1.本发明涉及药物分析领域,具体涉及一种推测药剂厂家来源的反向工程方法。
背景技术:
2.在原研药的专利保护失效之后,仿制药便纷纷出现,这些仿制药对医疗系统也做出了重要的贡献。但是仿制药的药效参差不齐。近年来,仿制药和原研药两者之间的差异一直受到关注,造成这种差异的原因除了生产设备、工艺等,很大程度来源于辅料的质量和构成。
3.2018年7月国家药品审评中心发布“新注册分类的皮肤外用仿制药的技术评价要求(征求意见稿)”,其中要求仿制药申请人须通过查阅参比制剂说明书、专利、文献或适当的处方解析手段(如反向工程等)等途径,对参比制剂处方进行解析,确定参比制剂中各辅料的型号和含量。虽然通过查阅参比制剂的说明书及专利,可能获得处方组成及用量,但是大部分辅料不是单一化合物,且不同厂家产品相差较大,同一厂家不同批次的产品也可能存在差异。
4.药物制剂中的辅料能保证药物选择地以一定方式运送到达相应的组织部位,并能保证药物以一定的时间和速度释放。因此不同来源的辅料可能会对药效产生较为显著的影响。
5.因此通过反向工程确定辅料的来源对于成功仿制出药效相当的药品是非常重要的。
技术实现要素:
6.本发明的目的在于克服现有技术的上述问题,提供一种推测药剂厂家来源的反向工程方法。本发明的方法能够较为准确地推测出需反向工程制剂中中链脂肪酸甘油酯的厂家来源,并能够更加准确地计算出中链脂肪酸甘油酯的含量;本发明为药品研发中的反向工程提供了有效的研究手段。
7.本发明的发明人发现,中链脂肪酸甘油酯是对药效影响比较显著的一种辅料,并且不同厂家生产的中链脂肪酸甘油酯的性质有显著差异。本发明的发明人经过进一步研究后发现,造成不同厂家的中链脂肪酸甘油酯的性质具有显著差异的主要原因在于:辛酸甘油三酯与癸酸甘油三酯的比例具有较大差异;这是因为中链脂肪酸甘油酯的来源是由椰子胚乳的坚硬干燥部分或油棕胚乳的干燥部分提取的脂肪油分离出的辛酸(c8h
16
o2)、癸酸(c
10
h
20
o2)等不饱和脂肪酸,再将己酸和癸酸按需要以一定的比例进行调整后,再与甘油酯化而得到的甘油三酯的混合物,这个过程使得不同工艺或原料所得的中链脂肪酸甘油酯中辛酸甘油三酯与癸酸甘油三酯的比例可以在较大范围内波动。
8.目前2020年版《中国药典》收载的中链脂肪酸甘油酯的指标规定含辛酸应为50.0~80.0%,癸酸应为20.0~50.0%,辛酸和癸酸的总和不得少于95.0%。这个规定的范围非常宽泛,无法用来区分不同来源的中链脂肪酸甘油酯。
9.本发明的发明人还发现,现有技术仅研究过如何测定中链脂肪酸甘油酯的含量,并且常常采用直接测定辛酸甘油三酯和癸酸甘油三酯以得到中链脂肪酸甘油酯的含量的方法,但是这种方法不适合药物制剂中中链脂肪酸甘油酯的检测,因为药物制剂中同时含有原料和多种不同种类的辅料,药物制剂中其他成分均可能干扰中链甘油三酯检测,并且这种方法也无法判断中链脂肪酸甘油酯的来源。
10.本发明的发明人经过进一步深入研究后发现,中链脂肪酸甘油酯的分子量与三分子的辛酸甲酯或三分子的癸酸甲酯具有大致相当的关系,从而可以通过将中链脂肪酸甘油酯转化为辛酸甲酯和癸酸甲酯,并通过检测辛酸甲酯和癸酸甲酯的含量而得到中链脂肪酸甘油酯。更重要的是,本发明的发明人意识到,辛酸甲酯和癸酸甲酯的含量之间的比例(本发明中称为辛癸比)在不同工艺或原料中具有显著的差异性,从而可以利用辛癸比有效推测出中链脂肪酸甘油酯的厂家来源。
11.本发明第一方面提供了一种推测药剂厂家来源的反向工程方法,所述方法包括如下步骤:
12.(1)将包括待反向药剂、作为内标物质的壬酸甲酯和第一无机碱的物料在第一溶剂中进行第一水浴回流,然后将所得物料用第二溶剂洗涤,取含有所述第二溶剂的液层并对其进行干燥,得到待反向药剂供试品溶液;
13.(2)将辛酸甲酯、癸酸甲酯、作为内标物质的壬酸甲酯和第三溶剂混合得到混合对照品溶液;
14.(3)将多个目标厂家的中链脂肪酸甘油酯分别参照步骤(1)的方法配制得到厂家供试品溶液;
15.(4)将所述待反向药剂供试品溶液、所述厂家供试品溶液和所述混合对照品溶液分别用气相色谱仪进行检测;
16.(5)基于检测图谱所得各成分的峰面积,进行第一计算、第二计算和第三评估;
17.所述第一计算包括:分别计算待反向药剂供试品溶液和各目标厂家的厂家供试品溶液中的辛酸甲酯的量和癸酸甲酯的量;
18.所述第二计算包括:分别计算待反向药剂供试品溶液和各目标厂家的厂家供试品溶液中的辛癸比,所述辛癸比为辛酸甲酯的量和癸酸甲酯的量的比例;
19.所述第三评估包括:对比待反向药剂供试品溶液的辛癸比与多个目标厂家的厂家供试品溶液的辛癸比的大小关系,当待反向药剂供试品溶液的辛癸比与某个厂家供试品溶液的辛癸比的相差在5%以内时,推测该厂家为待反向药剂的厂家来源;否则,做出所有目标厂家均不为厂家来源的结论;
20.所述步骤(1)、(2)和(3)顺序不限地进行或同时进行。
21.在步骤(1)中,所述第一溶剂例如为甲醇。
22.优选地,相对于1重量份的所述待反向药剂,所述第一溶剂的用量为20
‑
80重量份,更优选为30
‑
50重量份。
23.在步骤(1)中,所述第一无机碱例如为氢氧化钠和/或氢氧化钾。
24.优选地,所述第一无机碱以溶于所述第一溶剂所得的溶液的形式加入,相对于1g的所述待反向药剂,该溶液的加入量为0.3
‑
1ml;该溶液中第一无机碱的含量为3
‑
10重量%。
25.在步骤(1)中,优选地,所述第一水浴回流包括:将待反向药剂分散(例如通过超声)在第一溶剂中,并与所述第一无机碱溶解于所述第一溶剂所得的溶液、壬酸甲酯液体相混合,将混合物料进行水浴回流20
‑
50min。
26.经过所述第一水浴回流,使得中链脂肪酸甘油酯中主要成分辛酸甘油三酯和癸酸甘油三酯转化为三分子的辛酸甲酯和三分子的癸酸甲酯。
27.在步骤(1)中,所述第二溶剂为能够与所述第一溶剂分层,且能够溶解辛酸甲酯、癸酸甲酯和壬酸甲酯的有机溶剂。
28.优选地,所述第二溶剂例如为正己烷、正庚烷。
29.优选地,步骤(1)还包括:在所述所得物料用第二溶剂洗涤之前,先对物料进行冷却。
30.在步骤(1)中,所述干燥可以通过与固体干燥剂接触的方式进行;所述固体干燥剂例如为无水硫酸钠。
31.优选地,所述步骤(1)还包括:在干燥之后将固体干燥剂通过过滤除去。
32.根据一种具体实施方式,所述步骤(1)的过程包括:将待反向药剂置回流瓶中,加入第一溶剂(例如甲醇),超声使分散,并加入内标(即壬酸甲酯)液体、第一无机碱溶解于所述第一溶剂所得的溶液(例如氢氧化钠的甲醇溶液),置水浴回流后取出(例如转移到分液漏斗中),用第二溶剂(例如正己烷)洗涤,并合并洗涤液,用固体干燥剂(例如无水硫酸钠)对正己烷层进行干燥,过滤,即得到待反向药剂供试品溶液。
33.步骤(2)中的辛酸甲酯和癸酸甲酯以及本发明中作为内标物质的壬酸甲酯应为商购的分析纯。
34.在步骤(2)中,所述第三溶剂与所述第二溶剂相同或不同。优选地,所述第三溶剂和所述第二溶剂相同,均为正己烷。
35.根据一种具体实施方式,所述步骤(2)的过程包括:将辛酸甲酯、癸酸甲酯和作为内标物质的壬酸甲酯的混合对照品溶液:称取辛酸甲酯、癸酸甲酯置容量瓶中,加入正己烷溶剂并稀释至刻度,摇匀,作为辛酸甲酯与癸酸甲酯混合母液;精密量取内标溶液、辛酸甲酯与癸酸甲酯混合母液置量瓶中,用正己烷稀释至刻度,摇匀,即得混合对照品溶液。
36.在步骤(3)中,取可能是待反向药剂的厂家来源的多个目标厂家的药剂分别配制厂家供试品溶液,配制的方法参照步骤(1)中的方法进行,将步骤(1)中的待测药品制剂替换成目标厂家的药剂即可。步骤(3)与步骤(1)中各参数的选择不必须完全相同,在合适的范围内即可。
37.通过步骤(3),得到多个目标厂家的厂家供试品溶液。
38.在步骤(4)中,优选地,色谱柱的填充剂为固定性聚乙二醇,尺寸为(20
‑
60)m
×
(0.18
‑
0.53)mm
×
(0.25
‑
0.5)μm。
39.在步骤(4)中,优选地,所述气相色谱仪的条件包括:载气为惰性气体,流速为1
‑
4.5ml/min,进样量为1
‑
5μl,分流比为(10
‑
100):1;更优选地,所述气相色谱仪的条件包括:载气为氮气或氦气,流速为2
‑
4ml/min,进样量为1
‑
2μl,分流比为(40
‑
60):1。
40.优选地,所述气相色谱仪的条件还包括:使用的检测器为氢火焰离子化检测器,进样口温度为250~300℃,检测器温度为250~320℃;更优选地,进样口温度为250~260℃,检测器温度为250~270℃。
41.优选地,柱温采用程序升温,条件包括:起始温度40
‑
60℃,升温速度为30
‑
40℃/min,升温终止温度为250
‑
270℃,保持时间10
‑
20min。
42.根据一种优选的具体实施方式,所述气相色谱仪的条件包括:色谱柱为30m
×
0.32mm
×
0.25μm的agilent hp
‑
innowax,载气为氮气,流速为3ml/min,进样量为1μl,分流比为50:1,检测器为氢火焰离子化检测器,进样口温度为250℃,检测器温度为260℃;程序升温条件包括:起始温度50℃、35℃/min升至250℃,保持15min。
43.在步骤(5)中,基于检测图谱进行第一计算、第二计算和第三评估,其中第一计算的目的是确定待反向药剂供试品溶液和多个目标厂家的厂家供试品溶液中辛酸甲酯的量和癸酸甲酯的量,所述第二计算的目的是计算待反向药剂供试品溶液和多个目标厂家的厂家供试品溶液的辛癸比,所述第三评估的目的是通过对比确定待反向药剂的厂家来源。
44.所得色谱图中,溶剂峰、辛酸甲酯、壬酸甲酯(内标)、癸酸甲酯依次流出。
45.所述第一计算的方法可以采用本领域常规的基于检测图谱进行计算的方法,例如采用加内标的外标法来计算辛酸甲酯和癸酸甲酯的量;其中,通过设置内标物质壬酸甲酯,能够消除因操作而引入的误差。
46.所述第二计算为将所述第一计算所得的辛酸甲酯的量除以癸酸甲酯的量,所得的比例数值即为辛癸比。
47.由于工艺或原料的不同,因此不同厂家来源的中链脂肪酸甘油酯所测得到辛癸比有显著差异,从而能够作为推测厂家来源的重要依据。
48.所述第三评估通过对比待反向药剂供试品溶液的辛癸比与多个目标厂家的厂家供试品溶液的辛癸比的大小关系,选取与待反向药剂的辛癸比最为接近的目标厂家即为反向工程所推测的待反向药剂的厂家来源。
49.对比的结果非常接近时(例如相差在5%以内时)才能够认为推测的结论是有可能正确的。否则,则做出目标厂家均不符合结论,并继续寻找目标厂家。
50.在确定厂家来源之后,所述方法还包括第四计算,该第四计算包括:根据厂家来源中辛酸甲酯和癸酸甲酯的总百分比、待反向药剂中辛酸甲酯和癸酸甲酯的总和在待反向药剂中的百分比,计算待反向药剂中中链脂肪酸甘油酯的百分比;即将待反向药剂中辛酸甲酯和癸酸甲酯的总和在待反向药剂中的百分比除以厂家来源中辛酸甲酯和癸酸甲酯的总百分比,计算得到待反向药剂中中链脂肪酸甘油酯的百分比。
51.现有技术已经存在将中链脂肪酸甘油酯转化为辛酸甲酯和癸酸甲酯测定中链脂肪酸甘油酯的方法。相对于辛酸甘油三酯与癸酸甘油三酯,辛酸甲酯和癸酸甲酯的气相色谱的结果更不易于受其它成分的影响,因此检测结果更加准确。因此这种方法相对于直接测定辛酸甘油三酯与癸酸甘油三酯的方法更加准确。该方法所涉及的换算过程如下:辛酸甘油三酯分子量为470.69,三分子的辛酸甲酯分子量为158.24
×
3=474.72,辛酸甘油三酯与三分子的辛酸甲酯的分子量之比为1:1.008;癸酸甘油三酯分子量为554.85;三分子的癸酸甲酯分子量为186.29
×
3=558.87,癸酸甘油三酯与三分子的癸酸甲酯的分子量之比为1:1.007。辛酸甘油三酯与癸酸甘油三酯各自的分子量基本等于三分子的辛酸甲酯和三分子的癸酸甲酯。因此,认为测定供试品中的辛酸甲酯和癸酸甲酯的总量,即等于样品中中链脂肪酸甘油酯的量。
52.然而本发明的发明人发现,这种方法是建立在“中链脂肪酸甘油酯全部为辛酸甘
油三酯与癸酸甘油三酯”的假定的前提下的。而事实上,这是一种理想化的假定,而该假定几乎不可能实现。中链脂肪酸甘油酯中除了辛酸甘油三酯和癸酸甘油三酯这两种占比95%以上的主要成分之外,还可能存在己酸甘油三酯等其它成分。因此,这种通过测定辛酸甲酯和癸酸甲酯而得到中链脂肪酸甘油酯的含量的方法仍然不够准确。
53.本发明的发明人发现,通过上述第三估算而判断出厂家来源之后,能够进一步确定该厂家来源的中链脂肪酸甘油酯中辛酸甲酯和癸酸甲酯的占比(在此称为基准占比),基于此,将待反向药剂所测得的辛酸甲酯和癸酸甲酯的总含量除以上述基准占比,即能够较为精确地确定待反向药剂中中的链脂肪酸甘油酯的含量。该结果将己酸甘油三酯等成分都考虑在内,使结果更加精确。
54.通过上述技术方案,本发明与现有技术相比至少具有以下优势:
55.(1)本发明的方法能够较为准确地推测出需反向工程制剂中中链脂肪酸甘油酯的厂家来源;
56.(2)本发明的方法能够更加准确地计算出中链脂肪酸甘油酯的含量;
57.(3)本发明的方法不受药物制剂中其他成分的干扰;
58.(4)本发明的方法在操作过程中加入内标,能够消除因操作而引入的误差;
59.(5)本发明为药品研发中的反向工程提供了有效的研究手段。
60.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
附图说明
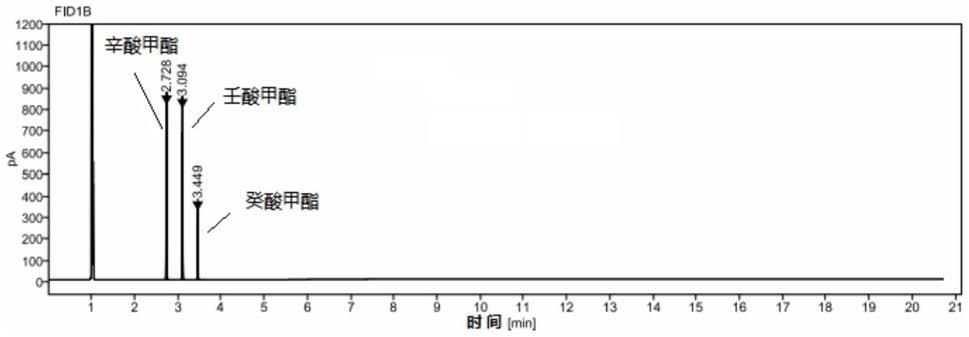
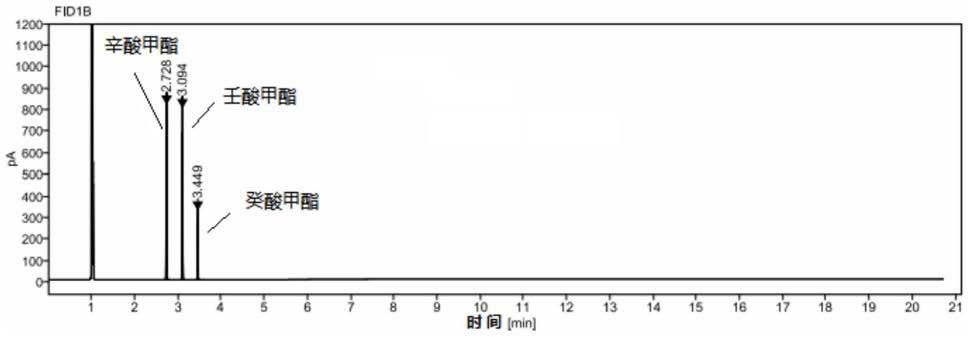
61.图1为实施例1所得混合对照品溶液的气相色谱图;
62.图2为实施例1所得一个厂家供试品溶液的气相色谱图;
63.图3为药物制剂供试品溶液的气相色谱图。
具体实施方式
64.以下将通过实施例对本发明进行详细描述。本发明所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
65.实施例1:
66.一种药物制剂中中链脂肪酸甘油酯含量测定的方法,包括如下步骤:
67.1、药物制剂供试品溶液配制:分别称取需进行反向工程的待测制剂约0.5g,置100ml回流瓶中,加甲醇40ml适当超声使分散,加入25mg/ml壬酸甲酯溶液(内标)2.0ml、6%氢氧化钠甲醇溶液0.5ml,水浴回流30min,取出,冷却至室温,转移至分液漏斗中,用正己烷20ml分三次洗涤回流瓶,洗液并入分液漏斗中,加水40ml,用力振摇提取,静置分层;水层再用正己烷20ml提取两次,并合并正己烷层;用水洗涤两次,每次20ml,取正己烷层;加入适量的无水硫酸钠干燥,滤过,取续滤液作为药物制剂供试品溶液。
68.2、辛酸甲酯、癸酸甲酯和内边的混合对照品溶液:称取辛酸甲酯约200mg、癸酸甲
酯约500mg置20ml容量瓶,正己烷溶剂并稀释至刻度,摇匀,作为辛酸甲酯与癸酸甲酯混合母液;精密量取内标溶液1ml、辛酸甲酯与癸酸甲酯混合母液1ml置10ml量瓶中,正己烷稀释至刻度,摇匀,即得。
69.3、辅料供试品溶液:分别称取各辅料厂家的中链脂肪酸甘油酯约100mg,置100ml回流瓶中,加甲醇40ml适当超声使分散,加入25mg/ml壬酸甲酯溶液(内标)2.0ml、6%氢氧化钠甲醇溶液0.5ml,水浴回流30min,取出,冷却至室温,转移至分液漏斗中,用正己烷20ml分三次洗涤回流瓶,洗液并入分液漏斗中,加水40ml,用力振摇提取,静置分层;水层再用正己烷20ml提取两次,并合并正己烷层;用水洗涤两次,每次20ml,取正己烷层;加入适量的无水硫酸钠干燥,滤过,即得。
70.利用如下色谱条件进行检测:
71.色谱柱:agilent hp
‑
innowax(30m
×
0.32mm
×
0.25μm);
72.载气:氮气;
73.流速:3ml/min;
74.进样量:1μl;
75.分流比:50:1;
76.检测器:氢火焰离子化检测器;
77.进样口温度:250℃;
78.检测器温度:260℃;
79.程序升温条件为:起始温度:50℃、35℃/min升至250℃,保持15min。
80.混合对照品溶液的气相色谱图如图1所示,一个厂家供试品溶液(经过提取,不含药剂,只含中链脂肪酸甘油酯)的气相色谱图如图2所示;待反向药剂供试品溶液的气相色谱图如图3所示。
81.在气相色谱仪中分别确定各溶液中各物质的峰面积,并通过加内标的外标法来计算辛酸甲酯和癸酸甲酯的量;其中,通过内标物质壬酸甲酯作为校正,能够消除因操作而引入的误差。
82.中链脂肪酸甘油酯的目标来源厂家选定两家:basf和croda,其中basf算得辛癸比(辛酸甲酯/癸酸甲酯的比例)为2.6,辛酸甲酯和癸酸甲酯的总量为99.8%;croda算得辛癸比(辛酸甲酯/癸酸甲酯的比例)为1.5,辛酸甲酯和癸酸甲酯的总量为99.1%。
83.待反向药剂1:
84.算得辛癸比(辛酸甲酯/癸酸甲酯的比例)为1.5,与croda的辛癸比相同,推测厂家来源为croda;
85.算得辛酸甲酯和癸酸甲酯的总量为7.9%,根据croda的该值为99.1%,计算得该待反向药剂1中中链脂肪酸甘油酯的含量为7.9%
÷
0.991=8.0%。
86.待反向药剂2:
87.算得辛癸比(辛酸甲酯/癸酸甲酯的比例)为2.6,与basf的辛癸比相同,推测厂家来源为basf;
88.算得辛酸甲酯和癸酸甲酯的总量为8.2%,根据basf的该值为99.8%,计算得该待反向药剂1中中链脂肪酸甘油酯的含量为8.2%
÷
0.998=8.2%。
89.实施例2
90.一种药物制剂中中链脂肪酸甘油酯来源及含量测定的方法,包括如下步骤:
91.1、药物制剂供试品溶液配制:分别称取需进行反向工程的待测制剂约1.0g,置100ml回流瓶中,加甲醇40ml适当超声使分散,加入25mg/ml壬酸甲酯溶液(内标)2.0ml、6%氢氧化钠甲醇溶液0.5ml,水浴回流40min,取出,冷却至室温,转移至分液漏斗中,用正己烷20ml分三次洗涤回流瓶,洗液并入分液漏斗中,加水40ml,用力振摇提取,静置分层;水层再用正己烷20ml提取两次,并合并正己烷层;用水洗涤两次,每次20ml,取正己烷层;加入适量的无水硫酸钠干燥,滤过,取续滤液作为药物制剂供试品溶液。
92.2、辛酸甲酯、癸酸甲酯和内边的混合对照品溶液:称取辛酸甲酯约200mg、癸酸甲酯约500mg置20ml容量瓶,正己烷溶剂并稀释至刻度,摇匀,作为辛酸甲酯与癸酸甲酯混合母液;精密量取内标溶液1ml、辛酸甲酯与癸酸甲酯混合母液1ml置10ml量瓶中,正己烷稀释至刻度,摇匀,即得。
93.3、辅料供试品溶液:分别称取各辅料厂家的中链脂肪酸甘油酯约100mg,置100ml回流瓶中,加甲醇40ml适当超声使分散,加入25mg/ml壬酸甲酯溶液(内标)2.0ml、6%氢氧化钠甲醇溶液0.5ml,水浴回流40min,取出,冷却至室温,转移至分液漏斗中,用正己烷20ml分三次洗涤回流瓶,洗液并入分液漏斗中,加水40ml,用力振摇提取,静置分层;水层再用正己烷20ml提取两次,并合并正己烷层;用水洗涤两次,每次20ml,取正己烷层;加入适量的无水硫酸钠干燥,滤过,即得。
94.利用如下色谱条件进行检测:
95.色谱柱:agilent hp
‑
innowax(30m
×
0.32mm
×
0.25μm);
96.载气:氮气;
97.流速:3ml/min;
98.进样量:1μl;
99.分流比:30:1;
100.检测器:氢火焰离子化检测器;
101.进样口温度:260℃;
102.检测器温度:280℃;
103.程序升温条件为:起始温度:50℃、35℃/min升至240℃,保持10min。
104.参照实施例1测定同样的混合对照品溶液、厂家供试品溶液和待反向药剂供试品溶液,得到与实施例1基本相同的气相色谱图。
105.选用与实施例1相同目标来源厂家和待反向药剂。所得结果为:
106.basf的辛癸比为2.6,辛癸总量(辛酸甲酯和癸酸甲酯的总量的简称)为99.8%;
107.croda的辛癸比为1.5,辛癸总量为99.0%。
108.待反向药剂1的辛癸比为1.5,推测来源croda;辛癸总量为7.7%,中链脂肪酸甘油酯的含量为7.8%。
109.待反向药剂2的辛癸比为2.6,推测来源basf;辛癸总量为8.1%,中链脂肪酸甘油酯的含量为8.1%
÷
0.998=8.1%。
110.通过上述实施例可以看出,本发明的方法能够较为准确地推测出需反向工程制剂中中链脂肪酸甘油酯的厂家来源;能够更加准确地计算出中链脂肪酸甘油酯的含量,且不受药物制剂中其他成分的干扰;通过设置内标,能够消除因操作而引入的误差。本发明为药
品研发中的反向工程提供了有效的研究手段。
111.以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。