离25mg的外消旋体,得到9mg的r-对映体以及9mg的s-对映体。
[0262]
实施例2a:lc-ms(qc):tr=0.518min,[m h]
=306.2。
[0263]
实施例3:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0264]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和叠氮苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=
0.727min,[m h]
=333.2.1h nmr(500mhz,dmso)δ:9.07(d,1h),8.97(s,1h),8.65(s,1h),7.92-7.94(m,2h),7.78(s,1h),7.60(m,2h),7.48-7.51(m,1h),6.89(d,j=3.5hz,1h),6.83(d,j=3.8hz,1h),2.48(m,1h),1.06-1.10(m,1h),0.94-1.01(m,3h)。
[0265]
实施例3a:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0266]
在手性固定相上分离外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(1-苯基-1h-[1,2,3]三唑-4-基)-甲醇:
[0267]
柱:chiralpak as-h 30x250mm,5μm;检测器波长:uv 227nm;洗脱液:75%co2以及25%etoh 0.1%dea;流量:160.00ml/min;bpr:100巴;温度:40℃.注射体积:1900μl。
[0268]
通过上述方法分离19mg的外消旋体,得到:5mg的r-对映体实施例3a以及6mg的s-对映体。
[0269]
实施例3a:lc-ms(qc):tr=0.727min,[m h]
=333.2。
[0270]
实施例4:外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0271]
使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇以及1-叠氮基-4-甲氧基苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.630min,[m h]
=362.2.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.52(s,1h),7.72-7.99(m,2h),7.49(d,j=9.4hz,1h),7.23-7.41(m,1h),7.13(m,2h),7.01(d,j=3.6hz,1h),6.67(m,2h),3.73-3.94(m,3h),2.16-2.30(m,1h),2.00-2.15(m,2h),0.97-1.07(m,2h),0.86-0.91(m,1h),0.71-0.83(m,1h)。
[0272]
实施例5:外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-(1-对甲苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0273]
使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和叠氮甲苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-(1-对甲苯基-1h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=0.678min,[m h]
=346.2.1h nmr(500mhz,dmso)δ:8.97(s,1h),8.48-8.51(m,1h),7.80(d,j=8.4hz,2h),7.48(d,j=9.4hz,1h),7.39(m,2h),7.32(s,1h),6.95-7.08(m,1h),6.67-6.71(m,1h),6.65(m,1h),2.38(s,3h),2.11-2.31(m,1h),0.95-1.13(m,2h),0.83-0.92(m,1h),0.74-0.83(m,1h)。
[0274]
实施例6:外消旋-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯基)-氨基甲酸甲酯
[0275]
步骤1:制备(4-叠氮基苯基)氨基甲酸甲酯
[0276]
使用4-甲氧基羰基氨基苯基硼酸根据实施例7步骤1所述的程序制备。
[0277]
步骤2:按照实施例1所述的程序并使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇以及(4-叠氮基苯基)氨基甲酸甲酯制备。通过prephplc(碱性条件)纯化得到外消旋-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯基)-氨基甲酸甲酯。lc-ms(qc):tr=0.589;[m h]
=405.2。
[0278]
实施例7:外消旋-2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲
基]-[1,2,3]三唑-1-基}-苯酚
[0279]
步骤1:制备4-叠氮基-2-氯苯酚
[0280]
在圆底烧瓶中,将(3-氯-4-羟基苯基)硼酸(508mg,2.83mmol)、叠氮化钠(279mg,4.24mmol)和乙酸铜(ii)(51mg,0.28mmol)悬浮在甲醇meoh(10ml)中。在空气气氛下于60℃将反应混合物搅拌3h。过滤混合物,减压除去溶剂,并将残余物通过fc(硅胶,et2o)纯化,得到4-叠氮基-2-氯苯酚。lc-ms(碱性):tr=0.40;[m h]
=未检测到。
[0281]
步骤2:使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和4-叠氮基-2-氯苯酚按照实施例1所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。lc-ms(qc):tr=0.601;[m h]
=382.2。
[0282]
实施例7a和7b:2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚和2-氯-4-{4-[(s)-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚
[0283]
在手性固定相上分离外消旋-2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。方法:柱:chiralpak ia30x250mm,5μm;检测器设置:uv-vis-1;210nm;洗脱液:55%co2和45%(ch2cl2/etoh 1:1);流量:160.00ml/min;bpr:100巴;温度:40℃.注射体积:1000μl。
[0284]
通过上述方法分离10.5mg的外消旋体,得到:2mg的r-对映体实施例7a和4mg的s-对映体实施例7b。
[0285]
实施例7a:lc-ms(qc):tr=0.602;[m h]
=382.2。
[0286]
实施例7b:lc-ms(qc):tr=0.601;[m h]
=382.2。
[0287]
实施例8:外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0288]
步骤1:制备4-叠氮基-2-氯-1-甲氧基苯
[0289]
于0℃向在1m aq.hcl(40ml)中的3-氯-4-甲氧基苯胺(500mg;3.11mmol)的溶液中加入在水(8ml)中的亚硝酸钠(217mg;3.11mmol)的溶液。将反应混合物搅拌20min并加入叠氮化钠(245mg;3.73mmol)。将反应混合物在rt下搅拌3h。用etoac稀释混合物,分离各层并将org.层用盐水洗涤,干燥(mgso4),过滤并减压浓缩,得到为棕色油的4-叠氮基-2-氯-1-甲氧基苯。1h nmr(400mhz,dmso)δ:7.23(d,j=2.7hz,1h),7.18(m,1h),7.10(dd,j1=2.7hz,j2=8.8hz,1h),3.85(s,3h)。
[0290]
步骤2:按照实施例1所述的程序并使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和4-叠氮基-2-氯-1-甲氧基苯制备。通过prephplc(碱性条件)纯化得到外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇。lc-ms(qc):tr=0.705;[m h]
=396.2.1h nmr(500mhz,dmso)δ:9.02(s,1h),8.35-8.72(m,1h),8.04-8.08(m,1h),7.90(dd,j1=8.9hz,j2=2.5hz,1h),7.48(d,j=9.3hz,1h),7.35(d,j=9.0hz,1h),7.49-7.28(br s,1h),7.01(d,j=3.9hz,1h),6.59-6.71(m,2h),3.93(s,3h),2.20(m,1h),0.94-1.10(m,2h),0.73-0.94(m,2h)。
[0291]
实施例8a:(r)-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0292]
在手性固定相上分离外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-甲醇。方法:柱:chiralpak as-h 30x250mm,5μm;检测器设置:uv 259nm;洗脱液:30%co2和70%mecn/etoh/dea 50:50:0.1;流量:160.00ml/min;bpr:120巴;温度:40℃.注射体积:3000μl。
[0293]
通过上述方法分离12.4mg的外消旋体,得到:1.9mg的r-对映体实施例8a和5.2mg的s-对映体。
[0294]
实施例8a:lc-ms(qc):tr=0.705;[m h]
=396.2。
[0295]
实施例9:外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(6-乙氧基-吡啶-3-基)-1h-[1,2,3]三唑-4-基]-甲醇
[0296]
步骤1:制备5-叠氮基-2-乙氧基吡啶
[0297]
使用2-乙氧基吡啶-5-硼酸根据实施例7步骤1所述的程序制备。
[0298]
步骤2:使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和5-叠氮基-2-乙氧基吡啶按照实施例1所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-[1-(6-乙氧基-吡啶-3-基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.652;[m h]
=377.2.1h nmr(500mhz,dmso)δ:9.00(s,1h),8.66(d,j=2.6hz,1h),8.39-8.61(m,1h),8.22(dd,j1=8.9hz,j2=2.8hz,1h),7.49(d,j=9.4hz,1h),7.22-7.44(m,1h),6.96-7.06(m,2h),6.70(d,j=4.0hz,1h),6.66(d,j=9.4hz,1h),4.37(q,j=7.0hz,2h),2.16-2.25(m,1h),1.35(t,j=7.0hz,3h),0.95-1.07(m,2h),0.76-0.91(m,2h)。
[0299]
实施例10:外消旋-2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚
[0300]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-氯苯酚按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-2-氯-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。lc-ms(qc):tr=0.703;[m h]
=383.1.1h nmr(500mhz,dmso)δ:8.94-8.96(m,2h),8.63(s,1h),7.95(d,j=2.7hz,1h),7.77(s,1h),7.70(dd,j1=8.8hz,j2=2.7hz,1h),7.12(d,j=8.8hz,1h),6.85(d,j=3.3hz,1h),6.77(d,j=3.9hz,1h),2.43-2.48(m,1h),1.05-1.10(m,1h),0.94-1.00(m,3h)。
[0301]
实施例10a:2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚
[0302]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-氯苯酚按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到2-氯-4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。lc-ms(qc):tr=0.702;[m h]
=383.1。
[0303]
实施例11:外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0304]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-氯-1-甲氧基苯按照实施例8所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]
吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.843;[m h]
=397.2.1h nmr(500mhz,dmso)δ:9.02(d,1h),8.96(s,1h),8.63(s,1h),8.06(d,j=2.7hz,1h),7.89(dd,j1=2.7hz,j2=8.9hz,1h),7.77(d,1h),7.35(d,j=9.1hz,1h),6.86(d,j=2.0hz,1h),6.78(d,j=2.7hz,1h),3.94(s,3h),2.44-2.48(m,1h),1.04-1.14(m,1h),0.91-1.05(m,3h)。
[0305]
实施例11a:(r)-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0306]
在手性固定相上分离外消旋-[1-(3-氯-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。方法:柱:chiralcel od-h 30x250mm,5μm;检测器波长:uv 223nm;洗脱液:35%co2和65%(mecn/etoh 1:1);流量:160.00ml/min;bpr:100巴;温度:40℃.注射体积:4000μl。
[0307]
通过上述方法分离21.1mg的外消旋体,得到:9mg的r-对映体实施例11a和8.8mg的s-对映体。
[0308]
实施例11a:lc-ms(qc):tr=0.843;[m h]
=397.2。
[0309]
实施例12:外消旋-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯基)-氨基甲酸甲酯
[0310]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和(4-叠氮基苯基)氨基甲酸甲酯按照实施例6所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯基)-氨基甲酸甲酯。lc-ms(qc):tr=0.685;[m h]
=406.2.1h nmr(500mhz,dmso)δ:9.94(s,1h),8.94-8.96(m,2h),8.63(s,1h),7.82(d,j=9.1hz,2h),7.77(s,1h),7.64(d,j=9.0hz,2h),6.86(d,j=3.8hz,1h),6.78(d,j=4.0hz,1h),3.70(s,3h),2.44-2.48(m,1h),1.05-1.11(m,1h),0.92-1.02(m,3h)。
[0311]
实施例13:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(6-乙氧基-吡啶-3-基)-1h-[1,2,3]三唑-4-基]-甲醇
[0312]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和5-叠氮基-2-乙氧基吡啶按照实施例9所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(6-乙氧基-吡啶-3-基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.782;[m h]
=378.2.1h nmr(500mhz,dmso)δ:9.00(d,1h),8.96(s,1h),8.69(dd,j=2.8hz,1h),8.61(s,1h),8.21(dd,j1=8.9hz,j2=2.8hz,1h),7.77(d,1h),7.02(dd,j=8.9hz,1h),6.87(d,j=3.8hz,1h),6.80(d,j=4.0hz,1h),4.37(q,j=7.0hz,2h),2.46(m,1h),1.35(t,j=7.0hz,3h),1.05-1.10(m,1h),0.97-1.02(m,1h),0.94-0.97(m,2h)。
[0313]
实施例14:外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚
[0314]
步骤1:制备4-叠氮基苯酚
[0315]
使用(4-羟基苯基)硼酸根据实施例7步骤1所述的程序制备。
[0316]
步骤2:使用中间体a外消旋-1-(6-环丙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和4-叠氮基苯酚按照实施例1所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。
lc-ms(qc):tr=0.518;[m h]
=348.2。
[0317]
实施例15:外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚
[0318]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基苯酚按照实施例14所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚。lc-ms(qc):tr=0.597;[m h]
=349.1。
[0319]
实施例16:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0320]
于50℃将在dmf(2ml)中的实施例15外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚(34.8mg,0.1mmol)、3-(溴甲基)-3-氟氧杂环丁烷(69mg,0.4mmol)、碘化钠(0.757mg,0.005mmol)和k2co3(55.3mg,0.4mmol)的混合物搅拌16h。然后将混合物过滤并通过制备型hplc(碱性条件)纯化,得到为白色固体的31.7mg的外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.760;[m h]
=437.2.1h nmr(500mhz,dmso)δ:8.96-8.97(m,2h),8.65(s,1h),7.86(d,j=9.1hz,2h),7.77(d,1h),7.20(d,j=9.1hz,2h),6.86(d,j=3.8hz,1h),6.77(d,j=4.0hz,1h),4.68-4.78(m,4h),4.52(d,j=22.1hz,2h),2.43-2.47(m,1h),1.05-1.13(m,1h),0.91-1.04(m,3h)。
[0321]
实施例17:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0322]
使用中间体b外消旋-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-4-甲氧基苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.747min,[m h]
=363.2.1h nmr(500mhz,dmso)δ:8.94-8.96(m,2h),8.64(s,1h),7.82(d,j=9.1hz,2h),7.77(d,1h),7.13(d,j=9.1hz,2h),6.86(s,1h),6.77(s,1h),3.83(s,3h),2.46(m,1h),1.06-1.09(m,1h),0.93-1.00(m,3h)。
[0323]
实施例18:外消旋-1-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯氧基)-2-甲基-丙-2-醇
[0324]
使用实施例15外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯酚和1-溴代-2-甲基丙-2-醇按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-1-(4-{4-[(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-苯氧基)-2-甲基-丙-2-醇。lc-ms(qc):tr=0.725min,[m h]
=421.3.1h nmr(500mhz,dmso)δ:8.94-8.96(m,2h),8.65(s,1h),7.81(d,j=9.1hz,2h),7.77(d,1h),7.13(m,2h),6.86(d,j=3.9hz,1h),6.76(d,j=4.0hz,1h),4.67(s,1h),3.79(s,2h),2.44-2.48(m,1h),1.22(s,6h),1.06-1.09(m,1h),0.94-1.00(m,3h)。
[0325]
实施例19:外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-{1-[4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0326]
使用实施例14外消旋-4-{4-[(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-羟基-甲
基]-[1,2,3]三唑-1-基}-苯酚和3-(溴甲基)-3-氟氧杂环丁烷按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡啶-5-基)-{1-[4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.645min,[m h]
=436.3.1h nmr(500mhz,dmso)δ:8.91-9.06(m,1h),8.39-8.57(m,1h),7.80-7.91(m,2h),7.44-7.53(m,1h),7.29-7.37(m,1h),7.13-7.26(m,2h),6.94-7.05(m,1h),6.63-6.69(m,2h),4.61-4.85(m,4h),4.44-4.60(m,2h),2.16-2.32(m,1h),0.94-1.17(m,2h),0.72-0.93(m,2h)。
[0327]
实施例20:外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇
[0328]
步骤1:制备1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-羧酸乙酯
[0329]
于50℃将在dmso(0.8ml)中的乙酰乙酸乙酯(0.21ml,1.67mmol)、1-叠氮基-4-甲氧基苯(193mg,1.29mmol)和k2co3(714mg,5.17mmol)的悬浮液搅拌1h30。将混合物冷却至rt,用水和etoac稀释,并用aq.1n hcl酸化。分离各层并用etoac(2x)萃取aq.层。将合并的org.萃取物干燥(mgso4),过滤并减压浓缩。将粗制固体在et2o/石油醚中研磨并过滤,得到为米色固体的165mg的1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-羧酸乙酯。lc-ms(酸性):tr=0.86,[m h]
=262.19。
[0330]
步骤2:制备(1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇
[0331]
在rt下向在etoh(8.7ml)中的1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-羧酸乙酯(165mg,0.63mmol)的溶液加入nabh4(218mg,5.76mmol)。在rt下将反应混合物搅拌48h。将反应混合物减压浓缩并将残余物在dcm和水之间分配。分离各层并用dcm(2x)萃取aq.层。将合并的org.萃取物干燥(mgso4),过滤并减压浓缩。将粗制固体用et2o/石油醚研制并过滤,得到为米色固体的126mg的(1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇。lc-ms(酸性):tr=0.60,[m h]
=220.27。
[0332]
步骤3:制备1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-甲醛
[0333]
向在ch2cl2(7ml)中的(1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇(126mg,0.58mmol)的溶液中加入dess-martin高碘烷(271mg,0.64mmol)。将反应混合物在rt下搅拌直至反应完成。将sat.aq.nahco3和sat.aq.na2s2o3加入并将混合物在rt下搅拌10min。分离各层并用ch2cl2萃取aq.层。将合并的org.萃取物用盐水洗涤,干燥(mgso4),过滤并减压浓缩,在制备型hplc(碱性条件)之后得到为白色固体的53mg的1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-甲醛。lc-ms(碱性):tr=0.76,[m h]
=218.16。
[0334]
步骤4:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇(实施例20)
[0335]
在-78℃下向在干thf(2.3ml)中的6-氯咪唑并[1,5-a]吡啶(30mg,0.20mmol)的溶液中加入n-buli(2.5m在己烷中,0.16ml,0.40mmol)。将所得棕色溶液在-78℃下搅拌45min。将在干thf(1ml)中的1-(4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-甲醛(45mg,0.21mmol)的溶液加入并将反应混合物在-78℃下搅拌1h并缓慢升温至0℃。将sat.aq.nh4cl溶液加入并用etoac(3x)萃取混合物。将合并的org.萃取物干燥(mgso4),过滤并减压浓缩。通过制备型hplc(酸性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=
0.695min,[m h]
=370.1。使用以下方法将标题化合物(38mg)在手性固定相上分离:柱:chiralpak ih 30x250mm,5μm;检测器波长:222nm;洗脱液:55%co2和45%(mecn/etoh/dea 50:50:0.1);流量:160.00ml/min;bpr:100巴;温度:40℃;注射体积:1000μl,得到13mg的(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇(实施例20a)和14mg的s-对映体。实施例20a:lc-ms(qc):tr=0.681min,[m h]
=370.3。
[0336]
实施例21:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(3-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0337]
步骤1:制备4-叠氮基-2-氟-1-甲氧基苯
[0338]
使用(3-氟-4-甲氧基苯基)硼酸根据实施例7步骤1所述的程序制备。
[0339]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-氟-1-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(3-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.780;[m h]
=381.2.1h nmr(500mhz,dmso)δ:9.00(d,j=0.6hz,1h),8.96(s,1h),8.62(s,1h),7.89(dd,j1=12.1hz,j2=2.6hz,1h),7.77(s,1h),7.74(ddd,j1=8.9hz,j2=2.6hz,j3=1.5hz,1h),7.38(t,j=9.1hz,1h),6.86(d,j=3.7hz,1h),6.79(d,j=4.0hz,1h),3.92(s,3h),2.47(m,1h),1.05-1.08(m,1h),0.93-1.00(m,3h)。
[0340]
实施例22:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0341]
步骤1:制备1-叠氮基-2,5-二氟-4-甲氧基苯
[0342]
使用(2,5-二氟-4-甲氧基苯基)硼酸根据实施例7步骤1所述的程序制备。
[0343]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-2,5-二氟-4-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.799;[m h]
=399.2.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.76(d,j=1.0hz,1h),8.62(s,1h),7.83(dd,j1=7.1hz,j2=11.2hz,1h),7.78(s,1h),7.52(dd,j1=7.6hz,j2=12.2hz,1h),6.88(d,j=3.6hz,1h),6.80(d,j=4.0hz,1h),3.94(s,3h),2.46-2.48(m,1h),1.05-1.08(m,1h),0.93-1.01(m,3h)。
[0344]
实施例23:(r)-[1-(3-溴代-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0345]
步骤1:制备4-叠氮基-2-溴代-1-甲氧基苯
[0346]
使用3-溴代-4-甲氧基苯胺根据实施例8步骤1所述的程序制备。
[0347]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-溴代-1-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-[1-(3-溴代-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.863;[m h]
=441.1.1h nmr(500mhz,dmso)δ:9.02(s,1h),8.96(s,1h),8.65(s,1h),8.19(d,j=2.7hz,1h),7.93(dd,j1=8.9hz,j2=
2.7hz,1h),7.78(br s,1h),7.31(d,j=9.1hz,1h),6.86(s,1h),6.78(br s,1h),3.93(s,3h),2.44-2.48(m,1h),1.06-1.09(m,1h),0.94-1.00(m,3h)。
[0348]
实施例24:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲氧基甲基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0349]
步骤1:制备1-叠氮基-4-(甲氧基甲基)苯
[0350]
使用4-(甲氧基甲基)苯胺根据实施例8步骤1所述的程序制备。
[0351]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-4-(甲氧基甲基)苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲氧基甲基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.741;[m h]
=377.2.1h nmr(500mhz,dmso)δ:9.05(d,1h),8.96(s,1h),8.65(s,1h),7.91(d,j=8.6hz,2h),7.78(s,1h),7.53(d,j=8.6hz,2h),6.88(d,j=3.8hz,1h),6.79(d,j=4.0hz,1h),4.49(s,2h),3.33(s,3h),2.47(m,1h),1.05-1.10(m,1h),0.94-1.01(m,3h)。
[0352]
实施例25:(r)-[1-(5-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0353]
步骤1:制备1-叠氮基-5-氯-2-氟-4-甲氧基苯
[0354]
使用5-氯-2-氟-4-甲氧基苯胺.根据实施例8步骤1所述的程序制备。
[0355]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-5-氯-2-氟-4-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-[1-(5-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.861;[m h]
=415.1.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.76(d,1h),8.63(s,1h),7.97(d,j=7.8hz,1h),7.76-7.79(m,1h),7.49(d,j=12.4hz,1h),6.88(s,1h),6.76-6.83(m,1h),3.97(s,3h),2.46-2.48(m,1h),1.05-1.08(m,1h),0.93-1.00(m,3h)。
[0356]
实施例26:(r)-[1-(3-氯-5-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0357]
步骤1:制备5-叠氮基-1-氯-3-氟-2-甲氧基苯
[0358]
使用3-氯-5-氟-4-甲氧基苯胺根据实施例8步骤1所述的程序制备。
[0359]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和5-叠氮基-1-氯-3-氟-2-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-[1-(3-氯-5-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.920;[m h]
=415.2.1h nmr(500mhz,dmso)δ:9.08(d,j=0.5hz,1h),8.96(s,1h),8.61(s,1h),7.99-8.02(m,2h),7.74-7.82(m,1h),6.87(m,1h),6.83(m,1h),3.96(d,j=1.4hz,3h),2.47(m,1h),1.06-1.09(m,1h),0.92-1.01(m,3h)。
[0360]
实施例27:4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚
[0361]
步骤1:制备4-叠氮基-2-氟苯酚
[0362]
使用2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧硼烷-2-基)苯酚根据实施例7步骤1所
述的程序制备。
[0363]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-2-氟苯酚按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚。lc-ms(qc):tr=0.642;[m h]
=367.2。
[0364]
实施例28:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0365]
使用实施例27,4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚和3-(溴甲基)-3-氟氧杂环丁烷按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.795min,[m h]
=455.2.1h nmr(500mhz,dmso)δ:9.01(d,j=0.6hz,1h),8.96(s,1h),8.62(s,1h),7.93(dd,j1=11.9hz,j2=2.6hz,1h),7.75-7.78(m,2h),7.45(t,j=9.0hz,1h),6.87(d,j=3.8hz,1h),6.80(d,j=4.0hz,1h),4.69-4.78(m,4h),4.61(d,j=22.2hz,2h),2.46(m,1h),1.06-1.09(m,1h),0.94-1.01(m,3h)。
[0366]
实施例29:1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丙-2-醇
[0367]
使用实施例27,4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚和1-溴代-2-甲基丙-2-醇按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丙-2-醇。lc-ms(qc):tr=0.767min,[m h]
=439.3.1h nmr(500mhz,dmso)δ:8.99(d,j=0.6hz,1h),8.96(s,1h),8.62(s,1h),7.88(dd,j1=11.9hz,j2=2.6hz,1h),7.77(d,j=0.5hz,1h),7.70(ddd,j1=8.9hz,j2=2.6hz,j3=1.5hz,1h),7.38(t,j=9.1hz,1h),6.86(d,j=3.8hz,1h),6.79(d,j=4.0hz,1h),4.71(s,1h),3.87(s,2h),2.47(m,1h),1.23(s,6h),1.06-1.08(m,1h),0.94-1.00(m,3h)。
[0368]
实施例30:(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0369]
使用中间体ca,(s)-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和1-叠氮基-4-甲氧基苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.683min,[m h]
=356.1.1h nmr(500mhz,dmso)δ:8.97(s,1h),8.62(s,1h),7.83(d,j=8.2hz,2h),7.65(d,j=9.3hz,1h),7.45-7.54(m,1h),7.13(d,j=8.2hz,2h),6.97(d,j=3.0hz,1h),6.90(d,j=9.4hz,1h),6.80(s,1h),3.83(s,3h)。
[0370]
实施例31:(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇使用中间体ca,(s)-1-(6-氯咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和1-叠氮基-2,5-二氟-4-甲氧基苯按照实施例22所述的程序制备。通过制备型hplc(碱性条件)纯化得到(r)-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.737min,[m h]
=392.1.1h nmr
(500mhz,dmso)δ:8.79(s,1h),8.59(s,1h),7.84(dd,j1=7.1hz,j2=11.2hz,1h),7.66(d,j=9.5hz,1h),7.47-7.54(m,2h),7.00(d,j=4.4hz,1h),6.91(d,j=9.5hz,1h),6.81(d,j=4.1hz,1h),3.95(s,3h)。
[0371]
实施例32:外消旋-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0372]
使用中间体f外消旋-1-(6-乙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-2,5-二氟-4-甲氧基苯按照实施例22所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.729min,[m h]
=387.2。
[0373]
实施例33:(r)-[1-(3-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇
[0374]
步骤1:制备1-叠氮基-3-氯-2-氟-4-甲氧基苯
[0375]
使用(3-氯-2-氟-4-甲氧基苯基)硼酸根据实施例7步骤1所述的程序制备。
[0376]
步骤2:使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-3-氯-2-氟-4-甲氧基苯按照实施例1所述的程序制备。通过prephplc(酸性条件)纯化得到(r)-[1-(3-氯-2-氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.863;[m h]
=415.2.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.79(d,j=1.1hz,1h),8.62(s,1h),7.77-7.78(m,2h),7.23(dd,j1=9.3hz,j2=1.8hz,1h),6.89(d,j=3.8hz,1h),6.81(d,j=4.0hz,1h),3.99(s,3h),2.47(m,1h),1.05-1.08(m,1h),0.93-1.01(m,3h)。
[0377]
实施例34:外消旋-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇使用中间体e,外消旋-1-(6-乙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和1-叠氮基-4-甲氧基苯按照实施例1所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.605min,[m h]
=350.2.1h nmr(500mhz,dmso)δ:8.95(d,1h),8.45-8.54(m,1h),7.83(m,2h),7.43-7.57(m,1h),7.32(s,1h),7.13(m,2h),6.76-6.79(m,1h),6.62(m,2h),3.83(s,3h),2.75-2.85(m,2h),1.09-1.31(m,3h)。
[0378]
实施例34a:(r)-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0379]
在手性固定相上分离外消旋-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-[1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。方法:柱:chiralpak ih 30x250mm,5μm;检测器波长:222nm;洗脱液:50%co2和50%(mecn/etoh/dea 50:50:0.1);流量:160.00ml/min;bpr:100巴;温度:40℃;注射体积:1000μl。
[0380]
通过上述方法分离18mg的外消旋体,得到:7.7mg的r-对映体实施例34a和9.6mg的s-对映体。
[0381]
实施例34a:lc-ms(qc):tr=0.606min,[m h]
=350.2.
[0382]
实施例35:外消旋-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0383]
使用中间体e外消旋-1-(6-乙基咪唑并[1,5-a]吡啶-5-基)丙-2-炔-1-醇和1-叠
氮基-2,5-二氟-4-甲氧基苯按照实施例22所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇。lc-ms(qc):tr=0.641min,[m h]
=386.2.1h nmr(500mhz,dmso)δ:8.74(s,1h),8.33-8.53(m,1h),7.82(dd,j1=11.2hz,j2=7.1hz,1h),7.46-7.56(m,2h),7.32(s,1h),6.77(d,j=9.2hz,1h),6.67(d,j=3.8hz,1h),6.56-6.62(m,1h),3.86-4.00(m,3h),2.80(q,j=7.5hz,2h),1.28(t,j=7.5hz,3h)。
[0384]
实施例35a:(r)-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0385]
在手性固定相上分离外消旋-[1-(2,5-二氟-4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-(6-乙基-咪唑并[1,5-a]吡啶-5-基)-甲醇。方法:柱:chiralpak ih 30x250mm,5μm;检测器波长:222nm;洗脱液:55%co2和45%(mecn/etoh/dea 50:50:0.1);流量:160.00ml/min;bpr:100巴;温度:40℃;注射体积:1000μl。
[0386]
通过上述方法分离16mg的外消旋体,得到:8.6mg的r-对映体实施例35a和5.5mg的s-对映体。
[0387]
实施例35a:lc-ms(qc):tr=0.641min,[m h]
=386.2。
[0388]
实施例36:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0389]
使用实施例27,4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2-氟-苯酚和3-(溴甲基)氧杂环丁烷按照实施例16所述的程序制备。通过制备型hplc(酸性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[3-氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.748min,[m h]
=437.2.1h nmr(500mhz,dmso)δ:9.00(d,j=0.5hz,1h),8.96(s,1h),8.62(s,1h),7.90(dd,j1=2.6hz,j2=12.0hz,1h),7.77(d,j=0.4hz,1h),7.74(ddd,j1=8.9hz,j2=2.6hz,j3=1.5hz,1h),7.43(t,j=9.1hz,1h),6.86(d,j=3.8hz,1h),6.80(d,j=4.0hz,1h),4.73(dd,j1=6.1hz,j2=7.9hz,2h),4.45(t,j=6.1hz,2h),4.37(d,j=6.7hz,2h),3.42-3.46(m,1h),2.40-2.47(m,1h),1.06-1.09(m,1h),0.94-1.00(m,3h)。
[0390]
实施例37:外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-乙基-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0391]
步骤1:制备2-重氮-3-氧代戊酸乙酯
[0392]
将在mecn(150ml)中的丙酰乙酸乙酯(4474mg,29.8mmol,1eq)和4-乙酰氨基苯磺酰基叠氮化物(7748mg,31.3mmol,1.05eq)的溶液在氩气气氛下在rt下搅拌。所有材料完全溶解,将tea(4.35ml,31.3mmol,1.05eq)加入到反应中,在室温下搅拌过夜。过滤白色沉淀,用ch2cl2洗涤并减压浓缩滤液,得到标题产物。
[0393]
步骤2:制备5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-羧酸乙酯
[0394]
在氩气气氛在rt下向在dmf(6.12ml)中的2-重氮-3-氧代戊酸乙酯(478mg,2.81mmol)的溶液中依次加入4-氨基-苯甲醚(0.363ml,3.09mmol,1.1eq)和氯化钛(iv)(~1.0m在甲苯中,2.81ml,2.81mmol,1eq)。将反应混合物在80℃下搅拌过夜。将浆液用水和etoac稀释并过滤。分离后,将水相用etoac萃取两次。将合并的有机萃取物用水和盐水洗涤,在mgso4上干燥,过滤并减压浓缩。通过硅胶快速色谱法(hept/etoac)纯化得到标题化
合物。lc-ms(酸性):tr=0.82min,[m h]
=276.02。
[0395]
步骤3:制备(5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇
[0396]
使用5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-羧酸乙酯按照实施例20步骤2所述的程序制备。lc-ms(酸性):tr=0.67min,[m h]
=234.41。
[0397]
步骤4:制备5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛
[0398]
使用(5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇按照实施例20步骤3所述的程序制备。lc-ms(酸性):tr=0.84min,[m h]
=232.03。
[0399]
步骤4:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇
[0400]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.83min,[m h]
=444.17。
[0401]
步骤5:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-乙基-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0402]
使用外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-乙基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-乙基-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.748min,[m h]
=384.2。
[0403]
实施例38:外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-甲基-1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0404]
步骤1:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-甲基-1-苯基-1h-1,2,3-三唑-4-基)甲醇
[0405]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和5-甲基-1-苯基-1h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.75min,[m h]
=400.23。
[0406]
步骤2:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-甲基-1-苯基-1h-[1,2,3]三唑-4-基)-甲醇
[0407]
使用外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-甲基-1-苯基-1h-1,2,3-三唑-4-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-甲基-1-苯基-1h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=0.651min,[m h]
=340.3.1h nmr(500mhz,dmso)δ:8.74(s,1h),7.61-7.66(m,6h),7.50(s,1h),6.87(d,j=9.5hz,1h),6.80(d,j=4.3hz,1h),6.74(d,j=4.2hz,1h),2.46(s,3h)。
[0408]
实施例39:外消旋-[1-(5-氯-2-氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-(6-氯-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0409]
步骤1:制备1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-羧酸乙酯
[0410]
将在dmf(1.2ml)中的1-叠氮基-5-氯-2-氟-4-甲氧基苯(248mg,1.23mmol,1eq)和丙炔酸乙酯(133mg,0.449mmol,1.5eq)的溶液冷却至0℃。在0℃下加入催化剂cuso4/抗坏血酸钠/bpds[通过将在水(658ul)和dmf(164μl)中的bpds(4.1mg,6.98 10e-3mmol,5.7e-3eq)加入到在水(205μl)中的cuso4(1.7mg,6.61 10e-3mmol,5.4 10e-3eq)和抗坏血酸钠
(1.7mg,8.24 10e-3mmol,6.7 10e-3eq)的溶液中来制备],并将混合物在rt下搅拌2d。将混合物过滤并用水洗涤,得到为黄色固体的标题产物(307mg,83%)。lc-ms(酸性):tr=0.92min,[m h]
=300.10。
[0411]
步骤2:制备(1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇
[0412]
使用1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-羧酸乙酯按照实施例20步骤2所述的程序制备。lc-ms(酸性):tr=0.69min,[m h]
=258.12。
[0413]
步骤3:制备1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛
[0414]
向在ch2cl2(11ml)中的(1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇(263mg,1mmol,1eq)的溶液中加入mno2(878mg,10.1mmol,10.1eq)。将混合物在rt下搅拌直至反应完成。将混合物过滤并减压浓缩。通过fc(hept/etoac)纯化得到为浅棕色固体的标题产物(137mg,54%)。lc-ms(酸性):tr=0.83min,[m h]
=256.22。
[0415]
步骤4:制备外消旋-(1-(5-氯-2-氟-4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-基)(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)甲醇
[0416]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和1-(5-氯-2-氟-4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.86min,[m h]
=482.08。
[0417]
步骤5:制备外消旋-[1-(5-氯-2-氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-(6-氯-咪唑并[1,5-a]吡啶-5-基)-甲醇
[0418]
使用外消旋-(1-(5-氯-2-氟-4-甲氧基苯基)-5-甲基-1h-1,2,3-三唑-4-基)(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-[1-(5-氯-2-氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-(6-氯-咪唑并[1,5-a]吡啶-5-基)-甲醇。lc-ms(qc):tr=0.782min,[m h]
=422.3.1h nmr(500mhz,dmso)δ:8.70(s,1h),7.93(d,j=7.8hz,1h),7.65(d,j=9.5hz,1h),7.50(m,2h),6.87(d,j=9.5hz,1h),6.84(d,j=4.4hz,1h),6.75(d,j=4.4hz,1h),3.98(s,3h),2.35(s,3h)。
[0419]
实施例40:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-吡咯烷-1-基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0420]
步骤1:制备1-(4-叠氮基苯基)吡咯烷
[0421]
使用(4-(吡咯烷-1-基)苯基)硼酸hcl根据实施例7步骤1所述的程序制备。
[0422]
步骤2:制备(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-吡咯烷-1-基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0423]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-(4-叠氮基苯基)吡咯烷按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-吡咯烷-1-基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.933min;[m h]
=402.4.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.82(d,1h),8.65(s,1h),7.77(s,1h),7.65(d,j=9.0hz,2h),6.84(d,j=3.9hz,1h),6.73(d,j=4.0hz,1h),6.65(d,j=9.1hz,2h),3.28(t,j=6.6hz,4h),2.45-2.47(m,1h),1.98(m,4h),1.05-1.08(m,1h),0.93-1.00(m,3h)。
[0424]
实施例41:(r)-[1-(4-氨基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并
[1,5-a]吡嗪-5-基)-甲醇
[0425]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基苯胺按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-[1-(4-氨基-苯基)-1h-[1,2,3]三唑-4-基]-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-甲醇。lc-ms(qc):tr=0.529min;[m h]
=348.3.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.75(d,1h),8.64(s,1h),7.76(d,1h),7.48(d,j=8.8hz,2h),6.83(d,j=3.8hz,1h),6.72(d,j=4.0hz,1h),6.67(d,j=8.9hz,2h),5.49(s,2h),2.44-2.48(m,1h),1.05-1.07(m,1h),0.93-0.99(m,3h)。
[0426]
实施例42:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0427]
步骤1:制备4-叠氮基-n-甲基苯胺
[0428]
使用n-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧硼烷-2-基)苯胺根据实施例7步骤1所述的程序制备。
[0429]
步骤2:制备(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0430]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-n-甲基苯胺按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.656min;[m h]
=362.3.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.78(d,1h),8.64(s,1h),7.77(s,1h),7.57(d,j=8.9hz,2h),6.83(d,j=3.8hz,1h),6.72(d,j=4.0hz,1h),6.65(d,j=9.0hz,2h),6.09(q,j=5.0hz,1h),2.72(d,j=5.0hz,3h),2.46(m,1h),1.05-1.08(m,1h),0.93-0.99(m,3h)。
[0431]
实施例43:外消旋-2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯酚
[0432]
步骤1:制备4-叠氮基-2-氯-1-(甲氧基甲氧基)苯
[0433]
使用(3-氯-4-(甲氧基甲氧基)苯基)硼酸根据实施例7步骤1所述的程序制备。
[0434]
步骤2:制备1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-羧酸乙酯
[0435]
使用4-叠氮基-2-氯-1-(甲氧基甲氧基)苯按照实施例20步骤1所述的程序制备。lc-ms(酸性):tr=0.92min,[m h]
=326.26。
[0436]
步骤3:制备(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇
[0437]
使用乙基1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-羧酸乙酯按照实施例20步骤2所述的程序制备。lc-ms(酸性):tr=0.70min,[m h]
=284.18。
[0438]
步骤4:制备1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-甲醛
[0439]
使用(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇按照实施例20,步骤3所述的程序制备。lc-ms(酸性):tr=0.85min,[m h]
=282.19。
[0440]
步骤5:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇
[0441]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.85min,[m h]
=494.24。
[0442]
步骤6:制备外消旋-(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇
[0443]
使用外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇。lc-ms(酸性):tr=0.73min,[m h]
=433.87。
[0444]
步骤7:制备外消旋-2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯酚
[0445]
向在etoac中的外消旋-(1-(3-氯-4-(甲氧基甲氧基)苯基)-5-甲基-1h-1,2,3-三唑-4-基)(6-氯咪唑并[1,5-a]吡啶-5-基)甲醇的溶液中加入在二噁烷(4m,4.5eq)中的hcl并将白色悬浮液在rt下搅拌直至反应完成。将悬浮液过滤并减压浓缩,得到外消旋-2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯酚。lc-ms(qc):tr=0.639min,[m h]
=390.3。
[0446]
实施例44:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-二甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0447]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和4-叠氮基-n,n-二甲基苯胺按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(4-二甲基氨基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.771min;[m h]
=376.3.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.84(d,1h),8.65(s,1h),7.77(s,1h),7.67(d,j=9.1hz,2h),6.84(m,3h),6.74(d,j=4.0hz,1h),2.97(s,6h),2.45-2.47(m,1h),1.06-1.08(m,1h),0.93-0.99(m,3h)。
[0448]
实施例45:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0449]
步骤1:制备6-(4-硝基苯基)-2-氧杂-6-氮杂螺旋[3.3]庚烷
[0450]
在rt下用2-氧杂-6-氮杂-螺旋-3-3-庚烷(278mg,2.72mmol,1eq)处理在ch3cn(3ml)中的1-氟-4-硝基苯(388mg,2.72mmol,1eq)和dipea(1.02ml,5.96mmol,2.19eq)的溶液。然后在75℃下将混合物搅拌24h。在rt下加入更多的2-氧杂-6-氮杂-螺旋-3-3-庚烷(278mg,2.72mmol,1eq)并在75℃下将混合物搅拌过夜。将混合物冷却至rt并减压浓缩。将残余物悬浮在dmf中并过滤以得到为黄色固体的标题产物(453mg,76%)。lc-ms(酸性):tr=0.80min,[m h]
=221.29。
[0451]
步骤2:制备4-(2-氧杂-6-氮杂螺旋[3.3]庚烷-6-基)苯胺
[0452]
将在meoh(8ml)中的6-(4-硝基苯基)-2-氧杂-6-氮杂螺旋[3.3]庚烷(453mg,2.04mmol,1eq)的溶液脱气3次并用n2惰性化。然后在rt下加入pd/c 10%(71mg)。将混合物脱气,置于氢气气氛下并在rt下搅拌4h。将混合物通过whatman 0.45μm玻璃微纤维过滤器过滤并在减压下浓缩以得到为紫色固体的标题产物(370mg,95%)。lc-ms(酸性):tr=
0.37min,[m h]
=190.31。
[0453]
步骤3:制备6-(4-叠氮基苯基)-2-氧杂-6-氮杂螺旋[3.3]庚烷
[0454]
使用4-(2-氧杂-6-氮杂螺旋[3.3]庚烷-6-基)苯胺根据实施例8步骤1所述的程序制备。
[0455]
步骤4:制备(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0456]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和6-(4-叠氮基苯基)-2-氧杂-6-氮杂螺旋[3.3]庚烷按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(2-氧杂-6-氮杂-螺旋[3.3]庚-6-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.700min;[m h]
=430.4.1h nmr(500mhz,dmso)δ:8.95(s,1h),8.84(s,1h),8.58-8.72(m,1h),7.73-7.87(m,1h),7.67(d,j=8.9hz,2h),6.84(d,j=3.9hz,1h),6.74(d,j=4.0hz,1h),6.57(d,j=8.9hz,2h),4.73(s,4h),4.05(s,4h),2.43-2.48(m,1h),1.05-1.09(m,1h),0.93-1.00(m,3h)。
[0457]
实施例46:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0458]
步骤1:制备1-(4-硝基苯基)-6-氧杂-1-氮杂螺旋[3.3]庚烷
[0459]
使用6-氧杂-1-氮杂螺旋[3.3]庚烷根据实施例45步骤1所述的程序制备。lc-ms(酸性):tr=0.75min,[m h]
=221.11。
[0460]
步骤2:制备4-(6-氧杂-1-氮杂螺旋[3.3]庚烷-1-基)苯胺
[0461]
使用1-(4-硝基苯基)-6-氧杂-1-氮杂螺旋[3.3]庚烷根据实施例45步骤2所述的程序制备。lc-ms(酸性):tr=0.36min,[m h]
=190.25。
[0462]
步骤3:制备1-(4-叠氮基苯基)-6-氧杂-1-氮杂螺旋[3.3]庚烷
[0463]
使用4-(6-氧杂-1-氮杂螺旋[3.3]庚烷-1-基)苯胺根据实施例8步骤1所述的程序制备。
[0464]
步骤4:制备(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0465]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-(4-叠氮基苯基)-6-氧杂-1-氮杂螺旋[3.3]庚烷按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[4-(6-氧杂-1-氮杂-螺旋[3.3]庚-1-基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.755min;[m h]
=430.4.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.86(d,1h),8.64(s,1h),7.75(m,3h),6.84(d,j=9.0hz,3h),6.76(s,1h),5.10(d,j=7.8hz,2h),4.72(d,j=8.0hz,2h),3.69(t,j=6.9hz,2h),2.54(m,2h),2.46(m,1h),1.06-1.08(m,1h),0.94-1.00(m,3h)。
[0466]
实施例47:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(1-甲基-2,3-二氢-1h-吲哚-5-基)-1h-[1,2,3]三唑-4-基]-甲醇
[0467]
步骤1:制备5-叠氮基-1-甲基吲哚啉
[0468]
使用1-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧硼烷-2-基)吲哚啉根据实施例7步
咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯酚。lc-ms(qc):tr=0.581min,[m h]
=374.3。
[0486]
实施例49:外消旋-4-(2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯氧基)-2-甲基-丁-2-醇
[0487]
使用实施例43外消旋-2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯酚和4-溴代-2-甲基丁-2-醇按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-4-(2-氯-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-苯氧基)-2-甲基-丁-2-醇。lc-ms(qc):tr=0.790min,[m h]
=476.2.1h nmr(500mhz,dmso)δ:8.72(s,1h),7.76(d,j=2.6hz,1h),7.65(d,j=9.5hz,1h),7.55(dd,j1=2.6hz,j2=8.8hz,1h),7.49(s,1h),7.37(d,j=8.9hz,1h),6.86(d,j=9.5hz,1h),6.78(d,j=4.4hz,1h),6.72(d,j=4.5hz,1h),4.43(s,1h),4.27(t,j=6.9hz,2h),2.37-2.44(m,3h),1.91(t,j=6.9hz,2h),1.20(s,6h)。
[0488]
实施例50:外消旋-4-(4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丁-2-醇
[0489]
使用实施例48外消旋-4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯酚和4-溴代-2-甲基丁-2-醇按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-4-(4-{4-[(6-氯-咪唑并[1,5-a]吡啶-5-基)-羟基-甲基]-5-甲基-[1,2,3]三唑-1-基}-2-氟-苯氧基)-2-甲基-丁-2-醇。lc-ms(qc):tr=0.732min,[m h]
=460.3.1h nmr(500mhz,dmso)δ:8.73(s,1h),7.65(d,j=9.5hz,1h),7.60-7.63(m,1h),7.49(s,1h),7.39-7.42(m,2h),6.86(d,j=9.6hz,1h),6.78(d,j=4.3hz,1h),6.72(d,j=4.3hz,1h),4.44(s,1h),4.26(t,j=7.0hz,2h),2.44(s,3h),1.90(t,j=7.0hz,2h),1.19(s,6h)。
[0490]
实施例51:外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-氯-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0491]
步骤1:制备(5-氨基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇
[0492]
向在thf(4.5ml)中的5-氨基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-羧酸乙酯(262mg,1.00mmol,1eq)的冷却(-70℃)溶液中加入二异丁基氢化铝溶液(1.0m在甲苯中,5ml,5.00mmol,5eq)。将所得橙色悬浮液在-70℃下搅拌2h以完成。将反应用sat.酒石酸钾钠溶液淬灭并用etoac萃取。将合并的org.层干燥(mgso4),过滤并减压浓缩,得到为米色固体的标题化合物(149mg,68%)。lc-ms(酸性):tr=0.49min,[m h]
=221.17。
[0493]
步骤2:制备(5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇
[0494]
于0℃向在mecn(2.4ml)中的(5-氨基-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇(179mg,0.813mmol,1eq)、氯化铜(i)(249mg,2.44mmol,3eq)和无水氯化铜(ii)(328mg,2.44mmol,3eq)的混合物中加入亚硝酸异戊酯(0.603ml,4.31mmol,5.3eq)。将所得溶液在rt下搅拌48h。将反应用水淬灭并用etoac萃取。将合并的org.层干燥(mgso4),过滤并减压浓缩,得到标题产物。lc-ms(酸性):tr=0.67min,[m h]
=240.29。
[0495]
步骤3:制备5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛
[0496]
使用(5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)甲醇按照实施例39步骤3所
述的程序制备。lc-ms(酸性):tr=0.82min,[m h]
=238.25。
[0497]
步骤4:制备外消旋-(5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)甲醇
[0498]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.85min,[m h]
=449.94。
[0499]
步骤5:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-氯-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇
[0500]
使用外消旋-(5-氯-1-(4-甲氧基苯基)-1h-1,2,3-三唑-4-基)(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-[5-氯-1-(4-甲氧基-苯基)-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.782min,[m h]
=390.2。
[0501]
实施例52:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0502]
步骤1:制备1-叠氮基-2,5-二氟-4-(甲氧基甲氧基)苯
[0503]
使用2,5-二氟-4-(甲氧基甲氧基)苯基硼酸根据实施例7步骤1所述的程序制备。
[0504]
步骤2制备(r)-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)(1-(2,5-二氟-4-(甲氧基甲氧基)苯基)-1h-1,2,3-三唑-4-基)甲醇
[0505]
使用中间体ba,(s)-1-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)丙-2-炔-1-醇和1-叠氮基-2,5-二氟-4-(甲氧基甲氧基)苯按照实施例7所述的程序制备。通过prephplc(碱性条件)纯化得到(r)-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)(1-(2,5-二氟-4-(甲氧基甲氧基)苯基)-1h-1,2,3-三唑-4-基)甲醇。lc-ms(酸性):tr=0.73;[m h]
=429.20。
[0506]
步骤3:制备(r)-4-(4-((6-环丙基咪唑并[1,5-a]吡嗪-5-基)(羟基)甲基)-1h-1,2,3-三唑-1-基)-2,5-二氟苯酚
[0507]
向在etoac中的(r)-(6-环丙基咪唑并[1,5-a]吡嗪-5-基)(1-(2,5-二氟-4-(甲氧基甲氧基)苯基)-1h-1,2,3-三唑-4-基)甲醇的溶液中加入在二噁烷(4m,3eq)中的hcl并将白色悬浮液在rt下搅拌直至反应完成。将悬浮液过滤并减压浓缩。通过prephplc(酸性条件)纯化得到(r)-4-(4-((6-环丙基咪唑并[1,5-a]吡嗪-5-基)(羟基)甲基)-1h-1,2,3-三唑-1-基)-2,5-二氟苯酚。lc-ms(酸性):tr=0.62min,[m h]
=385.19。
[0508]
步骤4:制备(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0509]
使用(r)-4-(4-((6-环丙基咪唑并[1,5-a]吡嗪-5-基)(羟基)甲基)-1h-1,2,3-三唑-1-基)-2,5-二氟苯酚和3-(溴甲基)-3-氟氧杂环丁烷按照实施例16所述的程序制备。通过制备型hplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(3-氟-氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.805min,[m h]
=473.3。
[0510]
实施例53:(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇
[0511]
使用(r)-4-(4-((6-环丙基咪唑并[1,5-a]吡嗪-5-基)(羟基)甲基)-1h-1,2,3-三
唑-1-基)-2,5-二氟苯酚和3-(溴甲基)-氧杂环丁烷按照实施例52所述的程序制备。通过制备型hplc(碱性条件)纯化得到(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-{1-[2,5-二氟-4-(氧杂环丁烷-3-基甲氧基)-苯基]-1h-[1,2,3]三唑-4-基}-甲醇。lc-ms(qc):tr=0.756min,[m h]
=455.3.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.78(d,j=1.0hz,1h),8.62(s,1h),7.88(dd,j1=11.1hz,j2=7.1hz,1h),7.78(d,1h),7.60(dd,j1=12.0hz,j2=7.5hz,1h),6.89(s,1h),6.76-6.87(m,1h),4.64-4.80(m,7h),2.47-2.49(m,1h),1.05-1.08(m,1h),0.92-1.01(m,3h)。
[0512]
实施例54:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,4-二氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇
[0513]
在rt下向在无水叔丁基甲基醚(0.128ml,1.07mmol,5.373eq)中的1-叠氮基-2,4-二氟苯(32.7mg,0.2mmol,1eq)的溶液中加入在thf(0.42ml,0.21mmol,1.05eq)中的1-丙炔基溴化镁溶液0.5m。将混合物在rt下搅拌3h。然后加入在thf(0.3ml)中的6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛(37.4mg,0.2mmol,1eq)的溶液并将混合物在rt下搅拌1h。混合物用水和acoet稀释。分离各层并进一步用etoac2x萃取水相。将合并的有机层干燥(mgso4),过滤并减压浓缩。通过制备型hplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,4-二氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.757min,[m h]
=383.3.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.66(s,1h),7.77-7.81(m,2h),7.70(m,1h),7.35-7.39(m,1h),6.86(d,j=4.0hz,1h),6.64(d,j=4.0hz,1h),2.38-2.42(m,1h),2.35(s,3h),1.01-1.05(m,1h),0.86-0.98(m,3h)。
[0514]
实施例55:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇
[0515]
使用1-叠氮基-2,5-二氟-4-甲氧基苯和6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛按照实施例54所述的程序制备。通过prephplc(碱性条件)纯化得到外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2,5-二氟-4-甲氧基-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.783min;[m h]
=413.3.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.66(s,1h),7.74-7.78(m,2h),7.52(dd,j1=11.7hz,j2=7.6hz,1h),6.84(d,j=4.0hz,1h),6.63(d,j=4.0hz,1h),3.96(s,3h),2.36-2.43(m,1h),2.34(s,3h),1.00-1.07(m,1h),0.86-0.99(m,3h)。
[0516]
实施例56:1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2,5-二氟-苯氧基)-2-甲基-丙-2-醇
[0517]
使用(r)-4-(4-((6-环丙基咪唑并[1,5-a]吡嗪-5-基)(羟基)甲基)-1h-1,2,3-三唑-1-基)-2,5-二氟苯酚和1-溴代-2-甲基丙-2-醇按照实施例52所述的程序制备。通过制备型hplc(碱性条件)纯化得到1-(4-{4-[(r)-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-羟基-甲基]-[1,2,3]三唑-1-基}-2,5-二氟-苯氧基)-2-甲基-丙-2-醇。lc-ms(qc):tr=0.772min,[m h]
=457.4.1h nmr(500mhz,dmso)δ:8.96(s,1h),8.75(d,j=1.0hz,1h),8.62(s,1h),7.78(d,2h),7.53(dd,j1=12.2hz,j2=7.5hz,1h),6.88(s,1h),6.80(s,1h),4.75(s,1h),3.92(s,2h),2.47(m,1h),1.22(s,6h),1.05-1.07(m,1h),0.93-1.00(m,3h)。
[0518]
实施例57:外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2-氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇
[0519]
使用1-叠氮基-2-氟苯和6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛按照实施例54所述的程序制备。通过prephplc(碱性条件)纯化为外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-[1-(2-氟-苯基)-5-甲基-1h-[1,2,3]三唑-4-基]-甲醇。lc-ms(qc):tr=0.722min;[m h]
=365.3.1h nmr(500mhz,dmso)δ:8.97(s,1h),8.67(s,1h),7.78(d,1h),7.66-7.72(m,2h),7.57-7.61(m,1h),7.46(td,j1=7.6hz,j2=1.1hz,1h),6.86(d,j=4.1hz,1h),6.64(d,j=4.1hz,1h),2.38-2.43(m,1h),2.35(d,3h),1.01-1.05(m,1h),0.88-1.01(m,3h)。
[0520]
参考实施例1(ref1):外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(5-苯基-噻吩-3-基)-甲醇
[0521]
在0℃下向异丙基氯化镁氯化锂(1.3m in thf,0.14ml,0.18mmol)的溶液中加入在thf(0.2ml)中的2-溴代-5-苯基-噻吩(45mg,0.18mmol)的溶液。将反应混合物在0℃下搅拌1h。然后将混合物冷却至-20℃并加入6-环丙基咪唑并[1,5-a]吡嗪-5-甲醛(30mg,0.16mmol)。允许反应混合物升温至rt并在该温度下搅拌1h。加入sat.aq.nh4cl和etoac,分离各层并用etoac(2x)萃取aq.层。将合并的org.萃取物干燥(mgso4),过滤并减压浓缩。将粗制产物通过制备型hplc(碱性条件)纯化,得到为白色固体的外消旋-(6-环丙基-咪唑并[1,5-a]吡嗪-5-基)-(5-苯基-噻吩-3-基)-甲醇。lc-ms(qc):tr=1.065min,[m h]
=348.10.1h nmr(400mhz,dmso)δ:8.98(s,1h),8.52(s,1h),7.78(s,1h),7.60(d,j=7.4hz,2h),7.29-7.41(m,4h),7.05(s,1h),6.88-6.90(m,2h),2.47(d,j=4.7hz,1h),0.94-1.07(m,4h)。
[0522]
参考实施例2(ref2):外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-吡唑-3-基)-甲醇
[0523]
步骤1:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(1-苯基-1h-吡唑-3-基)甲醇
[0524]
在-40℃下以逐滴的方式向在thf(1.6ml)中的二异丙基氨基锂溶液(1.0m in thf/己烷,0.80ml,0.80mmol)的溶液中加入在thf(1.6ml)中的6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶(85mg,0.40mmol)的溶液。将反应混合物在40℃下搅拌25min。然后将溶液冷却至-78℃和将1-苯基-1h-吡唑e-3-甲醛(145mg,0.80mmol)一次全部加入固体。将混合物在-78℃下搅拌5min,在-40℃下搅拌30min,然后允许达到rt过夜。将sat.aq.nh4cl和etoac加入,分离各层并用etoac(2x)萃取aq.层。将合并的org.萃取物干燥(mgso4),过滤并减压浓缩,得到外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(1-苯基-1h-吡唑-3-基)甲醇。lc-ms(酸性):tr=0.84min,[m h]
=385.2。
[0525]
步骤2:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-吡唑-3-基)-甲醇
[0526]
向在etoh(4ml)中的外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(1-苯基-1h-吡唑-3-基)甲醇(154mg,0.40mmol)的溶液中加入raney镍。将所得黑色悬浮液在45℃下搅拌直至反应完成。将混合物过滤并用dcm和etoh洗涤并减压浓缩。将粗制产物通过制备型hplc(碱性条件)纯化,得到为灰白色固体的外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(1-苯基-1h-吡唑-3-基)-甲醇。lc-ms(qc):tr=0.760min,[m h]
=325.10.1h nmr(500mhz,dmso)δ:8.57(m,1h),8.49(d,j=2.5hz,1h),7.66-7.68(m,2h),7.58-7.63(m,1h),7.42-7.46(m,3h),7.27(m,1h),6.86-6.90(m,1h),6.80-6.82(m,1h),6.72(d,j=
2.5hz,1h),6.69(d,j=4.4hz,1h)。
[0527]
参考实施例3(ref3):外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(2-苯基-2h-[1,2,3]三唑-4-基)-甲醇
[0528]
步骤1:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(2-苯基-2h-1,2,3-三唑-4-基)甲醇
[0529]
使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和2-苯基-2h-1,2,3-三唑-4-甲醛按照参考实施例2所述的程序制备。lc-ms(酸性):tr=0.88min,[m h]
=385.85。
[0530]
步骤2:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(2-苯基-2h-[1,2,3]三唑-4-基)-甲醇
[0531]
使用外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(2-苯基-2h-1,2,3-三唑-4-基)甲醇按照参考实施例2所述的程序制备。通过制备型hplc(碱性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(2-苯基-2h-[1,2,3]三唑-4-基)-甲醇。lc-ms(qc):tr=0.833min,[m h]
=326.10.1h nmr(500mhz,dmso)δ:8.57(s,1h),8.34(s,1h),7.84-7.89(m,2h),7.64-7.69(m,1h),7.48-7.55(m,3h),7.35-7.43(m,1h),7.11(d,j=4.4hz,1h),6.92(m,1h),6.80-6.87(m,1h)。
[0532]
参考实施例4(ref4):外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-苯基-噻吩-2-基)-甲醇
[0533]
步骤1:制备外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-苯基噻吩-2-基)甲醇
[0534]
按照参考实施例2所述的程序制备使用6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶和5-苯基噻吩-2-甲醛。lc-ms(酸性):tr=0.99min,[m h]
=401.09。
[0535]
步骤2:制备外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-苯基-噻吩-2-基)-甲醇
[0536]
按照参考实施例2所述的程序制备使用外消旋-(6-氯-3-(乙硫基)咪唑并[1,5-a]吡啶-5-基)(5-苯基噻吩-2-基)甲醇。通过制备型hplc(碱性条件)纯化得到外消旋-(6-氯-咪唑并[1,5-a]吡啶-5-基)-(5-苯基-噻吩-2-基)-甲醇。lc-ms(qc):tr=1.030min,[m h]
=341.00.1h nmr(500mhz,dmso)δ:8.50-8.53(m,1h),7.68(dd,j=9.6hz,1h),7.58-7.63(m,2h),7.50(s,1h),7.37-7.43(m,2h),7.35(m,1h),7.28-7.32(m,1h),7.25(d,j=4.6hz,1h),6.91-6.95(m,2h),6.79-6.81(m,1h)。
[0537]
实施例1a的化合物的绝对手性和结合模式通过相应化合物-酶共晶体的x-射线衍射分析使用以下实验程序确定:
[0538]
1.蛋白质纯化与共结晶:
[0539]
按照文献(biochem et biophysica acta 1814(2011)1947-1954)中描述的程序表达和纯化ido1蛋白。ido1蛋白在含有10mm mes(2-(n-吗啉代)乙磺酸)ph 6.50、100mm nacl和2mm tcep(三(2-羧乙基)膦盐酸盐)的缓冲液中浓缩至29mg/ml。将蛋白质溶液与终浓度为2mm的实施例1的化合物在277k下温育3小时。然后使用eppendorf5424r台式离心机在277k下以15,000rpm将溶液离心5分钟。将离心的溶液与储槽溶液混合,该储槽溶液含有30mm硫酸锂、30mm硫酸钠、30mm硫酸钾、100mm 3-吗啉代-2-羟基丙磺酸/双(2-羟乙基)氨基-三(羟甲基)甲烷ph 6.5、10%(w/v)peg 8000和20%(w/v)1,5-戊二醇。ido1和实施例1a
的化合物的共晶最终通过在293k下从坐滴的蒸气扩散获得。
[0540]
2.x射线数据采集与结构测定:
[0541]
将上述共晶使用尼龙环收获并直接置于液氮中。同步加速器数据是使用pilatus 2m-f检测器在瑞士维利根保罗谢勒研究所的瑞士光源(swiss light source at the paul scherrer institute,villigen,switzerland)的光束线x06da处收集的。使用程序xds(acta cryst.(2010)d66,125-132)处理衍射图像。使用程序相位器(j.appl.cryst.(2007)40,658-674)解析初步结构。结构的精化和重建分别使用程序refmac5(acta cryst.(2004)d60,2284-2295)和coot(acta cryst.(2010)d66,486-501)进行。使用从观察到的反射中随机选择的5%的总数据计算自由r(r-free)。基于测得的电子密度,明确确定实施例1a的化合物是(r)-对映体。
[0542]
数据收集和精化统计a括号中显示的值对应于最高分辨率壳b[0543]
基于更活跃的对映体的结合模式与实施例1的化合物的结合模式相同的假设,将根据本发明的化合物的(r)-或(s)-构型分配给实施例2a、3a、7a、8a、10a、11a、20a、21-31、33、34a、35a、36、40-42、44-47、52、53和56的化合物。
[0544]
生物测试
[0545]
1)在ido1酶促测定中测试化合物的ido抑制活性:
[0546]
在检测缓冲液中以2nm的最终浓度孵育重组全长人ido1,其中n端六组氨酸标签在大肠杆菌中表达并纯化至均质,该检测缓冲液由补充有10mm抗坏血酸的ph为6.5的37.5mm
磷酸盐缓冲液、0.45μm亚甲蓝、50u/ml催化酶、0.01%bsa和0.01%吐温20组成(方案修改自seegers等人,jbs 2014)。示例化合物在dmso中连续稀释,在磷酸盐缓冲液中进一步稀释,并以范围为10μm至0.5nm的最终浓度加入到酶中。最终dmso浓度为0.6%。在rt下预孵育30分钟后,通过在测定缓冲液中加入最终浓度为5μm的l-色氨酸开始反应。在rt下孵育30分钟后,将20μl反应混合物的3μl转移到含有25μl去离子水的384深孔板中。加入在冷100%甲醇中的200nm l-色氨酸-(吲哚-d5)中的100μl,接着在4℃下以3220x g离心10分钟。然后加入额外的75μl去离子水,接着在4℃下以3220x g离心10分钟。将反应的产物n'-甲酰基犬尿氨酸(nfk)通过lcms量化并归一化为l-色氨酸-(吲哚-d5)信号。具有0.6%dmso(0%效果)和tdo/ido抑制剂(100%效果)的样本用作对照样本,以设置确定每个化合物的半最大抑制浓度(ic50)所需的非线性回归参数。对于每个化合物浓度,与0%和100%效果相比的活性百分比计算为平均值
±
stdev(每个浓度以一式两份进行测量)。ic50值和曲线是使用剂量-响应单点模型203(四参数逻辑曲线模型)用xlfit软件(idbs)生成的。计算出的ic50值可根据日常检测性能而波动。这种波动是本领域技术人员已知的。当化合物被多次测量时,给出平均值。
[0547]
2)在tdo2酶促测定中测试化合物的tdo抑制活性:
[0548]
在检测缓冲液中以15nm的最终浓度孵育其中n端六组氨酸标签在大肠杆菌中表达并纯化至均质的包含氨基酸19-407的重组人tdo,该检测缓冲液由补充有100μm抗坏血酸的ph为7的75mm磷酸盐缓冲液、50u/ml催化酶、0.01%bsa和0.01%吐温20组成(方案修改自seegers等人,jbs 2014)。示例化合物在dmso中连续稀释,在磷酸盐缓冲液中进一步稀释,并以范围为10μm至0.5nm的最终浓度加入到反应混合物中。最终dmso浓度为0.6%。在rt下预孵育30分钟后,通过在测定缓冲液中加入最终浓度为200μm的l-色氨酸开始反应。在rt下孵育30分钟后,将3μl反应混合物转移到含有25μl去离子水的384深孔板中。加入在冷100%甲醇中的200nm l-色氨酸-(吲哚-d5)中的100μl,接着在4℃下以3220x g离心10分钟。然后加入额外的75μl去离子水,接着在4℃下以3220x g离心10分钟。将反应的产物n'-甲酰基犬尿氨酸(nfk)通过lcms量化并归一化为l-色氨酸-(吲哚-d5)信号。具有0.6%dmso(0%效果)和tdo/ido抑制剂(100%效果)的样本用作对照样本,以设置确定每个化合物的半最大抑制浓度(ic50)所需的非线性回归参数。对于每个化合物浓度,与0%和100%效果相比的活性百分比计算为平均值
±
stdev(每个浓度以一式两份进行测量)。ic50值和曲线是使用剂量-响应单点模型203(四参数逻辑曲线模型)用xlfit软件(idbs)生成的。计算出的ic50值可根据日常检测性能而波动。这种波动是本领域技术人员已知的。当化合物被多次测量时,给出平均值。
[0549]
3)在基于细胞的测定中测试化合物的ido/tdo抑制活性和毒性
[0550]
sw48细胞(atcc,ccl-231)用于测量化合物的tdo抑制活性,并常规保持在dmem高葡萄糖/glutamax
tm
/丙酮酸90%(v/v)、fcs 10%(v/v)、青霉素/链霉素中1%(v/v)。在用ifnγ刺激后上调ido1的skov3细胞(nci,编号0503405)用于测量化合物的ido抑制活性。skov3细胞通常保存在rpmi 90%(v/v)、fcs 10%(v/v)、青霉素/链霉素1%(v/v)中。sw48或skov3细胞分别以每孔45ul 8000个细胞或每孔45ul 4000个细胞的密度接种在384孔板中。板在37℃/5%co2下孵育24小时。第二天,加入连续稀释的10ul化合物(测试浓度范围10um-40nm)和200um l-色氨酸(skvo3另外接受最终浓度为50ng/ml的ifnγ)。在37℃/5%
co2下孵育24小时后,将每孔3ul的上清液转移到384深孔板中每孔的25ul h2o中,并将每孔25ul的上清液转移到废液中。每孔剩余25ul上清液的skov3和sw48细胞板用于测量活力(见下文)。将每孔含有3ul上清液和25ul h2o的384深孔板进一步处理用于lcms:在加入在甲醇中的200nm的100ul l-色氨酸-(吲哚-d5)(sigma 615862)后,在4℃下以3220xg将384深孔板离心10分钟,每孔加入75ul h2o并在4℃下以3220x g将板再次离心10分钟。n-甲酰基犬尿氨酸和犬尿氨酸通过lcms定量,归一化为内标l-色氨酸-(吲哚-d5)并计算总和。具有0.2%dmso(0%效果)和tdo/ido抑制剂(100%效果)的样本用作对照样本,以设置确定每个化合物ic50所需的非线性回归参数。对于每个化合物浓度,与0%和100%效果相比的活性百分比计算为平均值
±
stdev(每个浓度以一式两份进行测量)。ic50值和曲线是使用剂量-响应单点模型203用xlfit软件(idbs)生成的。计算出的ic50值可根据每日细胞检测性能而波动。这种波动是本领域技术人员已知的。当化合物被多次测量时,给出平均值。
[0551]
由于对nfk和kyn产生的抑制可只是细胞毒性的影响,因此平行进行活力测定(celltiter-glo 2.0发光细胞活力测定,promega目录#g9243)。将celltiter-glo试剂(25ul/孔)加入细胞板,在室温下避光孵育15分钟,并根据制造商的说明使用来自perkin elmer的envision多标签读取器测量发光。发光信号与存在的atp量成比例。atp的量与存在的活细胞数成正比。具有0.2%dmso(0%效果)和有毒化合物(100%效果)的样本用作对照样本以设置非线性回归的参数。对于每个化合物浓度,与0%和100%效果相比的活性百分比计算为平均值
±
stdev(每个浓度以一式两份进行测量)。tox ic50值和曲线是使用剂量-响应单点模型203使用xlfit软件(idbs)生成的。计算出的ic50值可根据每日细胞检测性能而波动。这种波动是本领域技术人员已知的。当化合物被多次测量时,给出平均值。
[0552]
针对实施例1至57和参考实施例1至4的化合物获得的生物学测试1、2和3的结果被总结在下表1中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。