一种hplc法测定痫愈胶囊中多种有效成分含量的方法
技术领域
1.本发明属于医药检测分析技术领域,尤其是一种hplc法测定痫愈胶囊中多种有效成分含量的方法。
背景技术:
2.癫痫包括原发性及继发性癫痫,癫痫可见于各个年龄段。儿童癫痫发病率较成人高,随着年龄的增长,癫痫发病率有所降低。进入老年期(65岁以后)由于脑血管病、老年痴呆和神经系统退行性病变增多,癫痫发病率又见上升。癫痫病做为一种慢性疾病,虽然短期内对患者没有多大的影响,但是长期频繁的发作可导致患者的身心、智力产生严重影响。癫痫患者经常会在任何时间、地点、环境下且不能自我控制地突然发作,容易出现摔伤、烫伤、溺水、交通事故等;经常被社会所歧视,在就业、婚姻、家庭生活等方面均遇到困难,患者精神压抑,身心健康受到很大影响。主要表现为患者记忆障碍、智力下降、性格改变等,最后逐渐丧失工作能力甚至生活能力。
3.癫痫中医认为属痫病范畴,痫病由痰、火、瘀,以及先天因素等,至气血逆乱,清窍蒙蔽而发病,以卒然昏仆,强直抽搐,移时自醒,醒后如常人为特征的发作性疾病。中医学认为癫痫的发生,大抵由于先天因素,后天七情失调,饮食不节,劳逸过度,或患其它病之后,造成脏腑失调、痰浊阻;《三因极一病证方论
·
癫痫伤痛论》:“夫癫痫病,皆由惊动,或少小感风寒暑湿,或饮食不节,逆于脏气。”《丹溪心法
·
癫痫》也指出本症之发生,非无痰涎壅塞,迷闷孔窍。《医学纲目
·
癫痫》:“癫痫者,痰邪逆上也。”也就是说,“痰逆心窍”是本症的发病机理。由于痰浊蒙蔽心窍,必然导致心主神明的功能失调,出现精神异常,所以癫痫症的主要病因是痰,重要病位在脑、心。痰是一种病理产物,由脏腑津液所化,而津液是人体来源于饮食的营养精微物质具有的营养组织、脏器的作用。由于情志不畅,或饮食不当,损伤脾胃,或素体虚弱,中气不足,皆能使脾失健运,不能为胃行其津液,停聚而成痰,正所谓“脾为生痰之源也”。临床上痫症发作时的轻重大抵与痰浊的深浅,正气的盛衰有关。初起时,正气未衰,痰浊不重,故发作持续时间短,间歇期长,如反复发作,正气渐衰,痰浊不化,愈发愈频,使正气更衰,这样互为因果,其病亦渐重,故此病常为虚实夹杂,本虚标实。虚者,多为气虚、血虚;实者多为痰火上扰,痰气郁结。一般说来癫痫病虚多于实,且由于其发病较慢,病程较长,正气渐衰,气血虚弱。治疗以益气养血,扶正固本,安神定惊,息风解痉,使机体正气恢复、风熄、痰消、心窍通而神志渐复。
4.目前,治疗癫痫疾病的药物多种多样,常规的治疗方法有药物治疗、饮食治疗以及手术治疗等,但每种方法都有各自的优缺点,每位癫痫病患者的发病因素与病情都不同,因此只有选择合适的治疗方法才是最有效的治疗方法。中成药治疗癫痫病的话,药性温和,副作用小,对身体无损伤等优点,常用的抗癫痫药物痫愈胶囊经过长期大量反复临床试验,并已上市销售多年,证实其对癫痫病的治疗是具有良好的治疗效果。
5.痫愈胶囊为西安千禾药业股份有限公司生产的独家品种,已上市销售多年,在治疗风痰闭阻所致的癫痫抽搐、小儿惊风、面肌痉挛方面取得了良好的效果,其主要成分:黄
芪、党参、丹参、柴胡、酸枣仁、远志、天麻、钩藤、石菖蒲、胆南星、当归、僵蚕、六神曲、郁金、甘草、制白附子。功效豁痰开窍、安神定惊、息风解痉。现有痫愈胶囊的检测方法中,仅对君药天麻、臣药甘草进行了含量测定,对臣药黄芪、丹参、党参进行了薄层鉴别,佐药僵蚕进行了显微鉴别,这些质量控制指标并不能全面反映药品的质量,不能对临床疗效性指标进行控制,所以,很有必要进一步完善该药品的检测方法,将控制指标与临床疗效进行关联,从而更进一步保证该产品的质量和疗效。
技术实现要素:
6.本发明的目的是克服现有技术的不足,提供一种hplc法测定痫愈胶囊中多种有效成分含量的方法,该分析方法对制剂中的天麻的有效成分天麻素和对羟基苯甲醇,远志的有效成分细叶远志皂苷、远志酮ⅲ和3,6
’‑
二芥子酰基蔗糖,柴胡的有效成分柴胡皂苷a和柴胡皂苷d共7种有效成分同时进行测定。该检测方法能够弥补现有方法的不足,提供一种更为全面、有效的药物控制质量标准,进一步有效保障产品质量。
7.为实现上述发明目的,本发明采用如下技术方案:
8.一种hplc法测定痫愈胶囊中多种有效成分含量的方法,所述痫愈胶囊由以下16味药材制备而成:黄芪、党参、丹参、柴胡、酸枣仁、远志、天麻、钩藤、石菖蒲、胆南星、当归、僵蚕、六神曲、郁金、甘草、制白附子;采用高效液相色谱法对天麻中的天麻素、对羟基苯甲醇,远志中的细叶远志皂苷、远志酮ⅲ、3,6
’‑
二芥子酰基蔗糖,柴胡中的柴胡皂苷a、柴胡皂苷d进行了含量检测,该检测方法的包含以下步骤:
9.(1)色谱条件与系统适用性试验
10.以十八烷基硅烷键合硅胶为填充剂;以有机溶剂为a相、一定浓度的稀酸水溶液为b相,梯度洗脱,所述有机溶剂为选自甲醇、乙腈或乙腈
‑
甲醇混合液,所述稀酸水溶液选自体积百分浓度0.01%
‑
1.0%的磷酸水溶液或体积百分浓度0.01%
‑
0.5%的醋酸水溶液;柱温20
‑
40℃;采用紫外检测器或二极管阵列检测器,检测波长200
‑
400nm;流速为0.8ml/min
‑
1.2ml/min;理论板数细叶远志皂苷峰计算应不低于3000;
11.优选的,所述a相为乙腈,b相为体积百分浓度0.05%的磷酸水溶液;流速为1.0ml/min;柱温为35℃;检测波长215nm。
12.优选的,梯度洗脱时,梯度洗脱程序为:0~16min,5%a;16~19min,5%~20%a;19~40min,20%a;40~50min,20%~5%a;50~60min,5%a;具体梯度洗脱程序为:
13.时间(分钟)流动相a(%)流动相b(%)0~1659516~195
→
2095
→
8019~40208040~5020
→
580
→
9550~60595
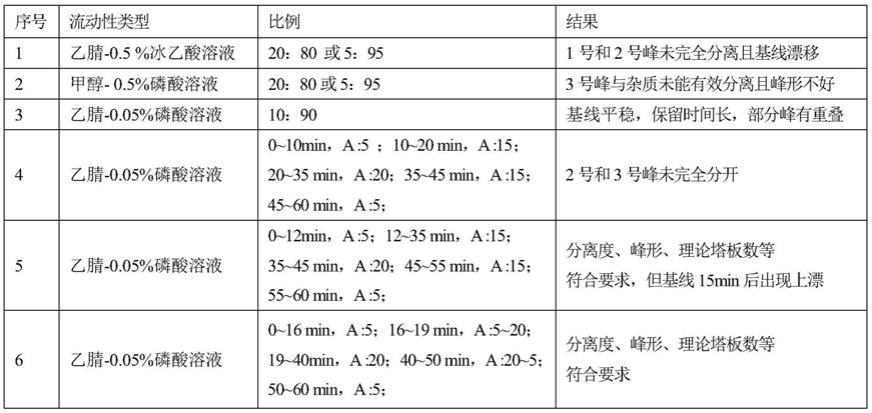
14.(2)对照品溶液的制备
15.取对照品天麻素、对羟基苯甲醇、细叶远志皂苷、远志酮ⅲ、3,6
’‑
二芥子酰基蔗糖、柴胡皂苷a、柴胡皂苷d,精密称定,加甲醇制成每lml含50μg天麻素、25μg对羟基苯甲醇、1.0mg细叶远志皂苷、0.15mg远志酮ⅲ、0.2mg 3,6
’‑
二芥子酰基蔗糖、0.4mg柴胡皂苷a、
0.5mg柴胡皂苷d的混合溶液,即得;
16.(3)供试品溶液的制备
17.取痫愈胶囊内容物,研细,称取1.0
‑
2.0g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50
‑
100ml,称定重量,超声处理(200w,40khz)30
‑
90min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
18.优选的,取痫愈胶囊内容物,研细,称取1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,称定重量,超声处理(200w,40khz)60min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
19.(4)测定
20.分别精密吸取对照品溶液与供试品溶液各10
‑
20μl,注入高效液相色谱仪,测定,即得测定结果。
21.本发明中所述痫愈胶囊具体处方为:黄芪、党参、丹参、柴胡、酸枣仁、远志、天麻、钩藤、石菖蒲、胆南星、当归、僵蚕、六神曲、郁金、甘草、制白附子。功效豁痰开窍、安神定惊、息风解痉。
22.与现有技术相比,本发明具有以下有益的技术效果:
23.本发明提供了一种同时测定痫愈胶囊中多种有效成分的检测方法,采用同一检测波长对君药天麻中的有效成分天麻素和对羟基苯甲醇、佐药远志中的有效成分细叶远志皂苷、远志酮ⅲ和3,6
’‑
二芥子酰基蔗糖、使药柴胡中的有效成分柴胡皂苷a和柴胡皂苷d进行了含量检测,该检测方法能够更全面有效地弥补现有检测方法的不足,可以在同一色谱条件下同时对痫愈胶囊中君药、佐药、使药中的7种有效成分进行测定,有效地控制了药物的质量,提高检验效率,降低检验成本,使药物质量与临床治疗有效成分进行关联,从而达到稳定、可控、高效及安全的标准,进一步有效保障了产品的质量,能够更好地满足临床治疗需求,为该产品的质量标准提升提供更多的依据。
附图说明
24.下面结合附图对本发明作进一步的说明:
25.图1为本发明实施例的混合对照品的色谱图;
26.图2为本发明实施例的供试品的色谱图;
27.图3为本发明实施例的天麻阴性样品色谱图;
28.图4为本发明实施例的远志阴性样品色谱图;
29.图5为本发明实施例的柴胡阴性样品的色谱图;
30.其中,图中1为天麻素峰、2为对羟基苯甲醇峰、3为细叶远志皂苷峰、4为柴胡皂苷a峰、5为3,6
’‑
二芥子酰基蔗糖峰、6为柴胡皂苷d峰、7为远志酮ⅲ峰。
31.图6为7种有效成分的线性关系图;图中,从上到下,从左到右,依次为天麻素、对羟基苯甲醇、细叶远志皂苷、远志酮ⅲ、3,6
’‑
二芥子酰基蔗糖、柴胡皂苷a和柴胡皂苷d的线性关系图。
具体实施方式
32.以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅
用于说明本发明,其不以任何方式限制本发明的范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原辅料、试剂材料等,如无特殊说明,均为市售购买产品。
33.实施例1:痫愈胶囊的处方及制备方法
34.处方:
35.黄芪140g、党参140g、丹参140g、柴胡70g、酸枣仁105g、远志70g、天麻105g、钩藤105g、石菖蒲70g、胆南星70g、当归140g、僵蚕105g、六神曲70g、郁金70g、甘草70g、制白附子35g;
36.制法:以上十六味,僵蚕、六神曲粉碎成细粉,过筛,备用;其余黄芪等十四味加水煎煮三次,第一次加10倍量水,煎煮2小时,第二次加8倍量水,煎煮1.5小时,第三次加6倍量水,煎煮1小时,合并煎液,滤过,滤液减压浓缩至相对密度为1.20~1.25(60℃)的稠膏,加入上述细粉,混匀,真空干燥(60~80℃),粉碎成细粉,加入碳酸钙22g,磷酸氢钙22g,混匀,用80~85%的乙醇适量制粒,干燥(75~80℃),装胶囊,制成1000粒,即得。
37.实施例2:痫愈胶囊中含量测定研究
38.2.1仪器与材料
39.岛津lc
‑
2030c plus型高效液相色谱仪;as5150a超声波清洗机,昆山超声波仪器有限公司;gb204电子天平,at201电子天平,梅特勒
‑
托利多;ld4
‑
2电动离心机,金坛市国旺实验仪器厂。
40.甲醇/乙腈(色谱纯)购于赛默飞世尔科技公司;其余试剂购买均为分析纯,购置于天津大茂化学试剂厂;
41.本实验所才用的痫愈胶囊均为西安千禾药业股份有限公司所生产。对照品均购置于陕西标普医药科技有限公司,由中国药品生物制品检定所生产。
42.2.2波长的选择
43.将天麻素、对羟基苯甲醇对照品溶液在200
‑
400nm范围内进行波长扫描,结果显示在215nm和220nm处均有最大吸收,考虑到有效成分在220nm处吸收度也较大,但分离效果不好,故选择干扰较少、分离效果较好的215nm做为检测波长。
44.将细叶远志皂苷对照品溶液在200
‑
400nm范围内进行波长扫描,结果显示在210nm和215nm处均有最大吸收,考虑到有效成分在210nm处吸收度也较大,但峰形拖尾,存在干扰较多,故选择干扰较少、峰形较好的215nm做为检测波长。
45.将远志酮ⅲ峰、3,6
’‑
二芥子酰基蔗糖对照品溶液在200
‑
400nm范围内进行波长扫描,结果显示在215nm和320nm处均有最大吸收,考虑到有效成分在320nm处吸收度也较大,但分离效果不好,故选择干扰较少、分离效果较好的215nm做为检测波长。
46.将柴胡皂苷a、柴胡皂苷d对照品溶液在200
‑
400nm范围内进行波长扫描,结果显示在210nm和215nm处均有最大吸收,考虑到处方中其他成分在210nm左右处吸收度也较大,但峰形拖尾,存在干扰较多,故选择干扰较少、峰形较好的215nm做为检测波长。
47.2.3流动相的选择
48.为使供试品中7种有效成分能得到有效的分离,同时峰形、理论塔板数等达到要求,预试验分别采用乙腈
‑
0.5%冰乙酸水溶液、甲醇
‑
0.5%磷酸水溶液、甲醇
‑
0.1%甲酸水溶液、甲醇
‑
乙腈
‑
0.05%磷酸水溶液、乙腈
‑
0.05%磷酸水溶液等不同比例的流动性进行试
验。以乙腈
‑
0.05%磷酸水溶液等度洗脱分离效果最好,基线最平稳,但保留时间长,部分峰有重叠;故进行流动相a相为乙腈,流动相b相为体积百分浓度0.05%磷酸水溶液进行梯度洗脱优化试验。
49.表1:流动相选择试验结果
[0050][0051]
经过反复多次试验,常规等度系统难以有效对这7种有效成分进行有效分离,要么保留时间太长,要么部分成分有重叠,为了兼顾药物中7种有效成分的含量测定,进行梯度系统,经过多次流动相发不同比例和时间的调整,结果表明:采用乙腈
‑
0.05%磷酸水溶液组成的流动相体系,在进行梯度洗脱的条件下分离效果最好,基线平稳,7种成分的色谱峰可完全分离,各组分出峰时间、峰型和分离度均符合要求,故该条件能满足分析测定的相关要求。
[0052]
3.专属性研究
[0053]
3.1各阴性样品的制备:
[0054]
按痫愈胶囊的处方及制备方法(实施例1),分别制备不含天麻、远志、柴胡的阴性样品;如天麻阴性样品的制备:取不含天麻的十五味药材,僵蚕、六神曲粉碎成细粉,过筛,备用;其余黄芪等十三味加水煎煮三次,第一次加10倍量水,煎煮2小时,第二次加8倍量水,煎煮1.5小时,第三次加6倍量水,煎煮1小时,合并煎液,滤过,滤液减压浓缩至相对密度为1.20~1.25(60℃)的稠膏,加入上述细粉,混匀,真空干燥(60~80℃),粉碎成细粉,加入碳酸钙22g,磷酸氢钙22g,混匀,用80~85%的乙醇适量制粒,干燥(75~80℃),装胶囊,制成1000粒,即得。
[0055]
3.2阴性样品溶液的制备:
[0056]
分别称取各阴性样品1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,称定重量,超声处理(200w,40khz)60min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
[0057]
3.3供试品溶液的制备:
[0058]
试验方法:取痫愈胶囊内容物,研细,称取1.5g,精密称定,置具塞锥形瓶中,精密加入50%甲醇、70%甲醇或100%甲醇50ml,称定重量,超声处理(200w,40khz)30
‑
90min,放冷,补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
[0059]
方法1:精密加甲醇50ml,精密称定,超声提取30min,放冷补足重量,过滤;
[0060]
方法2:精密加50%甲醇50ml,精密称定,超声提取30min,放冷补足重量,过滤;
[0061]
方法3:精密加70%甲醇50ml,精密称定,超声提取90min,放冷补足重量,过滤;
[0062]
方法4:精密加70%甲醇50ml,精密称定,超声提取60min,放冷补足重量,过滤;
[0063]
分别精密吸取对照品溶液与供试品溶液各10μl,注入高效液相色谱仪,测定,即得测定结果。结果详见表2。
[0064]
表2:供试品制备方法的选择试验结果
[0065][0066]
结果表明:采用方法1时天麻素提取不充分,峰面积明显低于其他方法;采用方法2时有效成分天麻素、对羟基苯甲醇、细叶远志皂苷、远志酮ⅲ和3,6
’‑
二芥子酰基蔗糖的峰面积明显低于方法4;采用方法3时除了柴胡皂苷a和柴胡皂苷d有效成分峰面积略低于方法4,其余有效成分转移率与方法4相当,可能是超声时间过长柴胡皂苷a和柴胡皂苷d有破碎或损失或转化。
[0067]
故确定最佳的供试品溶液的制备方法为:取痫愈胶囊内容物,研细,称取1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,称定重量,超声处理(200w,40khz)60min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
[0068]
3.4色谱条件与系统适用性试验
[0069]
以十八烷基硅烷键合硅胶为填充剂;按表1中规定流动相,以流速为1.0ml/min;柱温为35℃;检测波长为215nm;进样体积为10μl;理论板均应不低于3000;
[0070]
对照品溶液的制备:
[0071]
精密称取各对照品适量,分别加甲醇制成含天麻素0.0512mg/ml、对羟基苯甲醇0.0264mg/ml、细叶远志皂苷1.0251mg/ml、远志酮ⅲ峰0.1632mg/ml、3,6
’‑
二芥子酰基蔗糖0.2215mg/ml、柴胡皂苷a 0.5215mg/ml、柴胡皂苷d 0.6253mg/ml的贮备液;依次精密吸取上述7种贮备液各1ml置于同一10ml容量瓶中,加甲醇溶解并定容,摇匀,作为质量浓度分别为5.12,2.64,102.51,16.32,22.15,52.15,62.53μg/ml的混合对照品溶液。
[0072]
取痫愈胶囊内容物,研细,称取1.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇50ml,称定重量,超声处理(200w,40khz)60min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
[0073]
分别精密吸取各阴性样品溶液、混合对照品溶液、供试品溶液各10μl,注入高效液相色谱仪,测定。
[0074]
结果显示:供试品溶液色谱图中,在7种对照品色谱峰相应的位置上有相同的色谱峰;阴性无干扰,且分离度和理论板数均符合要求;表明该色谱条件专属性良好。具体见下附图1
‑
5。
[0075]
4.重复性试验
[0076]
4.1色谱条件与系统适用性试验同3.4。
[0077]
4.2精密吸取同一供试品溶液10μl,注入液相色谱仪,重复进样6次,计算峰面积rsd。具体结果见表3。
[0078]
表3:重复性实验7种有效成分峰面积数据表
[0079][0080]
4.3结果显示,7种有效成分峰面积的rsd值分别为0.12%、1.01%、0.76%、0.50%、0.34%、0.18%、1.07%,均小于1.5%;表明精密度良好。
[0081]
5.线性范围
[0082]
5.1色谱条件与系统适用性试验同3.4。
[0083]
5.2分别精密吸取各质量浓度对照品1.5ml置于10ml量瓶中用甲醇定容,得混合对照品溶液,再分别精密吸取对照品溶液各2μl、5μl、8μl、10μl、15μl、20μl、按实施例3所述色谱条件进行检测,记录色谱图,以峰面积(y)对浓度(x)进行线性回归分析,绘制线性关系图,报告线性方程。结果见表4,图6。
[0084]
表4:7种成分的标准曲线方程和相关系数及线性范围
[0085]
6.准确度(回收率)试验
[0086]
6.1色谱条件与系统适用性试验同3.4。
[0087]
6.2对照品溶液的制备:
[0088]
取天麻素对照品,精密称定,加甲醇制成浓度为52.36μg/ml的对照品溶液;
[0089]
6.3供试品溶液的制备:
[0090]
取痫愈胶囊内容物,研细,分别称取0.5g样品共9份,取血平胶囊内容物,研细,各称取0.5g,共9份,精密称定,置具塞锥形瓶中,再分别精密吸称取天麻素对照品0.5ml、1.0ml、1.5ml共9份,依次加入上述加样后的锥形瓶中,精密加入70%甲醇50ml,称定重量,超声处理(200w,40khz)60min,放冷,用70%甲醇补足减失的重量,摇匀,离心,滤过,取续滤液,即得。
[0091]
回收率%=(c
‑
a)/b
×
100%
[0092]
式中a为供试品所含被测成分量;b为加入对照品量;c为实测值。
[0093]
表5:回收率试验数据表
[0094][0095]
标准要求:回收率要求85%~110%,回收率rsd<4.0%;实验结果表明该方法准确度良好。
[0096]
7.中间精密度试验
[0097]
7.1色谱条件与系统适用性试验同3.4。
[0098]
7.2在同一实验室,由不同实验人员a和b在不同日期,使用不同色谱仪、进行实验,计算各有效成分含量。
[0099]
表6:中间精密度试验数据表
[0100][0101][0102]
结果见表6,结果表明发明专利方法精密度良好。
[0103]
8.耐用性试验
[0104]
8.1色谱条件与系统适用性试验
[0105]
表7:稳定性试验数据表
[0106][0107]
8.2取同一供试品溶液,室温下放置,分别于0、2、4、8、12、24小时,精密吸取10μl注入高效液相色谱仪;结果见表6,结果表明,供试品溶液24小时内保峰面积的rsd最大值仅为1.48%,小于标准2.0%;表明供试品溶液在室温条件下24h内稳定。
[0108]
综上所述:本发明痫愈胶囊中多种有效成分的含量测定方法,从专属性、重复、准确度(回收率)、中间精密度、耐用性等方面验证了本方法的科学性。通过对处方中3味药材中7中有效成分含量的测定,进一步提升了产品质量保证,保证了痫愈胶囊的安全性和有效性,弥补了现有技术的不足。
[0109]
9.稳定性试验
[0110]
9.1色谱条件与系统适用性试验同3.4。
[0111]
9.2取痫愈胶囊市售样品(批号:20190601、20190602、20190701,西安千禾药业股份有限公司生产)进行加速稳定性实验和室温长期稳定性实验。加速实验条件:将样品放置在相对湿度为75%
±
5%,温度为40℃
±
2℃的稳定性试验箱中进行6个月加速实验考察,分别在第0、1、2、3、6月取样进行检测;室温实验条件:将样品放置在相对湿度60%
±
5%,温度25℃
±
2℃的稳定性试验箱中进行长期24个月的实验考察,分别在第0、3、6、12、18月、24个月检查。试验结果见表8。
[0112]
表8:痫愈胶囊加速及长期稳定性试验数据
[0113][0114]
实验结果表明,加速试验6个月和长期实验24个月,所检测的有效成分天麻素、对羟基苯甲醇、细叶远志皂苷、远志酮ⅲ、3,6
’‑
二芥子酰基蔗糖、柴胡皂苷a、柴胡皂苷d含量均符合要求,说明该检测方法,能有效控制药物的质量,可考虑纳入质量标准中。另,从连续三批售产品(批号:20190601、20190602、20190701)24个月末的检测结果来看:本发明痫愈胶囊中7种有效成分的含量测定方法,从重复性色谱表现图、供试品溶液稳定性图、仪器精密度、回收率、线性、专属性等方面确定本方法的科学性。增加了制剂君药、贵重药的含量测定,降低检验成本,提高检验效率,保证了制剂的安全性和有效性,弥补现有技术的不足,同时在检测的精确性上进一步提升。
[0115]
以上给出的实施例是实现本发明较优的例子,本发明不限于上述实施例。本领域的技术人员根据本发明技术方案的技术特征所做出的任何非本质的添加、替换,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。