多手性位点化合物a及其手性异构体的分离检测方法及其在化合物a的合成工艺中的应用
技术领域
1.本发明涉及药物合成领域,尤其涉及一种多手性位点化合物a及其手性异构体的分离检测方法及其在化合物a的合成工艺中的应用。
背景技术:
2.在医药领域中,由叔丁(s)
‑2‑
((1r,2r)
‑3‑
((r)
‑4‑
苄基
‑2‑
恶唑烷酮
‑3‑
基)
‑1‑
羟基
‑2‑
甲基
‑3‑
氧代丙基)吡咯烷
‑1‑
羧化物为中间体可制备出高活性的抗癌药物,可用于治疗乳腺癌等癌症。
[0003][0004]
叔丁基(s)
‑2‑
((1r,2r)
‑3‑
((r)
‑4‑
苄基
‑2‑
恶唑烷酮
‑3‑
基)
‑1‑
羟基
‑2‑
甲基
‑3‑
氧代丙基)吡咯烷
‑1‑
羧化物的结构为上述的compound a(化合物a),其由n
‑
boc
‑
l
‑
脯氨醛和(r)
‑4‑
苄基
‑3‑
丙酰基
‑2‑
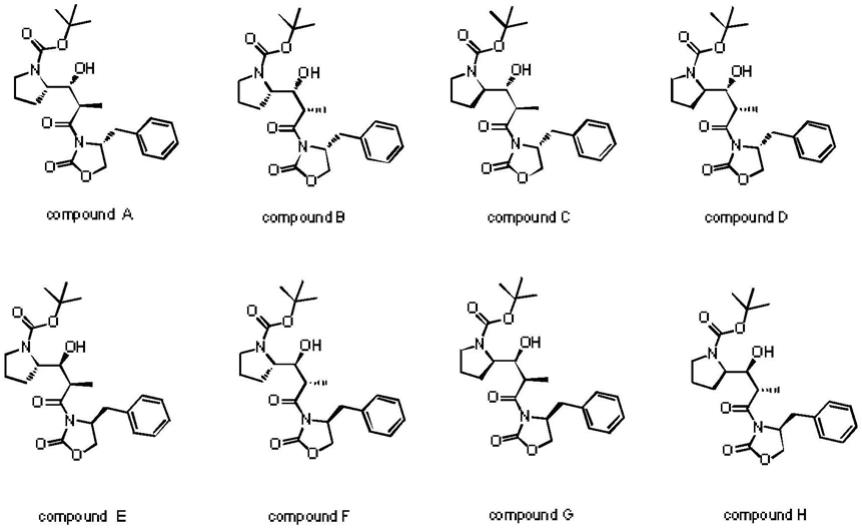
恶唑烷酮反应合成,但是,在该合成反应过程中由于条件控制不当或原料手性不纯,易产生上述另外七种结构所示化合物compound b(化合物b)、compound c(化合物c)、compound d(化合物d)、compound e(化合物e)、compound f(化合物f)、compound g(化合物g)、compound h(化合物h)中的一种或几种。
[0005]
化合物a作为重要的医药中间体往后会继续反应生成更为复杂的化合物,如果在化合物a的合成过程中手性不做控制,往后将极大的影响产品质量且后续的分离纯化会更麻烦,损失更大。因此对合成产物进行严格手性控制尤显重要。
[0006]
目前,需多个正向色谱结合,或正向色谱与反向色谱结合多个分析方法结合才能判断合成产物是否含有手性异构体和确定含有的是哪种手性异构体,过程比较繁琐和麻烦,且无法准确定量。
技术实现要素:
[0007]
本发明的目的在于提供一种既可以准确检测化合物a的含量和手性纯度,同时检测其手性异构体的具体结构和含量的快速、准确、可靠的分离检测多手性位点化合物a及其手性异构体的分离检测方法及其在化合物a的合成工艺中的应用。
[0008]
为实现以上目的,本发明提供如下技术方案:
[0009]
一种多手性位点化合物a及其手性异构体的分离检测方法,采用液相色谱法检测;
[0010]
所述化合物a为叔丁基(s)
‑2‑
((1r,2r)
‑3‑
((r)
‑4‑
苄基
‑2‑
恶唑烷酮
‑3‑
基)
‑1‑
羟基
‑2‑
甲基
‑3‑
氧代丙基)吡咯烷
‑1‑
羧化物;
[0011]
所述液相色谱法检测的条件包括:色谱柱为多糖涂敷型手性色谱柱或化学键合型手性色谱柱;流动相为第一溶剂和第二溶剂按体积比为(90
‑
97):(3
‑
10)的混合物,所述第一溶剂为正己烷,所述第二溶剂为异丙醇或乙醇。
[0012]
通过采用液相色谱法,选择采用多糖涂敷型手性色谱柱或化学键合型手性色谱柱,其类型为直链淀粉
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)或纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)手性色谱柱,及选择第一溶剂(正己烷)和第二溶剂(异丙醇或乙醇)按体积比为(90
‑
97):(3
‑
10)的混合物作为流动相;实现用同一色谱条件下有效分离和定量检测多手性位点化合物a及其手性异构体,还可以准确检测化合物a的含量和手性纯度,同时能够检测其手性异构体的具体结构和含量,该分离检测方法检测时间仅需30
‑
60min,快速、准确性强、操作简便。
[0013]
优选地,所述流动相为第一溶剂和第二溶剂按体积比为93:7的混合物,能够更充分溶解样品,并将手性异构体得到更好的分离,提高分离度和灵敏度。
[0014]
在一些实施方式中,所述流动相的流速为0.8
‑
1.2ml/min;优选地,所述流动相的流速为1.0ml/min,由此可以在最佳流速下将手性异构体得到良好的分离,提高分离度和耐用性。
[0015]
在一些实施方式中,用于所述液相色谱法检测的色谱柱柱温为15℃~30℃;优选地,所述色谱柱柱温为20℃,可以减小对色谱柱的损伤,同时进一步提高化合物a及其手性异构体的分离度。
[0016]
在一些实施方式中,用于所述液相色谱法检测的检测器为可变波长扫描紫外检测器,检测波长为200
‑
220nm;优选地,所述检测波长为210nm,可以更准确检测化合物a及其手性异构体的含量,检测限可达0.0003mg/ml,定量限可达0.001mg/ml。
[0017]
在一些实施方式中,用于所述液相色谱法检测的洗脱方式为流动相等度洗脱;所述的多糖涂敷型手性色谱柱为大赛璐chiralpak ad
‑
h柱或大赛璐chiralcel od
‑
h柱;所述化学键合型手性色谱柱为大赛璐chiralpak ia柱;优选地,所述大赛璐chiralpak ad
‑
h柱、大赛璐chiralcel od
‑
h柱及大赛璐chiralpak ia柱的规格为250mm*4.6mm,5um,可以有效实现化合物a及其手性异构体的同时检测,使手性异构体得到良好的分离且有更好的峰形,提高分离度和灵敏度,提高检测的稳定性、重复性和准确度,得到更准确的化合物a的手性纯度和手性异构体的含量结果。
[0018]
在一些实施方式中,用于所述液相色谱法检测的样品通过稀释剂溶解或稀释;优选地,所述稀释剂采用所述流动相。样品溶液的浓度为0.8
‑
1.2mg/ml;样品溶液的进样量为5
‑
10μl;优选地,所述样品溶液的浓度为1mg/ml。
[0019]
通过选择合适的进样量的同时使得响应值满足测定的需要,避免连续高浓度进样,采用合适的进样浓度,以延长色谱柱的使用寿命;并且在上述最优进样浓度1mg/ml条件下,主要成分化合物a与其他手性异构体的峰之间完全分离。
[0020]
本技术还提供了一种多手性位点化合物a及其手性异构体的分离检测方法在化合物a的合成工艺中的应用。
[0021]
本发明的有益效果:
[0022]
本技术通过采用液相色谱法,选择采用多糖涂敷型手性色谱柱或化学键合型手性色谱柱,及选择第一溶剂(正己烷)和第二溶剂(异丙醇或乙醇)按体积比为(90
‑
97):(3
‑
10)的混合物作为流动相;实现用同一色谱条件下有效分离和定量检测多手性位点化合物a及其手性异构体,还可以准确检测化合物a的含量和手性纯度,同时能够检测其手性异构体的具体结构和含量,该分离检测方法快速、准确性强、操作简便。
[0023]
另外,通过本技术的分离检测方法检测,若检测出化合物的某种手性异构体,可反推是由于那种原因条件控制不当导致,从而调整该条件以全面保障产品质量,并有效降低成本,提高产品收率,避免影响化合物a的后续应用。
附图说明
[0024]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对本发明范围的限定。
[0025]
图1为本技术实施例1的化合物a及其手性异构体定位的液相色谱图;
[0026]
图2为本技术实施例1的化合物a及其手性异构体定位的定量限溶液的液相色谱图;
[0027]
图3为本技术实施例1的合成化合物a样品的液相色谱图;
[0028]
图4为本技术实施例2的化合物a及其手性异构体定位的液相色谱图;
[0029]
图5为本技术实施例2的化合物a及其手性异构体定位的定量限溶液的液相色谱图;
[0030]
图6为本技术实施例2的合成化合物a样品的液相色谱图;
[0031]
图7为本技术实施例3的化合物a及其手性异构体定位的液相色谱图;
[0032]
图8为本技术实施例3的化合物a及其手性异构体定位的定量限溶液的液相色谱图;
[0033]
图9为本技术实施例3的合成化合物a样品的液相色谱图;
[0034]
图10为本技术实施例4的化合物a及其手性异构体定位的液相色谱图;
[0035]
图11为对比例1的化合物a及其手性异构体定位的液相色谱图;
[0036]
图12为对比例2的化合物a及其手性异构体定位的液相色谱图;
[0037]
图13为对比例3的化合物a及其手性异构体定位的液相色谱图。
[0038]
图14为对比例4的化合物a及其手性异构体定位的液相色谱图。
[0039]
图15为对比例5的化合物a及其手性异构体定位的液相色谱图。
具体实施方式
[0040]
下面将结合具体实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0041]
下述实施例中所用的合成化合物a样品由n
‑
boc
‑
l
‑
脯氨醛和(r)
‑4‑
苄基
‑3‑
丙酰基
‑2‑
恶唑烷酮反应合成。
[0042]
实施例1
[0043]
检测条件:
[0044]
仪器:agilent 1100液相色谱仪,vwd检测器
[0045]
色谱柱:大赛璐chiralcel od
‑
h(250mm*4.6mm,5um);
[0046]
流动相:正己烷:乙醇=93:7;
[0047]
洗脱方式:等度洗脱;
[0048]
流速:1.0ml/min
[0049]
柱温:20℃
[0050]
检测器波长:210nm
[0051]
进样体积:10μl
[0052]
稀释剂:正己烷:乙醇=93:7;
[0053]
运行时间:30min
[0054]
实验步骤:
[0055]
(1)定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0056]
(2)对照品定量限溶液配置:分别将上述8个对照品溶液,各取1ml分别用稀释剂溶解并稀释配制成0.001mg/ml的定量限溶液。
[0057]
(3)样品溶液配制:取合成的化合物a样品100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,配制成1mg/ml的样品溶液。
[0058]
(4)将上述步骤(1)配制的对照品溶液、步骤(2)配制的定位限溶液和步骤(3)配制的样品溶液置于进样器中,平衡后,将其注入液相色谱仪,采用上述检测条件检测,记录色谱图。
[0059]
实验结果:
[0060]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图1所示,对应数据结果如下表1所示。
[0061]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h的定量限溶液的液相色谱图如图2所示,对应数据结果如下表2所示;
[0062]
合成化合物a样品的液相色谱图如图3所示,对应数据结果如下表3所示。
[0063]
表1 化合物a及其手性异构体定位结果
[0064]
分析物保留时间分离度化合物b6.481min
‑
化合物g8.060min3.71化合物c9.081min2.00化合物f11.251min3.62化合物a12.202min1.60化合物e14.487min2.89化合物h16.060min1.70化合物d18.705min2.53
[0065]
表2 化合物a及其手性异构体定量限结果
[0066]
分析物保留时间信噪比化合物b6.481min73.4化合物g8.060min59.4化合物c9.081min46.6化合物f11.251min29.8化合物a12.202min37.8化合物e14.487min30.5化合物h16.060min26.6化合物d18.705min23.1
[0067]
表3 合成化合物a样品的手性纯度结果
[0068]
分析物保留时间纯度化合物b6.481min0.266%化合物g8.060min 化合物c9.081min0.270%化合物f11.251min0.435%化合物a12.202min99.029%化合物e14.487min 化合物h16.060min 化合物d18.705min [0069]
实施例2
[0070]
检测条件:
[0071]
仪器:agilent 1100液相色谱仪,vwd检测器
[0072]
色谱柱:大赛璐chiralpak ad
‑
h(250mm*4.6mm,5um);
[0073]
流动相:正己烷:异丙醇=90:10;
[0074]
洗脱方式:等度洗脱;
[0075]
流速:1.0ml/min
[0076]
柱温:20℃
[0077]
检测器波长:210nm
[0078]
进样体积:5μl
[0079]
稀释剂:正己烷:异丙醇=90:10;
[0080]
运行时间:40min
[0081]
实验步骤:
[0082]
(1)定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0083]
(2)对照品定量限溶液配置:分别将上述8个对照品溶液,各取1ml分别用稀释剂溶解并稀释配制成0.001mg/ml的定位限溶液。
[0084]
(3)样品溶液配制:取合成的化合物a样品100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,配制成1mg/ml的样品溶液。
[0085]
(4)将上述步骤(1)配制的对照品溶液、步骤(2)配制的定位限溶液和步骤(3)配制的样品溶液置于进样器中,平衡后,将其注入液相色谱仪,采用上述检测条件检测,记录色谱图。
[0086]
实验结果:
[0087]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图4所示,对应数据结果如下表4所示。
[0088]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h的定量限溶液的液相色谱图如图5所示,对应数据结果如下表5所示;
[0089]
合成化合物a样品的液相色谱图如图6所示,对应数据结果如下表6所示。
[0090]
表4 化合物a及其手性异构体定位结果
[0091]
分析物保留时间分离度化合物g7.937min
‑
化合物f8.336min0.71化合物b11.097min4.54化合物e16.476min8.10化合物h18.872min2.80化合物d20.552min1.87化合物c23.779min3.14化合物a29.881min2.09
[0092]
表5 化合物a及其手性异构体定量限结果
[0093]
分析物保留时间信噪比化合物g7.937min26.7化合物f8.336min15.6化合物b11.097min25.4化合物e16.476min14.7化合物h18.872min12.1化合物d20.552min11.7化合物c23.779min11.7化合物a29.881min
‑
[0094]
表6 合成化合物a样品的手性纯度结果
[0095][0096][0097]
实施例3
[0098]
检测条件:
[0099]
仪器:agilent 1100液相色谱仪,vwd检测器
[0100]
色谱柱:大赛璐chiralpak od
‑
h(250mm*4.6mm,5um);
[0101]
流动相:正己烷:异丙醇=97:3;
[0102]
洗脱方式:等度洗脱;
[0103]
流速:1.2ml/min
[0104]
柱温:30℃
[0105]
检测器波长:210nm
[0106]
进样体积:10μl
[0107]
稀释剂:正己烷:异丙醇=97:3;
[0108]
运行时间:100min
[0109]
实验步骤:
[0110]
(1)定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0111]
(2)对照品定量限溶液配置:分别将上述8个对照品溶液,各取1ml分别用稀释剂溶解并稀释配制成0.001mg/ml的定位限溶液。
[0112]
(3)样品溶液配制:取合成的化合物a样品100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,配制成1mg/ml的样品溶液。
[0113]
(4)将上述步骤(1)配制的对照品溶液、步骤(2)配制的定位限溶液和步骤(3)配制的样品溶液置于进样器中,平衡后,将其注入液相色谱仪,采用上述检测条件检测,记录色谱图。
[0114]
实验结果:
[0115]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图7所示,对应数据结果如下表7所示。
[0116]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合
物g、化合物h的定量限溶液的液相色谱图如图8所示,对应数据结果如下表8所示;
[0117]
合成化合物a样品的液相色谱图如图9所示,对应数据结果如下表9所示。
[0118]
表7 化合物a及其手性异构体定位结果
[0119]
分析物保留时间分离度化合物b15.346min
‑
化合物g20.261min3.91化合物c27.402min4.55化合物f30.976min1.85化合物a49.223min6.38化合物e56.263min1.77化合物h77.369min4.28化合物d81.059min0.60
[0120]
表8 化合物a及其手性异构体定量限结果
[0121][0122][0123]
表9 合成化合物a样品的手性纯度结果
[0124]
分析物保留时间纯度化合物b15.346min0.291%化合物g20.261min0.081%化合物c27.402min 化合物f30.976min0.277%化合物a49.223min98.816%化合物e56.263min 化合物h77.369min 化合物d81.059min [0125]
实施例4
[0126]
检测条件:
[0127]
仪器:agilent 1100液相色谱仪,vwd检测器
[0128]
色谱柱:大赛璐chiralpak ia(250mm*4.6mm,5um);
[0129]
流动相:正己烷:异丙醇=90:10;
[0130]
洗脱方式:等度洗脱;
[0131]
流速:0.8ml/min
[0132]
柱温:20℃
[0133]
检测器波长:220nm
[0134]
进样体积:10μl
[0135]
稀释剂:正己烷:异丙醇=90:10;
[0136]
运行时间:60min
[0137]
实验步骤:
[0138]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0139]
实验结果:
[0140]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图10所示,对应数据结果如下表10所示。
[0141]
表10 化合物a及其手性异构体定位结果
[0142]
分析物保留时间分离度化合物g12.171min
‑
化合物f12.822min0.88化合物b16.562min4.53化合物e25.184min9.81化合物h28.204min2.71化合物d32.263min3.26化合物c33.918min1.13化合物a45.040min2.60
[0143]
对比例1
[0144]
检测条件:
[0145]
仪器:agilent 1100液相色谱仪,vwd检测器
[0146]
色谱柱:大赛璐chiralpak od
‑
h(250mm*4.6mm,5um);
[0147]
流动相:正己烷:异丙醇=85:15;
[0148]
洗脱方式:等度洗脱;
[0149]
流速:1.0ml/min
[0150]
柱温:20℃
[0151]
检测器波长:210nm
[0152]
进样体积:10μl
[0153]
稀释剂:正己烷:异丙醇=85:15;
[0154]
运行时间:30min
[0155]
实验步骤:
[0156]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物
e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0157]
实验结果:
[0158]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图11所示,对应数据结果如下表11所示。
[0159]
表11 化合物a及其手性异构体定位结果
[0160]
(
“‑”
表示该峰为第一个峰;“0”表示该峰与前一个峰重合)
[0161]
分析物保留时间分离度化合物b4.890min
‑
化合物g6.055min4.01化合物c6.572min1.46化合物f8.630min3.27化合物a8.630min0化合物e9.261min0.83化合物h10.632min1.98化合物d12.593min2.56
[0162]
对比例2
[0163]
检测条件:
[0164]
仪器:agilent 1100液相色谱仪,vwd检测器
[0165]
色谱柱:大赛璐chiralpak od
‑
h(250mm*4.6mm,5um);
[0166]
流动相:正己烷:异丙醇=98:2;
[0167]
洗脱方式:等度洗脱;
[0168]
流速:1.0ml/min
[0169]
柱温:30℃
[0170]
检测器波长:210nm
[0171]
进样体积:10μl
[0172]
稀释剂:正己烷:异丙醇=98:2;
[0173]
运行时间:120min
[0174]
实验步骤:
[0175]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0176]
实验结果:
[0177]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图12所示,对应数据结果如下表12所示。
[0178]
表12 化合物a及其手性异构体定位结果
[0179]
(
“‑”
表示该峰为第一个峰;“0”表示该峰与前一个峰重合)
[0180][0181][0182]
对比例3
[0183]
仪器:agilent 1100液相色谱仪,vwd检测器
[0184]
色谱柱:chiralpak oj
‑
h(250mm*4.6mm,5um);
[0185]
流动相:正己烷:异丙醇=90:10;
[0186]
洗脱方式:等度洗脱;
[0187]
流速:1.0ml/min
[0188]
柱温:20℃
[0189]
检测器波长:210nm
[0190]
进样体积:5μl
[0191]
稀释剂:正己烷:异丙醇=90:10;
[0192]
运行时间:30min
[0193]
实验步骤:
[0194]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0195]
实验结果:
[0196]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图12所示,对应数据结果如下表13所示。
[0197]
表13 化合物a及其手性异构体定位结果
[0198]
(
“‑”
表示该峰为第一个峰;“0”表示该峰与前一个峰重合)
[0199]
分析物保留时间分离度化合物g7.119min
‑
化合物f7.119min0化合物b9.644min2.14化合物c10.013min0化合物a10.821min0.49化合物h10.821min0
化合物e12.194min0.79化合物d14.030min1.01
[0200]
对比例4
[0201]
仪器:agilent 1100液相色谱仪,vwd检测器
[0202]
色谱柱:chiralpak as
‑
h(150mm*4.6mm,5um);
[0203]
流动相:正己烷:异丙醇=98:2;
[0204]
洗脱方式:等度洗脱;
[0205]
流速:0.8ml/min
[0206]
柱温:20℃
[0207]
检测器波长:220nm
[0208]
进样体积:5μl
[0209]
稀释剂:正己烷:异丙醇=98:2;
[0210]
运行时间:40min
[0211]
实验步骤:
[0212]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0213]
实验结果:
[0214]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图14所示,对应数据结果如下表14所示。
[0215]
表14 化合物a及其手性异构体定位结果
[0216]
(
“‑”
表示该峰为第一个峰;“0”表示该峰与前一个峰重合)
[0217]
分析物保留时间分离度化合物g9.217min
‑
化合物f9.217min0化合物b10.794min1.51化合物h10.794min0化合物a18.870min4.56化合物c19.302min0化合物e24.178min2.04化合物d27.933min1.69
[0218]
对比例5
[0219]
仪器:agilent 1100液相色谱仪,vwd检测器
[0220]
色谱柱:chiralpak ic(250mm*4.6mm,5um);
[0221]
流动相:正己烷:异丙醇=80:20;
[0222]
洗脱方式:等度洗脱;
[0223]
流速:1.0ml/min
[0224]
柱温:30℃
[0225]
检测器波长:220nm
[0226]
进样体积:5μl
[0227]
稀释剂:正己烷:异丙醇=80:20;
[0228]
运行时间:30min
[0229]
实验步骤:
[0230]
定位对照品溶液配制:分别称取标准化合物a、化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h对照品各约100mg,精密称定,置100ml容量瓶中,用稀释剂溶解并稀释至刻度,摇匀,分别配制成1mg/ml的溶液作为定位对照品溶液。
[0231]
实验结果:
[0232]
化合物a及其7种手性异构体化合物b、化合物c、化合物d、化合物e、化合物f、化合物g、化合物h定位的液相色谱图如图15所示,对应数据结果如下表15所示。
[0233]
表15 化合物a及其手性异构体定位结果
[0234]
(
“‑”
表示该峰为第一个峰;“0”表示该峰与前一个峰重合)
[0235]
化合物g5.382min
‑
化合物f6.242min3.16化合物b6.421min0.59化合物e6.792min1.20化合物a8.004min2.49化合物c8.004min0化合物d8.786min1.40化合物h9.188min0.83
[0236]
结论:
[0237]
上述实施例1
‑
4实验结果表明,采用本技术的色谱分离检测方法,化合物a及其7个手性异构体可以在同一色谱条件下达到良好的分离,另外,该液相色谱分离检测方法还可以用作定性定量分析,化合物a及其手性异构体的定量限可达0.001mg/ml,可直接得出合成的化合物a样品含有哪些手性异构体,及得出化合物a的正向纯度和各手性异构体杂质的含量。其中,实施例1为最优条件,该色谱条件下,峰与峰之前均达到完全分离,分离度均大于1.5,且有效缩短分析时间,节省时间和流动相。
[0238]
上述对比例1实验结果表明,使用类型为纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)的手性色谱柱,但流动相中异丙醇含量过高,流动相比例不合适,导致该方法下手性异构体的各峰之间的分离不完全,有两个手性异构体峰完全重合,且与化合物a的分离也不完全,无法定性定量进行检测,表明对比例1方法不适合用于化合物a的手性纯度检测和手性异构体的检测。
[0239]
上述对比例2实验结果表明,使用类型为纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)的手性色谱柱,但流动相中正己烷含量过高,流动相比例不合适,导致该方法下手性异构体的各峰之间的分离不完全,有两个手性异构体峰完全重合,且分析时间过长,无法定性定量进行检测,表明对比例2方法不适合用于化合物a的手性纯度检测和手性异构体的检测。
[0240]
上述对比例3实验结果表明,由于未使用类型为直链淀粉
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)或纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)的手性色谱柱,其他条件均符合要
求,图谱显示该方法下多个手性异构体的峰出现重叠包裹现象,且与化合物a的分离也不完全,无法定性定量进行检测,表明该对比例3的方法不适合用于化合物a的手性纯度检测和手性异构体的检测。
[0241]
上述对比例4实验结果表明,由于未使用类型为直链淀粉
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)或纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)的手性色谱柱,且流动相中正己烷含量过高,流动相比例不合适,导致该方法下多个手性异构体的峰出现重叠包裹现象,且与化合物a的分离也不完全,存在包峰现象,无法定性定量进行检测,表明该对比例4的方法不适合用于化合物a的手性纯度检测和手性异构体的检测。
[0242]
上述对比例5实验结果表明,由于未使用类型为直链淀粉
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)或纤维素
‑
三
‑
(3,5
‑
二甲基苯基氨基甲酸酯)的手性色谱柱,且流动相中异丙醇含量过高,流动相比例不合适,导致该方法下手性异构体的各峰之间的分离不完全,且与化合物a之间的分离也不完全,无法定性定量进行检测,表明对比例5方法不适合用于化合物a的手性纯度检测和手性异构体的检测。
[0243]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
[0244]
此外,本领域的技术人员能够理解,尽管在此的一些实施例包括其它实施例中所包括的某些特征而不是其它特征,但是不同实施例的特征的组合意味着处于本发明的范围之内并且形成不同的实施例。例如,在上面的权利要求书中,所要求保护的实施例的任意之一都可以以任意的组合方式来使用。公开于该背景技术部分的信息仅仅旨在加深对本发明的总体背景技术的理解,而不应当被视为承认或以任何形式暗示该信息构成已为本领域技术人员所公知的现有技术。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。