基于iib型限制性内切酶特征的宏基因组测序数据处理系统及处理方法
技术领域
1.本发明属于生物信息学技术领域,具体涉及一种基于iib型限制性内切酶特征的宏基因组测序数据处理系统。
背景技术:
2.目前,在微生物多样性研究中,主要有两种高通量研究技术:扩增子测序(amplicon sequencing)和宏基因组测序(whole-metagenome sequencing,wms)。扩增子测序具有扩增偏好性、分辨率到属水平,无法有效区分种、株水平的差异。宏基因组测序分辨率可达到种、甚至株水平,但其测序成本太高。因此,简化基因组测序技术使得在有限的预算内进行大规模研究成为可能。
3.通过简化基因组测序分析微生物的研究报道比较少,目前已报道的研究中有采用rad、ddrad、gbs进行微生物鉴定,但这些简化基因组都存在片段选择的问题:一、会造成数据的损失;二、酶切后得到的片段长度不一,导致pcr扩增存在偏好性,测序深度不均一,定量不准确。
4.iib型限制性内切酶,是一种能够识别双链dna分子中的某种特定核苷酸序列,并在识别位点上游和下游的特定距离进行切割,形成等长短片段(20-35bp)的核酸内切酶。基于iib型限制性内切酶的简化基因组测序技术,由于其可获得等长的内切酶片段(后续称为“标签”),既解决了上述简化基因组存在的片段选择、扩增偏好、定量不准确等问题,又能以低成本、高分辨率检测微生物,但目前基于iib型限制性内切酶特征的宏基因组测序技术并没有检测微生物的数据处理方法。
5.因此,如何提供一种基于iib型限制性内切酶特征的宏基因组测序数据处理系统是本领域亟待解决的问题。
技术实现要素:
6.本发明公开了一种基于iib型限制性内切酶特征的宏基因组测序数据处理系统及方法。
7.为了实现上述目的,本发明采用如下技术方案:
8.一种基于iib型限制性内切酶特征的宏基因组测序数据处理系统,包括:数据预处理模块、定性模块、定量模块和多酶切结果定性/定量合并模块;
9.数据预处理模块,包括:数据质控单元和电子酶切单元;其中,
10.数据质控单元对iib型限制性内切酶酶切片段测序结果进行数据质控,提取质控后含有iib型限制性内切酶标签的数据作为高质量测序数据;优选地,将双端测序数据根据reads之间的重叠区进行拼接、对单端测序和双端测序的测序数据质量进行数据质控,提取质控后含有iib型限制性内切酶标签的数据作为高质量测序数据;
11.电子酶切单元:从含有微生物基因组的数据库下载基因组,对每个基因组采用iib
型限制性内切酶进行电子酶切,即对基因组正向和反向分别提取特定识别序列后排序输出,生成每个基因组含有iib型限制性内切酶识别位点的标签;
12.所述标签是整个基因组的随机子集,能反馈全基因组特征,因此不需要对整个基因组进行分析,降低成本。
13.定性模块包括:第一微生物唯一标签数据库和定性分析单元;其中,
14.第一微生物唯一标签数据库:将电子酶切单元获得标签,在界、门、纲、目、科、属、种、株不同物种分类水平上判断上述每个标签的唯一性,输出不同物种分类水平、每个基因组的唯一标签,这些唯一标签组成第一微生物唯一标签数据库;
15.所述标签的唯一性是指在不同物种分类水平上,只在某一微生物中存在的标签;
16.定性分析单元:将样品的高质量测序数据和第一微生物唯一标签数据库比对,即通过构建哈希表的方式,检测高质量测序数据在第一微生物唯一标签数据库中是否有序列完全一致的标签,获得不同物种分类水平鉴定到的唯一标签,统计微生物不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度,并计算获得候选微生物定性信息;
17.所述定性是指根据是否检测到某微生物的唯一标签来确定是否存在该微生物;定性是根据是否检测到某微生物的唯一标签,如果有唯一标签,则判定该微生物存在;如果没有该微生物的唯一标签,则判定该微生物不存在;
18.唯一标签数为:唯一标签种类的数量;唯一标签数的深度为:唯一标签被检测到的数量;
19.定量模块包括:第二微生物唯一标签数据库和定量分析单元;其中,
20.第二微生物唯一标签数据库:通过对候选微生物进行假阳性过滤,然后对过滤后微生物基因组信息通过电子酶切单元和第一微生物唯一标签数据库,判断每个标签的唯一性,输出不同物种分类水平、每个基因组的唯一标签,这些唯一标签组成第二微生物唯一标签数据库。
21.定量分析单元:将样品的高质量测序数据和第二微生物唯一标签数据库比对,即通过构建哈希表的方式,检测高质量测序数据在第二微生物唯一标签数据库中是否有序列完全一致的标签,获得不同物种分类水平鉴定到的唯一标签,统计微生物不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度,并计算统计微生物的相对含量;
22.在不同分类水平上微生物基因组存在特异性的dna分子标记,即可以用唯一标签来检测某种微生物是否存在;而且这些特异性dna分子标记可以达到种水平、甚至株水平的分辨率,因此可以用来进行定性和定量分析。
23.多酶切结果定性/定量合并模块,包括:多酶切结果定性合并单元和多酶切结果定量合并单元;
24.多酶切结果定性合并单元:将一种以上iib型限制性内切酶的不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度进行累加合并,判断是否检测到某微生物的唯一标签,并重新计算微生物定性信息;
25.多酶切结果定量合并单元:将一种以上iib型限制性内切酶酶切的定量结果进行累加合并,作为最终的相对定量结果;
26.第一微生物唯一标签数据库和第二微生物唯一标签数据库中,某一物种分类在界、门、纲、目、科、属、种、株水平下,每个基因组的唯一序列标签必须只在某微生物基因组
存在且在其他微生物基因组中均没有的序列;
27.第一微生物唯一标签数据库和第二微生物唯一标签数据库中,合并某一物种分类水平下所有基因组唯一标签作为该物种分类水平下唯一标签数据库,不同物种分类水平对应不同的微生物唯一标签数据库,即每个物种分类水平都有一个微生物唯一标签数据库;例如:在种水平下,仅在a物种的基因组中存在且在除a物种外其他任何物种的基因组中都不存在的序列为a物种的唯一序列标签;
28.数据预处理模块中,微生物基因组或测序数据输入格式为fasta、fastq或fasta/fastq的压缩文件;
29.数据质控单元用于对任意单端测序数据进行质控和酶切或对任意双端测序数据进行拼接、质控和酶切,以获得含有iib型限制性内切酶酶切特异性位点的20bp至38bp的碱基片段;优选地,碱基片段的长度为25bp至35bp;
30.数据质控单元用于对重叠区碱基数≥10bp的双端测序数据进行拼接、去除质量值低于q20的碱基数超过整条reads碱基数20%的低质量reads、去除n碱基含量比例大于10%的的reads、去除不含iib型限制性内切酶酶切位点的reads。
31.第二微生物唯一标签数据库过滤假阳性采用gscore≥3作为过滤假阳性结果指标,其计算公式为:
[0032][0033]
所述si:一个样本中物种i的所有唯一标签的测序深度和;
[0034]
所述ti:一个样本中物种i被测到的唯一标签数的数目。
[0035]
第二微生物唯一标签数据库过滤假阳性采用设定某基因组检测到的唯一标签的数目≥2来过滤假阳性;
[0036]
定量分析,微生物的相对含量的计算公式为:
[0037][0038]
si:一个样本中物种i的所有唯一标签的测序深度和;
[0039]
ti:iib型限制性内切酶对物种i基因组进行电子酶切可获得的标签数。
[0040]
基于以上系统,本发明还提出了一种基于iib型限制性内切酶特征的宏基因组测序数据处理方法,所述方法包括以下步骤:
[0041]
步骤1:对iib型限制性内切酶酶切片段测序结果进行数据质控,提取质控后含有iib型限制性内切酶标签的数据作为高质量测序数据;
[0042]
步骤2:将样品的高质量测序数据和第一微生物唯一标签数据库比对,获得不同物种分类水平鉴定到的唯一标签,统计微生物不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度,并计算获得候选微生物定性信息、gscore值;
[0043]
步骤3:将一种以上iib型限制性内切酶的不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度进行累加合并,并重新计算获得候选微生物定性信息、gscore值;
[0044]
步骤4:将样品的高质量测序数据和第二微生物唯一标签数据库比对,获得不同物种分类水平鉴定到的唯一标签,统计微生物不同物种分类水平鉴定到的唯一标签数、唯一标签数的深度,并计算统计微生物的相对含量,即relative abundance值;
[0045]
步骤5:将一种以上iib型限制性内切酶酶切的定量结果进行累加合并,并重新计算统计微生物的相对含量,作为最终的相对定量结果。
[0046]
电子酶切单元,对于任何一种采用限制性内切酶的方法来说,都可以对基因组进行电子酶切获得酶切结果。但是iib型限制性内切酶得到的片段是等长的短片段,这是其他类型的限制性内切酶所不具有的特征。这些等长标签,即内切酶片段在基因组中均匀分布,且不同分类水平、不同基因组中存在特异性的分子标记,因此这些特异性的分子标记就组成了微生物唯一标签数据库。这是其他技术所不具有的。
[0047]
基于iib型限制性内切酶得到的酶切片段(标签)只是整个微生物基因组的子集,这些酶切片段沿微生物基因组均匀分布,而且这些标签的g c含量与全基因组的g c含量非常相似(pearson’s相关系数r=0.992)。此外,标签的数量与给定微生物的基因组大小高度相关(pearson’s相关系数r=0.976)。这表明,基于iib型限制性内切酶得到的标签足可以用来反馈微生物的全基因组特征,而不需要整个基因组测序。
[0048]
接下来,本发明通过寻找特异性的dna标记进行分类并通过测序reads深度对这些标记进行量化。本发明所需要的dna标记是特定于某个分类群、等长和每个基因组只出现一次的短dna片段。总的来说,分类级别越高,可用的标签就越多。在界水平,几乎所有的标签对界水平来说都是特异的,因此,无论哪种iib型限制性内切酶,细菌、真菌、古菌之间很少有共享的标签。这表明,本发明可以很容易地使用基于iib型限制性内切酶的数据计算出界水平之间的丰度比率,而在整个基因组水平上很难做到。因此,本发明首先将所有的特异性dna标记构建成参考数据库,即唯一标签数据库,然后采用“两步定量法”对微生物群落进行分析,第一步是定性,即检测样品中有哪些可能的微生物;第二步是对候选微生物进行相对定量。该策略能够最大化地、准确检出微生物,而且可以同时检出细菌、真菌和古菌。
[0049]
本发明的有益效果包括:本发明提出的基于iib型限制性内切酶特征的宏基因组测序数据处理系统基于唯一标签的“两步定量法”对具有iib型限制性内切酶特征的宏基因组测序数据进行分析处理,能够提高微生物检出率和准确性,降低假阳性,同时也减少了所需的计算资源和运行时间,提高效率,能够在短时间内快速、准确地获得样本中的物种信息和相对定量信息;进一步,本发明所述方法和系统能以低成本、高分辨率实现同时对细菌和真菌等微生物的鉴定和获得相对含量信息,填补了目前该技术在微生物检测上的空白;更进一步,本发明所述方法和装置解决了降解和痕量样品的微生物检测,拓宽了其应用范围。本发明通过构建唯一标签数据库,需要对从所有基因组得到的所有标签对应到不同的分类水平,然后判断每个标签在特定分类水平、特定基因组中的唯一性,才能构成唯一标签数据库;此外,本发明将定性和定量分开执行,并且自创相对定量公式,快速实现准确定性和定量。
附图说明
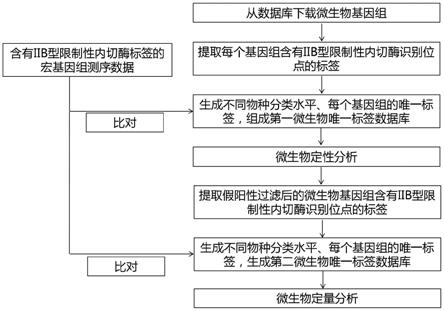
[0050]
图1是本发明实施例的基于iib型限制性内切酶特征的宏基因组测序数据处理示意图;
[0051]
图2是本发明基于iib型限制性内切酶特征的宏基因组测序数据处理系统示意图;
[0052]
图3是基于iib型限制性内切酶特征的宏基因组数据处理方法、宏基因组测序方法(wms)和16s rrna测序方法对美国人体微生物计划(hmp)所用标准品mock-msa1002的微生
物群落分类结果的比较图。
具体实施方式
[0053]
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0054]
实施例1
[0055]
以美国人体微生物计划(hmp)所用标准品mock-msa1002为对象(该标准品混合了20种等16s比例的细菌,技术方案如图1、图2所示)进行实验检验:选择iib型限制性内切酶如bcgi进行酶切建库,然后在hiseqxten se50平台上进行测序。
[0056]
1)针对每一个样品的测序数据,首先进行数据预处理,包括去除接头、去除含有n碱基比例大于8%的reads,去除低质量reads(质量值低于q30的碱基数超过整条reads碱基数15%),去除不含有bcgi酶切位点的reads,最终获得高质量reads。
[0057]
2)从ncbi refseq数据库下载180412个微生物基因组,包括细菌、真菌、古菌和病毒。
[0058]
3)采用bcgi对180412个微生物基因组进行电子酶切,采用哈希表记录每个微生物基因组酶切后的标签数目和种类,然后判断每个标签的物种唯一性,输出种水平、每个基因组的唯一标签,这些唯一标签组成第一微生物唯一标签数据库。
[0059]
4)将高质量reads比对至第一微生物唯一标签数据库,基于哈希表获得初步物种鉴定结果。
[0060]
5)针对初步物种鉴定结果,统计第一微生物唯一标签数据库中的唯一标签数、有支持序列的唯一标签数和其深度,并计算每种微生物的gscore。如果gscore≥5,则选择满足条件的所有微生物用于构建第二微生物唯一标签数据库。
[0061]
6)采用bcgi酶对步骤5的基因组进行电子酶切,采用哈希表记录每个微生物基因组酶切后的标签数目和种类,然后判断每个标签的唯一性,输出不同物种分类水平、每个基因组的唯一标签,这些唯一标签组成第二微生物唯一标签数据库。
[0062]
7)将步骤1获得的高质量测序数据与步骤6的第二微生物唯一标签数据库进行比对,统计比对到微生物唯一物种标签数据库2的唯一标签数目、有支持序列的唯一标签数和深度,从而计算物种相对含量。
[0063]
表1微生物物种相对含量
[0064][0065][0066]
实施例2
[0067]
以人粪便样本为实验材料(技术方案如图1、图2所示),采用iib型限制性内切酶如bcgi对5个人粪便基因组dna进行酶切、建库,然后在illumina nova pe150平台上进行测序。
[0068]
1)针对每一个样品的测序数据,首先进行数据预处理,包括使用flash对数据进行拼接,然后去除接头、去除含有n碱基比例大于8%的reads,去除低质量reads(质量值低于q30的碱基数超过整条reads碱基数15%),去除不含有bcgi酶切位点的reads,最终获得高质量reads。
[0069]
2)从ncbi refseq数据库下载180412个微生物基因组,包括细菌、真菌、古菌和病毒。
[0070]
3)采用bcgi对180412个微生物基因组进行电子酶切,采用哈希表记录每个微生物基因组酶切后的标签数目和种类,然后判断每个标签的物种唯一性,输出种水平、每个基因组的唯一标签,这些唯一标签组成第一微生物唯一标签数据库。
[0071]
4)将高质量reads比对至第一微生物唯一标签数据库,基于哈希表获得初步物种鉴定结果。
pe150平台上进行测序。
[0079]
1)针对每一个样品的测序数据,首先进行数据预处理,包括使用flash对数据进行拼接,然后去除接头、去除含有n碱基比例大于8%的reads,去除低质量reads(质量值低于q30的碱基数超过整条reads碱基数15%),去除不含有bcgi和bsaxi酶切位点的reads,最终获得高质量reads。
[0080]
2)从ncbi refseq数据库下载180412个微生物基因组,包括细菌、真菌、古菌和病毒。
[0081]
3)分别采用bcgi和bsaxi对180412个微生物基因组进行电子酶切,采用哈希表记录每个微生物基因组酶切后的标签数目和种类,然后判断每个标签的物种唯一性,输出种水平、每个基因组的唯一标签,这些唯一标签组成第一微生物唯一标签数据库。
[0082]
4)将高质量reads比对至第一微生物唯一标签数据库,基于哈希表获得初步物种鉴定结果。
[0083]
5)将bcgi和bsaxi的定性结果进行累加和合并。
[0084]
6)针对初步物种鉴定结果,统计第一微生物唯一标签数据库中的唯一标签数、有支持序列的唯一标签数和其深度,并计算每种微生物的gscore。如果gscore≥5,则选择满足条件的所有微生物用于构建第二微生物唯一标签数据库。
[0085]
7)采用bcgi和bsaxi酶对步骤6的基因组进行电子酶切,采用哈希表记录每个微生物基因组酶切后的标签数目和种类,然后判断每个标签的唯一性,输出不同物种分类水平、每个基因组的唯一标签,这些唯一标签组成第二微生物唯一标签数据库。
[0086]
8)将步骤1获得的高质量测序数据与步骤7的第二微生物唯一标签数据库进行比对,统计比对到第二微生物唯一标签数据库的唯一标签数目、有支持序列的唯一标签数和深度,然后计算相对含量。
[0087]
9)将bcgi和bsaxi的定量结果进行累加和合并,作为最终的相对定量结果。
[0088]
表3人腋下样本top20微生物物种相对含量
[0089]
[0090][0091]
实施例4
[0092]
基于iib型限制性内切酶特征的宏基因组数据处理方法、宏基因组测序方法(wms)和16s rrna测序方法对美国人体微生物计划(hmp)所用标准品mock-msa1002的微生物群落分类结果的比较。图3左侧面板中的堆积柱形图显示了三种方法生成的分类结果;在相应的右侧面板中,每一行(样本)的柱形图显示了与预期的分类结果相比,对应方法分类结果的精确度、召回率和相似性。本发明的方法在dna起始量为0.01ng的时候准确度为40.0%,与宏基因组测序方法(29.8%)和16s rrna测序方法(17.3%)的准确度形成鲜明对比。在召回率方面,本发明的方法可以达到宏基因组测序方法和16s rrna测序方法的灵敏度。另外,本发明的方法在dna起始量为0.01ng的时候与预期结果的相似性为84.8%,和宏基因组测序方法(86.3%)相当,但优于16s rrna测序方法(70.0%)。因此,在准确评估种水平微生物群落和相对丰度方面,宏基因组测序方法和16s rrna测序方法难以处理的样本,本发明的方法也具有优越的表现。
[0093]
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。
[0094]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对上述实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本发明中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
