1.本发明涉及属于化工医药技术领域,具体涉及新型化合物在制备预防和/或治疗冠状病毒感染的药物中的应用。
背景技术:
2.冠状病毒是一个大型病毒家族,严重程度从感冒到重症疾病不等,如中东呼吸综合征(mers)、严重急性呼吸综合征(sars)以及目前全球大流行的新型冠状病毒肺炎(corona virus disease 2019,covid-19)。
3.sars病毒β属b亚群冠状病毒,是一种起病急、传播快、病死率高的传染病,被传染的病人多数都与患者直接或间接接触,或生活在流行区内。
4.mers病毒是一种β属c亚群冠状病毒,全名为中东呼吸综合征冠状病毒(middle east respiratory syndrome coronavirus,简称mers-cov),感染后引发中东呼吸综合征(middle east respiratory syndrome,简称mers)。
5.新型冠状病毒(sars-cov-2)是人畜共患的正单链rna病毒,有包膜,属冠状病毒科。该科分为α、β、δ和γ四个属,sars-cov-2属于β属,被列为第7类冠状病毒,可引起新型冠状病毒性肺炎(covid-19)。临床表现多为发热、乏力,或有呼吸道症状;约半数患者多在一周后出现呼吸困难,严重者快速进展为急性呼吸窘迫综合征、脓毒症休克、难以纠正的代谢性酸中毒和出凝血功能障碍。早发现、早诊断、早隔离是有效控制covid-19疫情的关键。
6.冠状病毒s蛋白是病毒感染和疾病发生中的关键蛋白。已有研究证实,sars-cov可与人体血管紧张素转换酶2(ace2)结合并降低其表达水平,最终导致疾病发生。研究发现,与sars-cov相比,sars-cov-2的s蛋白中与人体ace2结合的5个关键氨基酸中有4个发生了变化,变化后的氨基酸在整体上维持了sars-cov的s蛋白与ace2相互作用的原结构构象,甚至亲和力更高,这可能导致sars-cov-2比sars-cov具有更强的感染力。由此推测sars-cov-2依然是通过ace2感染人体并导致疾病发生。
7.针对sars-cov-2感染的患者的治疗,许多学者都参考sars-cov和mers-cov的治疗经验进行研究。目前临床主要的治疗药物是抗病毒药物,例如hiv蛋白酶抑制剂洛匹那韦/利托那韦、广谱核苷类抗病毒药利巴韦林、氯喹,以及抗疟药磷酸氯喹、保肝抗炎药甘草酸二胺以及一些疏风解表、清热解毒、宣肺泄热,抗流感的中成药,但是现有临床数据表明,上述药物并非适用于所有人群,有些药物应用价格较高。
8.申请人在先授权专利,授权公告号cn108676067b,专利名称为一种预防hiv感染的新化合物及其制备方法,公开了化合物iii的具体结构通式如下:
[0009][0010]
其中,n为1≤n≤5的整数;
[0011]
r5表示如下基团:
[0012][0013]
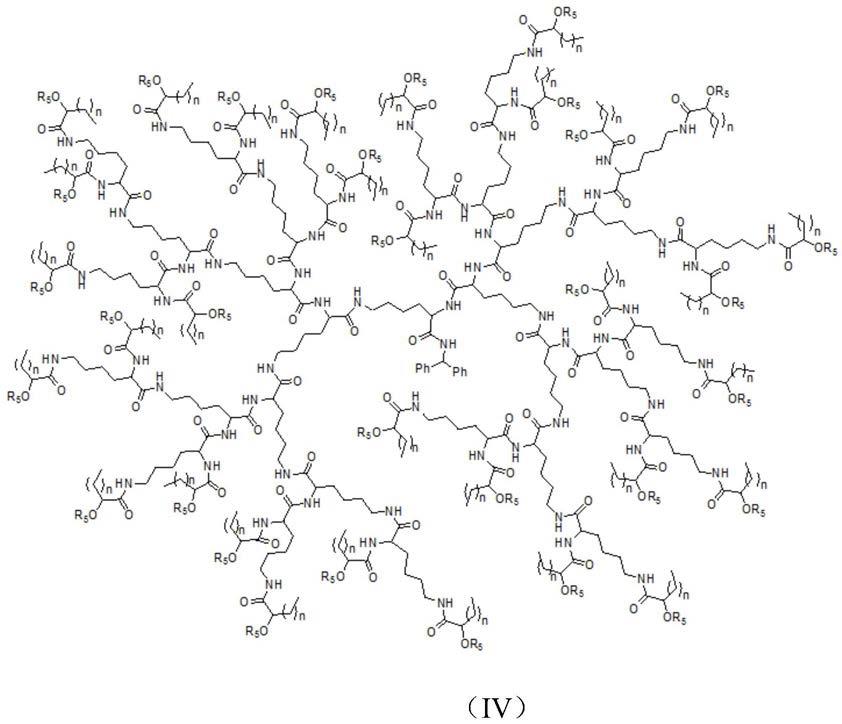
所述化合物iv的结构通式如式(iv)所示:
[0014]
其中,n为0≤n≤4的整数;
[0015]
r5表示如下基团:
[0016][0017]
该专利还公开了化合物iii和化合物iv的具体制备方法,其制备成本低,制备方法简单,酰溴活性高,与氨基反应迅速彻底,产生溴化氢被碱吸收,后处理简单且收率高;且封端基团片段3,6-二磺酸钠-1-萘酚取代α-溴或ε-溴原子反应温和,仅生成溴化氢,无缩合剂引入的多余片段,后处理简单,使用重结晶纯化的方法即可获得高纯度目标物,无需使用柱层析,非常适合工业化放大生产。在该申请中公开了化合物iii和化合物iv均具有很好的抗hiv活性,可有效预防hiv感染。
[0018]
目前全球新冠病毒肺炎感染人数的不断增加,针对该病毒的临床药物也在紧锣密鼓的研发中。无论是现在大流行的新冠肺炎,还是之前的sars、mers等病毒,要完全攻克都需要漫长的时间;作为抗击流行病毒的一员,申请人针对化合物iii和化合物iv做了新用途的开发,利用sars-cov、sars-cov-2以及mers-cov的假病毒,做了体外抑制试验,并证明了化合物iii、化合物iv及其衍生物对上述假病毒均具有较好的抑制作用,具有预防和/或治
疗冠状病毒的应用前景。
技术实现要素:
[0019]
本发明旨在为预防和/或治疗冠状病毒提供更多的治疗选择;本发明提供化合物iii在制备预防和/或治疗冠状病毒感染的药物中的应用。
[0020]
本发明的另一目的在于,提供化合物iv在制备预防和/或治疗冠状病毒感染的药物中的应用。
[0021]
本发明的另一目的在于,提供一种包括化合物iii、化合物iv以及药学上可以接受的辅料制备而成的药物,其中化合物iii的含量为0.01wt%~10wt%;其中化合物iv的含量为0.01wt%~10wt%。
[0022]
本发明针对的冠状病毒至少包括sars-cov、sars-cov-2以及mers-cov。
[0023]
本发明采取的技术方案是,本发明提供化合物iii、化合物iv在制备治疗因冠状病毒感染引起的呼吸道综合征的药物中的应用,所述呼吸道综合征包括中东呼吸综合征、严重急性呼吸综合征、新型冠状病毒肺炎等疾病。
[0024]
所述化合物iii的结构通式如式(iii)所示:
[0025]
其中,n为1≤n≤5的整数;
[0026]
r5表示如下基团:
[0027][0028]
因此,化合物iii分别代表了式(iii)所示结构通式中n=1、n=2、n=3、n=4和n=5的五个具体的化合物。
[0029]
所述化合物iv的结构通式如式(iv)所示:
[0030]
其中,n为0≤n≤4的整数;
[0031]
r5表示如下基团:
[0032][0033]
因此,化合物iv分别代表了式(iv)所示结构通式中n=0、n=1、n=2、n=3和n=4的五个具体的化合物。
[0034]
优选的,所述化合物iii的结构通式中,n=3或1。
[0035]
更为优选的,所述化合物iii的结构通式中,n=1。
[0036]
优选的,所述化合物iv的结构通式中,n=0或1。
[0037]
更为优选的,所述化合物iv的结构通式中,n=0。
[0038]
进一步的,化合物iii,r5基团上的na离子可以替换成其他金属离子;化合物iv,r5基团中,na离子可以替换成其他金属离子。
[0039]
所述金属离子可以选自:钾、锂、镁、钙、锌、铝。
[0040]
优选的,本发明制得的预防和/或治疗sars-cov感染、sars-cov-2感染以及mers-cov感染的药物,包含化合物iii和/或其衍生物以及药物学可接受的载体,其中化合物iii的含量为0.01wt%~10wt%,优选的为0.05wt%~1wt%。
[0041]
同样的,本发明制得的具体预防和/或治疗sars-cov感染、sars-cov-2感染以及mers-cov感染的药物,所述药物包含化合物iv和/或其衍生物以及药物学可接受的载体,其中化合物iv的含量为0.05wt%~1wt%。
[0042]
优选的,所述药物的剂型为片剂、颗粒剂、胶囊剂、液体制剂、凝胶剂、软膏剂、乳膏剂或注射剂。其中化合物iii或化合物iv的含量为0.01wt%~10wt%。
[0043]
更为优选的,所述药物的剂型为液体制剂,其中化合物iv的含量为0.05wt%~1wt%。
[0044]
所述液体制剂可以是喷雾剂、气雾剂、搽剂、洗涤剂或者消毒液等。
[0045]
进一步的,所述药物的剂型为片剂,其中化合物iii或化合物iv的最小单位剂量为1mg~100mg。
[0046]
进一步的,所述药物的剂型为颗粒剂,其中化合物iii或化合物iv的最小单位剂量为5mg~1000mg。
[0047]
进一步的,所述药物的剂型为胶囊剂,其中化合物iii或化合物iv的最小单位剂量为1mg~100mg。
[0048]
进一步的,所述药物的剂型为粉针剂,其中化合物iii或化合物iv的最小单位剂量为0.05mg~100mg。
[0049]
进一步的,所述药物为液体注射剂,注射剂总量范围在0.5~50ml,其中化合物iii或化合物iv的含量为0.5mg/ml-100mg/ml。
[0050]
优选的,所述药物为喷雾剂,其中化合物iii或化合物iv的含量为10~100mg/ml,更为优选的,药物iii或药物iv的含量为30~50mg/ml。
[0051]
优选的,所述药物的给药方式为口服给药、粘膜给药或注射给药。
[0052]
由于本发明的化合物分子量较大,一般的口服制剂的制备方法可能导致药效较低,因此,在一般口服制剂的制备过程中可以添加现有技术中的酶抑制剂或吸收促进剂,以增加肠道对药物的吸收。
[0053]
更为优选的为喷雾给药。
[0054]
另一个优选方案为,所述化合物为化合物iii,其中n=1,所述药物的剂型为液体制剂,化合物iii的含量为0.05wt%~1wt%。
[0055]
另一个优选方案为,所述化合物为化合物iv,其中n=0,所述药物的剂型为液体制剂,化合物iv的含量为0.05wt%~1wt%。
[0056]
本发明的另一目的在于,还提供化合物iii、化合物iv在制备治疗中东呼吸综合征、严重急性呼吸综合征、新型冠状病毒肺炎等呼吸系统疾病药物中的用途。包括covid-19、sars、中东呼吸综合征等。
[0057]
具体的,根据在先申请的专利cn108676067b可知,所述化合物iii的具体制备方法为:
[0058]
(1)制备化合物i,所述化合物i的结构通式如式(i)所示:
[0059]
其中,n为1≤n≤5的整数。
[0060]
其中式(i)所示化合物的制备方法为,将溴代酰溴溶解于有机溶剂并降温至-60~0℃,缓慢加入碱,然后再加入催化剂dmap,得到溴代酰溴/碱/dmap的有机溶剂体系;取bha-lys-lys2-lys4-lys8-16tfa溶解于有机溶剂的溶液,预冷至-40~-20℃后,滴加入溴代酰溴/碱/dmap的有机溶剂体系中,缓慢升温至-10~10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出有机溶剂层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除有机溶剂,即得式(i)所示化合物。
[0061]
本发明式(iii)所示化合物是以式(i)所示化合物为原料制备得到的;当式(i)所示化合物结构通式中n=1、n=2、n=3、n=4和n=5时,分别制备得到式(iii)所示结构通式中n=1、n=2、n=3、n=4和n=5的化合物。
[0062]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入碱和3,6-二磺酸钠-1-萘酚,升温至40~90℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入有机溶剂中,析出固体沉淀,过滤;将过滤所得固体使用c1、c2或c3醇与水的混合溶剂进行重结晶,即得如式(iii)所示的化合物。
[0063]
具体的,根据在先申请的专利cn110305188b可知,所述化合物iv的具体制备方法为:
[0064]
(1)制备化合物ii,所述化合物ii的结构通式如式(ii)所示:
[0065]
其中,n为0≤n≤4的整数。
[0066]
其中式(ii)所示化合物的制备方法,包括以下步骤:将溴代酰溴溶解于有机溶剂中并降温至-60~0℃,缓慢加入碱,然后再加入催化剂dmap,得到溴代酰溴/碱/dmap的有机溶剂体系,备用;取bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于有机溶剂的溶液,预冷至-40~-20℃后,滴加入溴代酰溴/碱/dmap的有机溶剂体系中,缓慢升温至-10~10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出有机溶剂层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除有机溶剂,即得式(ii)所示的化合物。
[0067]
优选地,将溴代酰溴2溶解于有机溶剂并降温至-40~-20℃,缓慢加入碱;所述溴代酰溴2、碱、dmap和bha-lys-lys2-lys4-lys8-lys16-32tfa的摩尔比为(32~64):(64~128):0.1:1;优选地,所述溴代酰溴、碱、dmap和bha-lys-lys2-lys4-lys8-lys16-32tfa的摩尔比为(38.4~48):(70.4~80):0.1:1;所述有机溶剂选自二氯甲烷,三氯甲烷,乙酸乙酯,乙酸异丙酯,甲苯或二甲苯;所述碱选自碳酸氢钠,碳酸钠,氢化钠,甲醇钠,乙醇钠,叔丁醇钠,三乙胺,二异丙基乙胺或吡啶。
[0068]
优选地,所述bha-lys-lys2-lys4-lys8-lys16-32tfa的制备方法同专利cn110305188b。
[0069]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入碱和3,6-二磺酸钠-1-萘酚,升温至40~90℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入有机溶剂中,析出固体沉淀,过滤;将过滤所得固体使用醇与水的混合溶剂进行重结晶,即得如式(iv)所示的化合物。
[0070]
优选地,所述步骤(2)碱和3,6-二磺酸钠-1-萘酚的摩尔比为(32~64):(32~64),优选地,所述碱和3,6-二磺酸钠-1-萘酚的摩尔比为(38~48):(35.2~41.6);所述碱和3,6-二磺酸钠-1-萘酚升温至50~70℃,搅拌反应;所述碱选自碳酸氢钠,碳酸钠,氢化钠,甲醇钠,乙醇钠,叔丁醇钠,三乙胺,二异丙基乙胺或吡啶;所述有机溶剂选自乙酸乙酯、乙酸异丙酯、乙腈、丙酮、四氢呋喃或异丙醇;所述醇选自甲醇、乙醇、正丙醇或异丙醇。
[0071]
与现有技术相比,本发明的有益效果为:本发明化合物iii、化合物iv应用在制备治疗sars-cov-2感染、sars感染或mers感染的药物中,活性较高,抗病毒效果较好。且本发明化合物制备成本低,制备方法简单,适合放大生产,且便于后期的临床推广应用。
具体实施方式
[0072]
为使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施方式,对本发明进行进一步的详细说明。应当理解的是,此处所描述的具体实施方式仅用以解释本发明,并不限定本发明的保护范围。实施例1
[0073]
本实施例提供n=1时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0074]
(1)将16mmol 4-溴丁酰溴溶解于二氯甲烷并降温至-60℃,缓慢加入32mmol的碳酸氢钠,然后再加入0.1mmol的催化剂dmap,得到4-溴丁酰溴/碳酸钠/dmap的三氯甲烷体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于二氯甲烷的溶液,预冷至-40℃后,滴加入4-溴丁酰溴/碳酸钠/dmap的三氯甲烷体系中,缓慢升温至-10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二氯甲烷层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二氯甲烷,即得n=1的式(i)所示的化合物,记为化合物1;
[0075]
(2)用无水dmf溶解步骤(1)获得的式(i)所示化合物,加入16mmol的甲醇钠和16mmol的3,6-二磺酸钠-1-萘酚,升温至40℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸乙酯中,析出固体沉淀,过滤;将过滤所得固体使用甲醇与水的混合溶剂进行重结晶,即得n=1的式(iii)所示化合物,记为化合物11。
[0076]
药物制备:称取10g上述化合物11,加入常规注射剂辅料,按照制作注射剂的一般工艺制成粉针剂;得到药物含量为100mg的药物1。实施例2
[0077]
本实施例提供n=2时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0078]
(1)将32mmol的3-溴丙酰溴溶解于三氯甲烷并降温至0℃,缓慢加入64mmol的碳酸钠,然后再加入0.1mmol的催化剂dmap,得到3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于三氯甲烷的溶液,预冷至-20℃后,滴加入3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系中,缓慢升温至10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出三氯甲
烷层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除三氯甲烷,即得n=2的式(i)所示化合物,记为化合物2;
[0079]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入32mmol的氢氧化钾和32mmol的3,6-二磺酸钾-1-萘酚,升温至90℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用乙醇与水的混合溶剂进行重结晶,即得n=2的式(iii)所示化合物,记为化合物12。
[0080]
药物制备:称取10g上述化合物12,加入常规片剂辅料,包括常规的吸收促进剂,按照制作片剂的一般工艺制成片剂;得到药物含量为50mg/片的药物2。实施例3
[0081]
本实施例提供n=3时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0082]
(1)将19mmol的5-溴戊酰溴溶解于乙酸乙酯并降温至-40℃,缓慢加入35mmol的氢化钠,然后再加入0.1mmol的催化剂dmap,得到5-溴戊酰溴/氢化钠/dmap的乙酸乙酯体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于乙酸乙酯的溶液,预冷至-30℃后,滴加入5-溴戊酰溴/氢化钠/dmap的乙酸乙酯体系中,缓慢升温至-5℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出乙酸乙酯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除乙酸乙酯,即得n=3的式(i)所示化合物,记为化合物3;
[0083]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入19mmol的氢氧化钠和17.6mmol的3,6-二磺酸钠-1-萘酚,升温至50℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙腈中,析出固体沉淀,过滤;将过滤所得固体使用正丙醇与水的混合溶剂进行重结晶,即得n=3的式(iii)所示化合物,记为化合物13。
[0084]
药物制备:称取1g上述化合物13,按照实施例2中的片剂制备方法制成片剂;得到药物含量为100mg/片的药物3。实施例4
[0085]
本实施例提供n=4时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0086]
(1)将24mmol的6-溴己酰溴溶解于乙酸异丙酯并降温至-20℃,缓慢加入40mmol的碳酸氢钠,然后再加入0.1mmol的催化剂dmap,得到6-溴己酰溴/碳酸钠/dmap的乙酸异丙酯体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于乙酸异丙酯的溶液,预冷至-35℃后,滴加入6-溴己酰溴/碳酸钠/dmap的乙酸异丙酯体系中,缓慢升温至0℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出乙酸异丙酯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除乙酸异丙酯,即得n=4的式(i)所示化合物,记为化合物4;
[0087]
(2)用无水dmf溶解步骤(1)获得的式(i)所示化合物,加入24mmol的氢化钙和20.8mmol的3,6-二磺酸钙-1-萘酚,升温至70℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入丙酮中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂
进行重结晶,即得n=4的式(iii)所示化合物,记为化合物14。
[0088]
药物制备:称取50g上述化合物14,按照实施例2中的片剂制备方法制成片剂;得到药物含量为5mg/片的药物4。实施例5
[0089]
本实施例提供n=5时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0090]
(1)将30mmol的7-溴庚酰溴溶解于乙酸异丙酯并降温至-30℃,缓慢加入45mmol的碳酸钠,然后再加入0.1mmol的催化剂dmap,得到7-溴庚酰溴/碳酸钠/dmap的乙酸乙酯体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于乙酸乙酯的溶液,预冷至-30℃后,滴加入7-溴庚酰溴/碳酸钠/dmap的乙酸乙酯体系中,缓慢升温至5℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出乙酸乙酯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除乙酸乙酯,即得式n=5的式(i)所示化合物,记为化合物5;
[0091]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入30mmol的氢氧化铝和25mmol的3,6-二磺酸铝-1-萘酚,升温至80℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入丙酮中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=5的式(iii)所示化合物,记为化合物15。
[0092]
药物制备:称取100g上述化合物15,按照实施例2中的片剂制备方法制成片剂;得到药物含量为1mg/片的药物5。实施例6
[0093]
本实施例提供n=1时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0094]
(1)将16mmol 3-溴丙酰溴溶解于二氯甲烷并降温至-60℃,缓慢加入32mmol的碳酸氢钠,然后再加入0.1mmol的催化剂dmap,得到3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于二氯甲烷的溶液,预冷至-40℃后,滴加入3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系中,缓慢升温至-10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二氯甲烷层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二氯甲烷,即得n=1的式(i)所示的化合物,记为化合物6;
[0095]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入16mmol的氢氧化锌和16mmol的3,6-二磺酸锌-1-萘酚,升温至40℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸乙酯中,析出固体沉淀,过滤;将过滤所得固体使用甲醇与水的混合溶剂进行重结晶,即得n=1的式(iii)所示化合物,记为化合物16。
[0096]
药物制备:称取1g上述化合物16,加入常规颗粒剂辅料,包括常规的吸收促进剂,按照制作颗粒剂的一般工艺制成颗粒剂;得到药物含量为1000mg/包的药物6,每包重量为10g。实施例7
[0097]
本实施例提供n=1时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0098]
(1)将16mmol 3-溴丙酰溴溶解于二氯甲烷并降温至-60℃,缓慢加入32mmol的碳酸氢钠,然后再加入0.1mmol的催化剂dmap,得到3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于二氯甲烷的溶液,预冷至-40℃后,滴加入3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系中,缓慢升温至-10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二氯甲烷层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二氯甲烷,即得n=1的式(i)所示的化合物,记为化合物7;
[0099]
(2)用无水dmf溶解步骤(1)获得的式(i)所示化合物,加入16mmol的甲醇钠和16mmol的3,6-二磺酸钠-1-萘酚,升温至40℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸乙酯中,析出固体沉淀,过滤;将过滤所得固体使用甲醇与水的混合溶剂进行重结晶,即得n=1的式(iii)所示化合物,记为化合物17。
[0100]
药物制备:称取1g上述化合物17,加入常规胶囊剂辅料,包括常规的吸收促进剂,按照制作胶囊剂的一般工艺制成胶囊剂;得到药物含量为100mg/颗的药物7。实施例8
[0101]
本实施例提供n=1时,式(i)和式(iii)所示化合物的一种制备方法,包括以下步骤:
[0102]
(1)将16mmol 3-溴丙酰溴溶解于二氯甲烷并降温至-60℃,缓慢加入32mmol的碳酸氢钠,然后再加入0.1mmol的催化剂dmap,得到3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-16tfa,取1mmol的bha-lys-lys2-lys4-lys8-16tfa溶解于二氯甲烷的溶液,预冷至-40℃后,滴加入3-溴丙酰溴/碳酸氢钠/dmap的二氯甲烷体系中,缓慢升温至-10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-16tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二氯甲烷层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二氯甲烷,即得n=1的式(i)所示的化合物,记为化合物8;
[0103]
(2)用无水dmso溶解步骤(1)获得的式(i)所示化合物,加入16mmol的氢氧化钾和16mmol的3,6-二磺酸钾-1-萘酚,升温至40℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸乙酯中,析出固体沉淀,过滤;将过滤所得固体使用甲醇与水的混合溶剂进行重结晶,即得n=1的式(iii)所示化合物,记为化合物18。
[0104]
药物制备:称取1g上述化合物18,加入常规注射剂辅料,按照制作注射剂的一般工艺制成粉针剂;得到药物含量为0.05mg/支的药物8。实施例9
[0105]
本实施例提供n=0时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0106]
(1)将32mmol的2-溴丙酰溴溶解于甲苯中并降温至-60℃,缓慢加入64mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴丙酰溴/三乙胺/dmap的甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol
的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于甲苯的溶液,预冷至-40℃后,滴加入2-溴丙酰溴/三乙胺/dmap的甲苯体系中,缓慢升温至-10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除甲苯,即得n=0的式(ii)所示化合物,记为化合物9;
[0107]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入32mmol的碳酸钠和32mmol的3,6-二磺酸钠-1-萘酚,升温至40℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入四氢呋喃中,析出固体沉淀,过滤;将过滤所得固体使用甲醇与水的混合溶剂进行重结晶,即得n=0的式(iv)所示化合物,记为化合物19。
[0108]
药物制备:称取10g上述化合物19,加入常规片剂辅料,按照实施例2中的片剂制备方法制成片剂;得到药物含量为50mg/片的药物9。实施例10
[0109]
本实施例提供n=1时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0110]
(1)将64mmol的2-溴丁酰溴溶解于二甲苯中并降温至0℃,缓慢加入128mmol的二异丙基乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴丁酰溴/二异丙基乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-20℃后,滴加入2-溴丁酰溴/二异丙基乙胺/dmap的二甲苯体系中,缓慢升温至10℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=1的式(ii)所示化合物,记为化合物100;
[0111]
(2)用无水dmf溶解步骤(1)获得的式(ii)所示化合物,加入64mmol的氢氧化钠锂和64mmol的3,6-二磺酸锂-1-萘酚,升温至90℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入异丙醇中,析出固体沉淀,过滤;将过滤所得固体使用乙醇与水的混合溶剂进行重结晶,即得n=1的式(iv)所示化合物,记为化合物10。
[0112]
药物制备:称取10g上述化合物10,加入常规片剂辅料,按照实施例2中的片剂制备方法制成片剂;得到药物含量为100mg/片的药物10。实施例11
[0113]
本实施例提供n=2时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0114]
(1)将38.4mmol的2-溴戊酰溴溶解于甲苯中并降温至-40℃,缓慢加入70.4的吡啶,然后再加入0.1mmol的催化剂dmap,得到2-溴戊酰溴/吡啶/dmap的甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于甲苯的溶液,预冷至-35℃后,滴加入2-溴戊酰溴/吡啶/dmap的甲苯体系中,缓慢升温至-5℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除甲苯,即得n=2的式(ii)所示化合物,记为化合物200;
[0115]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入38mmol的氢氧化铝和35.2mmol的3,6-二磺酸铝-1-萘酚,升温至55℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸乙酯中,析出固体沉淀,过滤;将过滤所得固体使用正丙醇与水的混合溶剂进行重结晶,即得n=2的式(iv)所示化合物,记为化合物20。
[0116]
药物制备:称取10g上述化合物20,加入常规片剂辅料,按照实施例2中的片剂制备方法制成片剂;得到药物含量为30mg/片的药物11。实施例12
[0117]
本实施例提供n=3时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0118]
(1)将48mmol的2-溴己酰溴溶解于二甲苯中并降温至-20℃,缓慢加入80mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴己酰溴/三乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-30℃后,滴加入2-溴己酰溴/三乙胺/dmap的二甲苯体系中,缓慢升温至5℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=3的式(ii)所示化合物,记为化合物300;
[0119]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入48mmol的氢氧化锌和41.6mmol的3,6-二磺酸锌-1-萘酚,升温至75℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=3的式(iv)所示化合物,记为化合物30。
[0120]
药物制备:称取50g上述化合物30,加入常规片剂辅料,按照实施例2中的片剂制备方法制成片剂;得到药物含量为10mg/片的药物12。实施例13
[0121]
本实施例提供n=4时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0122]
(1)将40mmol的2-溴庚酰溴溶解于二甲苯中并降温至-10℃,缓慢加入90mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴庚酰溴/三乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-25℃后,滴加入2-溴庚酰溴/三乙胺/dmap的二甲苯体系中,缓慢升温至3℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=4的式(ii)所示化合物,记为化合物400;
[0123]
(2)用无水dmf溶解步骤(1)获得的式(ii)所示化合物,加入60mmol的碳酸钠和60mmol的3,6-二磺酸钠-1-萘酚,升温至65℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=4的式(iv)所示化合物,记为化合物40。
[0124]
药物制备:称取100g上述化合物40,加入常规片剂辅料,按照实施例2中的片剂制
备方法制成片剂;得到药物含量为1mg/片的药物13。实施例14
[0125]
本实施例提供n=0时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0126]
(1)将40mmol的2-溴庚酰溴溶解于二甲苯中并降温至-10℃,缓慢加入90mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴庚酰溴/三乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-25℃后,滴加入2-溴庚酰溴/三乙胺/dmap的二甲苯体系中,缓慢升温至3℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=4的式(ii)所示化合物,记为化合物500;
[0127]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入60mmol的碳酸钠和60mmol的3,6-二磺酸钠-1-萘酚,升温至65℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=0的式(iv)所示化合物,记为化合物50。
[0128]
药物制备:称取1g上述化合物50,加入常规颗粒剂辅料,包括常规的吸收促进剂,按照制作颗粒剂的一般工艺制成颗粒剂;得到药物含量为5mg/包的药物14,每包总重量为1g。实施例15
[0129]
本实施例提供n=0时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0130]
(1)将48mmol的2-溴己酰溴溶解于二甲苯中并降温至-20℃,缓慢加入80mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴己酰溴/三乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-30℃后,滴加入2-溴己酰溴/三乙胺/dmap的二甲苯体系中,缓慢升温至5℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=0的式(ii)所示化合物,记为化合物600;
[0131]
(2)用无水dmso溶解步骤(1)获得的式(ii)所示化合物,加入60mmol的氢氧化锂和60mmol的3,6-二磺酸锂-1-萘酚,升温至65℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=0的式(iv)所示化合物,记为化合物60。
[0132]
药物制备:称取1g上述化合物60,加入常规胶囊剂辅料,包括常规的吸收促进剂,按照制作胶囊剂的一般工艺制成胶囊剂;得到药物含量为1mg/颗的药物15。实施例16
[0133]
本实施例提供n=0时,式(ii)和式(iv)所示化合物的一种制备方法,包括以下步骤:
[0134]
(1)将40mmol的2-溴庚酰溴溶解于二甲苯中并降温至-10℃,缓慢加入90mmol的三乙胺,然后再加入0.1mmol的催化剂dmap,得到2-溴庚酰溴/三乙胺/dmap的二甲苯体系,备用;根据专利cn110305188b所述方法合成得到bha-lys-lys2-lys4-lys8-lys16-32tfa,取1mmol的bha-lys-lys2-lys4-lys8-lys16-32tfa溶解于二甲苯的溶液,预冷至-25℃后,滴加入2-溴庚酰溴/三乙胺/dmap的二甲苯体系中,缓慢升温至3℃反应;检测体系中无bha-lys-lys2-lys4-lys8-lys16-32tfa剩余后,缓慢往体系中滴加饱和食盐水淬灭反应,搅拌,分液,分出二甲苯层,分别用0.5mol/l稀盐酸和饱和食盐水各洗涤第一次,减压蒸除二甲苯,即得n=0的式(ii)所示化合物,记为化合物700;
[0135]
(2)用无水dmf溶解步骤(1)获得的式(ii)所示化合物,加入60mmol的氢化钙和60mmol的3,6-二磺酸钙-1-萘酚,升温至65℃,搅拌反应;反应结束,过滤除去不溶物,将滤液缓慢滴加入乙酸异丙酯中,析出固体沉淀,过滤;将过滤所得固体使用异丙醇与水的混合溶剂进行重结晶,即得n=0的式(iv)所示化合物,记为化合物70。
[0136]
药物制备:称取1g上述化合物70,加入常规注射剂辅料,按照制作注射剂的一般工艺制成液体注射剂;得到药物含量为100mg/ml的药物16。实施例17
[0137]
本实施例与实施例9的不同之处在于,本实施例将实施例9中的化合物19制备成常规喷雾剂。
[0138]
药物制备:称取10g上述化合物9,加入常规喷雾剂辅料,按照制作喷雾剂的一般工艺制成喷雾剂;得到药物含量为50mg/ml的药物17。实施例18
[0139]
本实施例研究实施例1制备得到的化合物11、粉针剂药物1,实施例9制备得到的化合物19和片剂药物9以及实施例17制备得到的喷雾剂药物17的抗sars-cov-2、sars以及mers活性。
[0140]
1、试验方法
[0141]
1.1sars-cov-2假病毒的制备
[0142]
①
sars-cov-2s基因设计、合成与表达质粒构建
[0143]
根据genbank(accession number mn975262)中的序列信息,合成sars-cov-2的s基因序列,并对序列中胞浆区肽段(kfdeddsepvlkgvklhyt)进行局部缺失突变,该基因被命名为sopti。将上述基因通过酶切、连接插入到质粒载体pcdna3.1( )和pci-neo中,用酶切、连接,联合同源重组方法将s基因置换phcmv-e1e2载体里的hcv包膜蛋白e1e2基因。
[0144]
②
sars-cov-2s基因表达产物鉴定
[0145]
将293t细胞接种于24孔板,将上述s基因表达质粒用lipofectamine 2000试剂转染293t细胞,转染24h后,将细胞重新接种于96孔板,继续培养24h,用免疫荧光法检测s蛋白的表达。
[0146]
③
获得感染性的sars-cov-2假病毒
[0147]
将sars-cov-2s基因表达质粒与慢病毒骨架质粒pcmv-gag/pol、pcmv-rev和plenti-egfp共转染293t细胞。转染60h后,收集细胞培养上清,用0.45μm微量滤器过滤除去上清中可能残留的293t细胞后,用于靶细胞感染。vero细胞提前12h接种于96孔板,每孔8000个细胞。用于假病毒感染时,每孔先吸除20μl培养液,再加入假病毒20μl,混匀,置于细
胞培养箱内,6h后吸除培养液,每孔加入完全dmem培养液100μl。置于细胞培养箱内,18h后每间隔12h于荧光显微镜下观察细胞内是否出现绿色荧光,并用细胞成像及分析系统(biotek cytation 5imaging reader)对egfp阳性细胞进行计数,计算感染滴度(ffu/ml,ffu为focus forming unit),得到感染滴度约200ffu的sars-cov-2假病毒液。
[0148]
1.2sars假病毒的制备
[0149]
参考现有技术sars假病毒制备方法构建了不依赖于bsl-3级生物安全条件的sars假病毒。
[0150]
具体可以为,按照4
×
105~6
×
105/ml的浓度接种293t细胞至培养皿或培养瓶中,待长至80%-90%时进行转染,按照lipo2000提供说明书进行操作,转染24h后,加入vsvδg-s病毒稀释液,37℃孵育1h后,倒掉,用含2%新生牛血清的pbs清洗两遍,然后加入新鲜培养液,继续培养。收取细胞培养液上清,1500rpm离心5min,取上清,0.45μm的滤器过滤,分装,冻于-80℃备用。
[0151]
vero e6细胞按2
×
104个/孔比例接种96孔细胞培养板,每孔100μl,12h后,观察生长情况,待长成单层用于假病毒感染实验。取病毒液进行3倍系列稀释,每孔加入100μl稀释液,继续培养24h。化学发光检测,吸弃100μl培养基,加入100μl发光底物,检测相对荧光强度(rlu),reed-meuench法计算病毒滴度。得到感染滴度1
×
106tcid
50
/ml的sars假病毒(vsvδg-s)。
[0152]
1.3mers假病毒的制备
[0153]
参考现有技术mers假病毒制备方法,构建了不依赖于bsl-3级生物安全条件的mers假病毒。
[0154]
具体可以为,按照4
×
105~6
×
105/ml的浓度接种293t细胞至培养皿或培养瓶中,待长至80%-90%时进行转染,按照lipo2000提供说明书进行操作,转染24h后,加入vsvδg-m病毒稀释液,37℃孵育1h后,倒掉,用含2%新生牛血清的pbs清洗两遍,然后加入新鲜培养液,继续培养。收取细胞培养液上清,1500rpm离心5min,取上清,0.45μm的滤器过滤,分装,冻于-80℃备用。
[0155]
vero e6细胞按2
×
104个/孔比例接种96孔细胞培养板,每孔100μl,12h后,观察生长情况,待长成单层用于mers假病毒感染实验。取vsvδg-m病毒进行3倍系列稀释,每孔加入100μl稀释液,继续培养24h。化学发光检测,吸弃100μl培养基,加入100μl发光底物,检测相对荧光强度(rlu),reed-meuench法计算病毒滴度。得到感染滴度1
×
106tcid
50
/ml mers假病毒(vsvδg-m)。
[0156]
1.4受试样品及实验分组
[0157]
选择化合物11、药物1,化合物19和药物9以及药物17作为受试样品,依次命名为供试品1、供试品2、供试品3、供试品4以及供试品5,阳性药选择现有药物氯喹。空白对照组为假病毒感染,未加药物处理的细胞;细胞对照组为正常生长,未感染、也未加药物处理的细胞;阳性对照组为假病毒感染,加入了抗病毒药物氯喹处理细胞。
[0158]
1.5药物的感染抑制率
[0159]
将vero e6细胞接种于96孔板,12h后用于sars-cov-2假病毒、sars假病毒以及mers假病毒感染。受检测样品(阳性药和供试品1~4)分别用dmem培养基从最高测试浓度起连续3倍梯度稀释8个浓度,取不同浓度稀释的样品100μl分别与100μl的病毒液混匀后置于
37℃培养箱孵育30min。然后吸除vero e6细胞的培养液,将病毒/药物混合液加入细胞培养孔中,6h后换培养液,继续培养30h,进行细胞计数。
[0160]
计算公式:感染抑制率(%)=100-(样品组-细胞对照组)/(空白对照组-细胞对照组)
×
100。
[0161]
2、实验结果
[0162]
受试样品对sars-cov-2假病毒、sars假病毒以及mers假病毒侵入vero e6细胞的抑制活性检测结果如表1所示。注:ic50:50%抑制效果时抑制剂的浓度;表中的所有浓度均为样品与病毒孵育时浓度;表1阳性药的浓度是指制剂样品根据其活性成分含量折算的浓度。表1样品对3种假病毒入侵细胞的抑制作用
[0163]
3、实验结论
[0164]
由上表可知,本发明的化合物iii及其制得的药物,其ic50的值整体上低于阳性对照组,说明化合物iii对sars-cov、sars-cov-2以及mers-cov假病毒均有一定的抑制作用,特别是针对sars-cov-2病毒表现出更好的抑制效果。同时,本发明的化合物iv以及其制得的药物9、药物17的ic50的值均低于阳性对照组,说明化合物iv对于sars-cov、sars-cov-2以及mers-cov假病毒有较好的抑制作用,且效果明显优于阳性对照组,特别是喷雾剂药物17,ic50值最低,说明化合物iv制备得到的喷雾剂对sars-cov-2假病毒的抑制效果更好。综上,本发明的化合物iii、化合物iv制得的药物可以作为预防和/或治疗sars-cov、sars-cov-2以及mers-cov病毒引起的相关疾病较好的候选药物。可以将本发明制得的药物用于sars-cov、sars-cov-2以及mers-cov等冠状病毒引起的严重急性呼吸综合征、新型冠状病毒肺炎、中东呼吸综合征等疾病。
[0165]
显然,本发明的上述实施例仅仅是为清楚地说明本发明技术方案所作的举例,而并非是对本发明的具体实施方式的限定。凡在本发明权利要求书的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。