1.本技术涉及中药成分鉴定技术领域,特别是涉及一种血必净注射液中化学成分鉴定的方法。
背景技术:
2.血必净注射液由红花、赤芍、川芎、丹参、当归五味中药组成,功能为化瘀解毒,用于温热类疾病,症见发热、喘促、心悸、烦躁等瘀毒互结证。为了阐明血必净注射液的化学物质基础,寻找其活性成分,需要对血必净注射液的化学成分进行检测和鉴定。然而,由于现有分析技术的局限,难以对血必净注射液的化学成分进行全面、准确的检测与鉴定,因此亟需一种分析方法,以对血必净注射液的化学成分进行更全面并且有效的鉴定。
技术实现要素:
3.本技术的目的在于提供一种血必净注射液中化学成分鉴定的方法,采用超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱联用,鉴定血必净注射液的化学成分。
4.本技术提供了一种血必净注射液中化学成分鉴定的方法,所述方法包括:
5.(1)取待测血必净注射液,经0.22
‑
0.45μm微孔滤膜过滤,得到待测样品溶液;
6.(2)通过超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱,获得各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值;
7.其中,超高效液相色谱的色谱条件包括:
8.色谱柱:十八烷基硅烷键合硅胶色谱柱;
9.流动相:a相为体积分数为0.05
‑
0.15%的甲酸水溶液,b相为乙腈;采用体积分数5
‑
99%a相,1
‑
95%b相,梯度洗脱;柱温:38
‑
42℃;流速:0.2
‑
0.4ml/分钟;进样量v1:2
‑
5μl;
10.(3)基于各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值,确定待测样品中的化学成分。
11.本技术提供的一种血必净注射液中化学成分鉴定的方法,采用超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱联用,通过合理选择色谱条件和质谱条件,可以实现血必净注射液中多类别化学成分的鉴定,且所述方法具有简便、灵敏度高、分析速度快、专属性强等优势,为进一步研究血必净注射液的药效物质基础提供了依据。
附图说明
12.为了更清楚地说明本技术实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域普通技术人员来讲,还可以根据这些附图获得其他的实施例。
13.图1为表1中标准品1
‑
37的结构式。
14.图2为表1中标准品38
‑
71的结构式。
15.图3为分别采用10根色谱柱的基峰离子图和分离出的离子的散点图,其中各色谱柱的左图为基峰离子图,纵坐标为相对强度(%),横坐标为保留时间;右图为分离出的离子的散点图,纵坐标为质荷比,横坐标为保留时间。
16.图4为分别采用3种采集方式检测的总离子流图和离子数鉴定的统计结果,其中a图为3种采集方式的总离子流图;b图为3种采集方式分别检测得到被鉴定和未被鉴定的离子数的统计结果。
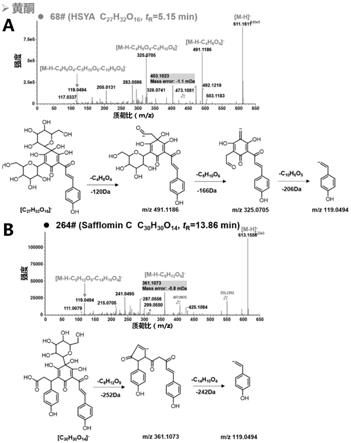
17.图5为血必净注射液中黄酮类成分鉴定的二级质谱图和裂解示意图;其中a图为68#羟基红花黄色素a(hsya)的二级质谱图和裂解示意图;b图为264#羟基红花黄色素c(safflomin c)或其异构体的二级质谱图和裂解示意图。
18.图6为血必净注射液中萜类成分鉴定的二级质谱图和裂解示意图;其中a图为288#苯甲酰氧化芍药苷(benzoyloxypaeoniflorin)的二级质谱图和裂解示意图;b图为148#芍药苷(paeoniflotin)或其同分异构体的二级质谱图和裂解示意图。
19.图7为血必净注射液中有机酸类成分鉴定的二级质谱图和裂解示意图;其中a图为283#丹酚酸b(salvianolic acid b)的二级质谱图和裂解示意图;b图为44#绿原酸(chlorogenic acid)或其异构体的二级质谱图和裂解示意图。
20.图8为血必净注射液中菲醌类成分鉴定的二级质谱图和裂解示意图;其中a图为370#丹参酮i(tanshinone i)的二级质谱图和裂解示意图;b图为319#丹参醛(tanshinaldehyde)或其同分异构体的二级质谱图和裂解示意图。
具体实施方式
21.下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本技术一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员基于本技术所获得的所有其他实施例,都属于本技术保护的范围。
22.本技术提供了一种血必净注射液中化学成分鉴定的方法,所述方法包括:
23.(1)取待测血必净注射液,经0.22
‑
0.45μm微孔滤膜过滤,得到待测样品溶液;
24.(2)通过超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱,获得各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值;
25.其中,超高效液相色谱的色谱条件包括:
26.色谱柱:十八烷基硅烷键合硅胶色谱柱;
27.流动相:a相为体积分数为0.05
‑
0.15%的甲酸水溶液,b相为乙腈;采用体积分数5
‑
99%a相,1
‑
95%b相,梯度洗脱;柱温:38
‑
42℃;流速:0.2
‑
0.4ml/分钟;进样量v1:2
‑
5μl;
28.(3)基于各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值,确定待测样品中的化学成分。
29.本领域技术人员可以根据本技术的方法分离的化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值等信息,与商业数据库、已发表的文章中已公开的红花、赤芍、川芎、丹参和当归中的化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值等信息进行比对,或者通过将已知的红花、赤芍、川芎、丹参和当归中的化学成分的标准品采用与待测样品溶液相同的方法进行鉴定,获得的红花、赤芍、
川芎、丹参和当归中的化学成分的标准品的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值等信息,与分离的化学成分的信息进行比对,以确定待测样品溶液中的化学成分。
30.本技术提供的血必净注射液中化学成分鉴定的方法,采用超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱联用,通过合理选择色谱条件和质谱条件,实现血必净注射液中多类别化学成分的鉴定,且所述方法具有简便、灵敏度高、分析速度快、专属性强等优势。
31.发明人在研究中发现,采用本技术的梯度洗脱,能够使血必净注射液中的化学成分获得更好的分离效果,优选地,在本技术的一些实施方式中,所述梯度洗脱具体为:0
‑
3分钟,1
‑
5%b相;3
‑
4分钟,5
‑
15%b相;4
‑
7分钟,15
‑
15%b相;7
‑
12分钟,15
‑
20%b相;12
‑
16分钟,20
‑
21%b相;16
‑
17分钟,21
‑
55%b相;17
‑
20分钟,55
‑
70%b相;20
‑
22分钟,70
‑
80%b相;22
‑
24分钟,80
‑
95%b相;24
‑
26分钟,95
‑
95%b相。
32.在本技术的一些实施方式中,所述色谱柱选自hss c18 sb、zorbax extend c18、zorbax eclipse plus c18或zorbax sb
‑
c18。发明人在研究中发现,所述色谱柱选自上述色谱柱时,待测样品能够获得较多的离子色谱峰,化学成分能够获得更好的分离效果。
33.在本技术的一些实施方式中,步骤(3)中所述基于各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值,确定待测样品中的化学成分包括:将获得的待测样品中化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值,与红花、赤芍、川芎、丹参和当归中的已知成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值进行比对,确定待测样品中的化学成分。
34.在本技术的一些实施方式中,离子淌度
‑
四级杆飞行时间质谱的质谱条件包括:
35.采用电喷雾离子源,正、负离子全扫描检测模式,质谱参数为:
36.毛细管电压:esi : 1~ 3kv,esi
‑
:
‑
1.0~
‑
3.0kv;锥孔电压:esi :20
‑
100v,esi
‑
:20
‑
100v;源偏移:60
‑
100v;源温度:100
‑
140℃;去溶剂气体温度:氮气,450
‑
550℃;去溶剂气体流量:n2,700
‑
900l/h;锥形气体流量:n2,45
‑
55l/h;数据采集模式为hdms
e
,hdms
e
设定每次扫描时间0.2
‑
0.4s,扫描范围m/z为50
‑
1500;数据采集模式为hddda,hddda设定每次扫描时间0.1
‑
0.2s,低碰撞能量设定为4
‑
8ev;高能裂解能量:ms
e
为esi :20
‑
70ev,esi
‑
:30
‑
70ev;dda为esi :20
‑
40ev/30
‑
50ev,esi
‑
:20
‑
50ev/30
‑
80ev;dda采集一级响应前n强离子的二级信息,3≤n≤10。
37.为了高效鉴定色谱分离后的化学成分,以便于获得更准确的化学成分的鉴定结果,在本技术的一些实施方式中,所述红花、赤芍、川芎、丹参和当归中的已知成分,包括没食子酸、没食子酸甲酯、没食子酸乙酯、没食子酸丙酯、香草酸、香兰素、阿魏酸、阿魏酸乙酯、丹酚酸b、9
’‑
丹酚酸b单甲酯、丹酚酸b二甲酯、鞣花酸、丹参酮i、丹参酮iia、隐丹参酮、对香豆酸、咖啡酸、紫草酸、迷迭香酸、二氢丹参酮i、丹酚酸c、丹酚酸f、丹酚酸a、丹参素、丹参素钠、原儿茶酸、原儿茶醛、盐酸川芎嗪、尿苷、腺苷、氧化芍药苷、苯甲酰芍药苷、苯甲酰氧化芍药苷、愈创木酚、厚朴酚、β
‑
谷甾醇、常春藤皂苷元、5
‑
羟甲基糠醛、欧当归内酯a、洋川芎内酯i、洋川芎内酯h、丁烯基苯酞、蒿本内酯、二氢欧山芹醇当归酸酯、佛手柑内酯、大黄酚、西红花苷ⅰ、( )
‑
儿茶素、芹菜素、杨梅黄酮、木犀草素、槲皮素、山柰酚、山奈酚
‑7‑
o
‑
葡萄糖苷、山柰酚
‑3‑
o
‑
β
‑
d
‑
葡萄糖苷、山奈酚
‑3‑
o
‑
芸香糖苷、山柰酚
‑3‑
o
‑
β
‑
d
‑
葡萄糖醛酸苷、槲皮素
‑7‑
o
‑
β
‑
d
‑
葡萄糖苷、芦丁、异槲皮苷、槲皮素
‑3‑
o
‑
β
‑
d
‑
葡萄糖
‑7‑
o
‑
β
‑
d
‑
龙胆
双糖苷、异鼠李素
‑3‑
o
‑
β
‑
d
‑
葡萄糖
‑7‑
o
‑
β
‑
d
‑
龙胆双糖苷、木犀草苷、木犀草素
‑7‑
o
‑
葡萄糖醛酸苷、异鼠李素
‑3‑
o
‑
葡萄糖苷、异鼠李素
‑7‑
o
‑
葡萄糖苷、金丝桃苷、5,7,4'
‑
三羟基
‑6‑
甲氧基黄酮
‑3‑
o
‑
β
‑
d
‑
芸香苷、(2s)
‑
4',5,6,7
‑
四羟基黄烷酮6
‑
o
‑
β
‑
d
‑
葡萄糖、羟基红花黄色素a和脱水羟基红花黄色素b。
38.在本技术的一些实施方式中,所述已知成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值是按照以下过程获得的:
39.制备5
‑
20μg/ml的所述已知成分的标准品溶液,溶剂为0
‑
100vol%甲醇水溶液;
40.通过超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱,获得各已知成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值;
41.其中,超高效液相色谱的色谱条件包括:
42.色谱柱:十八烷基硅烷键合硅胶色谱柱;
43.流动相:a相为体积分数为0.05
‑
0.15%的甲酸水溶液,b相为乙腈;采用体积分数5
‑
99%a相,1
‑
95%b相,梯度洗脱;柱温:38
‑
42℃;流速:0.2
‑
0.4ml/分钟;进样量v1:2
‑
5μl;
44.离子淌度
‑
四级杆飞行时间质谱的质谱条件包括:
45.采用电喷雾离子源,正、负离子检测模式,质谱参数为:
46.毛细管电压:esi : 1~ 3kv,esi
‑
:
‑
1.0~
‑
3.0kv;锥孔电压:esi :20
‑
100v,esi
‑
:20
‑
100v;源偏移:60
‑
100v;源温度:100
‑
140℃;去溶剂气体温度:氮气,450
‑
550℃;去溶剂气体流量:n2,700
‑
900l/h;锥形气体流量:n2,45
‑
55l/h;数据采集模式为hdms
e
,hdms
e
设定每次扫描时间0.2
‑
0.4s,扫描范围m/z为50
‑
1500;数据采集模式为hddda,hddda设定每次扫描时间0.1
‑
0.2s,低碰撞能量设定为4
‑
8ev;高能裂解能量:ms
e
为esi :20
‑
70ev,esi
‑
:30
‑
70ev;dda为esi :20
‑
40ev/30
‑
50ev,esi
‑
:20
‑
50ev/30
‑
80ev;dda采集一级响应前n强离子的二级信息,3≤n≤10。
47.本技术中所述溶剂为0
‑
100vol%甲醇水溶液,其可以为纯水或任意比例的甲醇水溶液或甲醇。
48.在本技术中,hdms
e
为离子淌度分离、数据非依赖性采集(dia)的高分辨二级质谱;hddda为离子淌度分离、数据依赖性采集(dda)的高分辨二级质谱;hdms
e
‑
hddda组合式采集方法为离子淌度分离功能、单次进样交替进行数据非依赖性采集(dia)与数据依赖性采集(dda)的高分辨二级质谱高效采集的方法。
49.下面对本技术所需的仪器与试剂进行说明。
50.1、仪器
51.agilent 1290高效液相系统(agilent,waldbronn,germany);超高效液相系统acquity uplc i
‑
class(waters,milford,ma,usa);高分辨质谱仪vion im
‑
qtof(waters,milford,ma,usa);eppendorf高速离心机(eppendorf中国有限公司,北京,中国);sb
‑
4200dts/p超声波提取仪(宁波新芝生物科技股份有限公司,浙江,中国);十万分之一天平(mettler toledo,switzerland);万分之一天平(mettler toledo,switzerland);vortex
‑
2旋涡混匀仪(上海泸析实业有限公司,上海,中国);waters xevo g2
‑
xs qtof高分辨质谱。
52.2、试剂和材料
53.乙腈、甲酸(fisher,fair lawn,nj,usa),均为色谱/质谱级;去离子水经milli
‑
q integral5系统(millipore,bedford,ma,usa)纯化。血必净注射液,购自天津红日药业股份
有限公司,批号:1911291。71个标准品购自上海诗丹德生物技术有限公司或成都德思特生物技术有限公司(化合物英文命名、中文命名、分子式、质量数及类型如表1所示,标准品1
‑
37的结构式见图1,标准品38
‑
71的结构式见图2)。
54.表1
55.56.[0057][0058]
如无特殊说明,本技术中所述的标准品均以上述的命名及对应关系为准。
[0059]
以下实施例中所涉及的试剂与药材如无特殊说明均可来源于市售或按照本领域公知方法取得。
[0060]
超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱的条件优化
[0061]
色谱柱优化
[0062]
取待测血必净注射液,经0.22μm微孔滤膜过滤,得到待测样品溶液;
[0063]
采用超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱进行检测,其中,超高效液相色谱的色谱条件包括:
[0064]
色谱柱:分别采用10根色谱柱hss t3、hss c18 sb、beh c18、kinetex 2.6u xb
‑
c18zorbax extend c18、zorbax eclipse plus c18、zorbax sb
‑
c18、zorbax sb
‑
aq、cortecs uplc c18 、uplc beh shield rp18,
[0065]
流动相:a相为体积分数为0.1%的甲酸水溶液,b相为乙腈;柱温:40℃;流速:0.3ml/分钟;进样量v1:3μl;梯度洗脱:0
‑
3分钟,1
‑
5%b相;3
‑
4分钟,5
‑
15%b相;4
‑
7分钟,15
‑
15%b相;7
‑
12分钟,15
‑
20%b相;12
‑
16分钟,20
‑
21%b相;16
‑
17分钟,21
‑
55%b相;17
‑
20分钟,55
‑
70%b相;20
‑
22分钟,70
‑
80%b相;22
‑
24分钟,80
‑
95%b相;24
‑
26分钟,95
‑
95%b相;
[0066]
离子淌度
‑
四级杆飞行时间质谱的质谱条件包括:
[0067]
采用电喷雾离子源,正、负离子全扫描检测模式,质谱参数为:
[0068]
毛细管电压:esi : 1.0kv,esi
‑
:
‑
1.0kv;锥孔电压:esi :60v,esi
‑
:80v;源偏移:80v;源温度:120℃;去溶剂气体温度:氮气,500℃;去溶剂气体流量:n2,800l/h;锥形气体流量:n2,50l/h;数据采集模式为hdms
e
,hdms
e
设定每次扫描时间0.3s,扫描范围m/z为50
‑
1500;数据采集模式为hddda,hddda设定每次扫描时间0.15s,低碰撞能量设定为6ev;高能裂解能量:ms
e
为esi :20
‑
70ev,esi
‑
:30
‑
70ev;dda为esi :20
‑
40ev/30
‑
50ev,esi
‑
:20
‑
50ev/30
‑
80ev;dda采集一级响应前3强离子的二级信息;
[0069]
经检测分别得到10根色谱柱的总离子流图(tic)和质谱图,通过观察各tic图中色谱峰的分离度,并用unifi软件分析提取的离子色谱峰个数,如图3所示,各色谱柱的检测结果中左图为基峰离子图,右图为分离出的离子的散点图;综合判断提取离子色谱峰的个数以及色谱峰的分离度和峰形,本技术采用的色谱柱选自hss c18 sb、zorbax eclipse plus c18、zorbax sb
‑
c18或zorbax extend c18,提取离子色谱峰的个数较多,色谱峰分离度和峰形较好;优选的,色谱柱为zorbax eclipse plus c18。
[0070]
柱温优化
[0071]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件进行检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),柱温分别采用25℃、30℃、35℃、40℃,经检测得到四个柱温的总离子流图和质谱图,通过比较最强离子峰的分离情况,确定待测样品于40℃可以获得更好的分离效果,且基线平稳、峰形对称性好、响应值高。
[0072]
锥孔电压与毛细管电压的优化
[0073]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),毛细管电压分别采用esi( ):1.0、1.5、2.0、2.5、3.0kv,经检测得到待测样品溶液的总离子流图和质谱图;毛细管电压分别采用:esi(
‑
):
‑
1.0、
‑
1.5、
‑
2.0、
‑
2.5、
‑
3.0kv,经检测得到待测样品溶液的总离子流图和质谱图;以5个标准品羟基红花黄色素a、芦丁、没食子酸丙酯、厚朴酚和苯甲酰芍药苷为指标,对峰面积进行分析,得到5个标准品的相对标准偏差(rsd)均小于2.1%。该结果表明当采用毛细管电压esi 为 1~ 3kv、esi
‑
为
‑
1~
‑
3kv时,该方法具有良好的准确性和可重复性,灵敏度较高;优选的,毛细管电压esi 为 1kv、esi
‑
为
‑
1kv时,该方法的准确性更好,灵敏度更高。
[0074]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),锥孔电压分别采用:esi( ):20、40、60、80、100v,经检测得到待测样品溶液的总离子流图和质谱图;锥孔电压分别采用:esi(
‑
):20、40、60、80、100v,经检测得到待测样品溶液的总离子流图和质谱图;以5个标准品(正离子模式:杨梅黄酮、佛手苷内酯、隐丹参酮、二氢丹参酮i和厚朴酚;负离子模式:羟基红花黄色素a和厚朴酚、芦丁、没食子酸丙酯、苯甲酰芍药苷)为指标,对峰面积进行分析,得到5个标准品的相对标准偏差(rsd)均小于5%。该结果表明当采用锥孔电压esi 为20
‑
100v、esi
‑
为20
‑
100v时,该方法具有良好的准确性和可重复性,灵敏度较高;优选的,锥孔电压esi 为60v、esi
‑
为80v时,该方法的准确性更好,灵敏度更高。
[0075]
高能裂解能量的优化
[0076]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条
件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),分别采用5个梯度的高能裂解能量:ms
e
为esi
‑
:20
‑
60ev、30
‑
70ev、20
‑
80ev、40
‑
80ev、60
‑
100ev,经检测得到5个梯度条件下的总离子流图和质谱图,以红花醌苷d(saffloquinoside d)、伞形酮(umbelliferone)、洋川芎内酯a(senkyunolide a)、羟基山奈酚
‑3‑
o
‑
β
‑
芸香糖
‑6‑
o
‑
β
‑
葡萄糖苷(hydroxykaempferol
‑3‑
o
‑
β
‑
rutinoside
‑6‑
o
‑
β
‑
d
‑
glucoside)为指标,观察其母离子峰及二级碎片的信息,saffloquinoside d(醌式查尔酮化合物)在不同能量下,表现出强烈的母离子峰,且碎片较多;而senkyunolide a(苯酞类化合物)产生碎片较多,且随能量增大,母离子丰度逐渐减弱;其它类型的化合物在不同的能量下,均可以产生特征碎片,且随能量增大,umbelliferone(香豆素类化合物)和hydroxykaempferol
‑3‑
o
‑
β
‑
rutinoside
‑6‑
o
‑
β
‑
d
‑
gl ucoside(黄酮类化合物)前体离子减弱较明显。综合判断,确定当高能裂解能量:ms
e
为e si
‑
:30
‑
70ev时,各二级质谱图能够比较清晰地显示出准分子离子峰以及多级碎片信息,提高可鉴定成分的数量和鉴定结果的可靠性。
[0077]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),分别采用5个梯度的高能裂解能量:ms
e
为esi :20
‑
60ev、20
‑
70ev、30
‑
90ev、40
‑
80ev、60
‑
100ev,经检测得到5个梯度条件下的总离子流图和质谱图,以洋川芎内酯a(senkyunolide a)、3
‑
亚丁基
‑7‑
羟基苯酞(3
‑
butylidene
‑7‑
hydroxyphthalide)、(8z)
‑
癸烷
‑
4,6
‑
二炔
‑1‑
醇
‑1‑
o
‑
β
‑
d
‑
葡萄糖醛酸
‑
(1
‑
2')
‑
β
‑
d
‑
吡喃葡萄糖苷((8z)
‑
decaene
‑
4,6
‑
diyne
‑1‑
ol
‑1‑
o
‑
β
‑
d
‑
glucuronyl
‑
(1
‑
2')
‑
β
‑
d
‑
glucopyranoside)、伞形酮(umbelliferone)为指标,观察其母离子峰及二级碎片的信息,综合判断,确定当高能裂解能量:ms
e
为esi :20
‑
70ev时,各二级质谱图能够比较清晰地显示出准分子离子峰以及多级碎片信息,提高可鉴定成分的数量和鉴定结果的可靠性。
[0078]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),分别采用5组高能裂解能量:dda为esi :10
‑
30ev/20
‑
40ev、20
‑
40ev/30
‑
50ev、20
‑
60ev/30
‑
50ev、10
‑
40ev/20
‑
60ev和30
‑
50ev/40
‑
60ev,经检测得到5组能量下的总离子流图和质谱图,以异隐丹参酮ii(isocryptotanshione ii)、脱水羟基红花黄色素b(anhydrosafflor yellow b,a nhsyb)、二氢丹参酮i(dihydroisotanshinone i)、槲皮素(quercetin)和丹参酮iia(tans hinone iia)为指标,观察其母离子峰及二级碎片的信息,并结合各成分的特征离子与母离子响应强度比率,综合判断,确定当高能裂解能量:dda为esi :20
‑
40ev/30
‑
50ev时,能够获得具有母离子宽质量跨度扫描的高质量ms2谱图,提高可鉴定成分的数量和鉴定结果的可靠性。
[0079]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),分别采用4组高能裂解能量:dda为esi
‑
:10
‑
30ev/20
‑
40ev、10
‑
40ev/20
‑
60ev、20
‑
50ev/30
‑
80ev和30
‑
60ev/40
‑
90ev,经检测得到4组能量下的总离子流图和质谱图,以红花醌苷d(saffloquino side d)、水杨酸芍药苷(salicylpaeoniflorin)、6
‑
羟基乙酸
‑
3,6,7
‑
三
‑
o
‑
β
‑
d
‑
葡萄糖(6
‑
hydro xykae
‑
3,6,7
‑
tri
‑
o
‑
β
‑
d
‑
glucoside)、salmiltiorin d和没食子酰芍药苷(galloylpaeoniflorin)为指标,观察其母离子峰及二级碎片的信息,并结合各成分
的特征离子与母离子响应强度比率,综合判断,确定当高能裂解能量:dda为esi
‑
:20
‑
50ev/30
‑
80ev时,能够获得具有母离子宽质量跨度扫描的高质量ms2谱图,提高可鉴定成分的数量和鉴定结果的可靠性。
[0080]
dda采集方式的优化
[0081]
按色谱柱优化中的方法制备待测样品溶液,按色谱柱优化中的色谱条件和质谱条件检测,其中,色谱柱采用zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm),分别采用dda采集一级响应前1强离子的二级信息(top1)、一级响应前2强离子的二级信息(top2)、一级响应前3强离子的二级信息(top3),经检测得到3种采集方式下的总离子流图和质谱图,3种采集方式的总离子流图如图4的a图所示,经unifi软件处理后得到3种采集方式分别检测出的离子数,被鉴定的离子标记为“identified”,未被鉴定出的离子标记为“unknown”,统计结果如图4的b图所示;根据图4的b图可知,“被鉴定”和“未被鉴定”的离子数,top3均多于top1、top2,表明dda采集一级响应前3强离子的二级信息,检测和鉴定的离子数均更多,检测和鉴定的结果更全面。
[0082]
红花、赤芍、川芎、丹参和当归中的已知成分的数据库的建立
[0083]
(1)汇总红花、赤芍、川芎、丹参、当归5种药材的已报道成分(至2020年底),其中红花263种,赤芍126种,川芎195种,丹参238种,当归96种,导入unifi软件,建立已报道成分的数据库,包括化合物的英文名、分子式、质量数及结构式。
[0084]
(2)获得71个标准品的数据:称取表1中71个标准品各1mg,用甲醇溶解,配制成1mg/ml的各标准品储备液,分别取适量合并,并用甲醇稀释得到标准品浓度均为10μg/ml的标准品混合溶液,通过超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱,得到标准品混合溶液的总离子流图和质谱图,采用unifi软件进行数据处理,获得各标准品的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值;
[0085]
其中,超高效液相色谱的色谱条件包括:
[0086]
色谱柱:zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm);
[0087]
流动相:a相为体积分数为0.1%的甲酸水溶液,b相为乙腈;柱温:40℃;流速:0.3ml/分钟;进样量v1:3μl;梯度洗脱:0
‑
3分钟,1
‑
5%b相;3
‑
4分钟,5
‑
15%b相;4
‑
7分钟,15
‑
15%b相;7
‑
12分钟,15
‑
20%b相;12
‑
16分钟,20
‑
21%b相;16
‑
17分钟,21
‑
55%b相;17
‑
20分钟,55
‑
70%b相;20
‑
22分钟,70
‑
80%b相;22
‑
24分钟,80
‑
95%b相;24
‑
26分钟,95
‑
95%b相;
[0088]
离子淌度
‑
四级杆飞行时间质谱的质谱条件包括:
[0089]
采用电喷雾离子源,正、负离子全扫描检测模式,质谱参数为:
[0090]
毛细管电压:esi : 1.0kv,esi
‑
:
‑
1.0kv;锥孔电压:esi :60v,esi
‑
:80v;源偏移:80v;源温度:120℃;去溶剂气体温度:氮气,500℃;去溶剂气体流量:n2,800l/h;锥形气体流量:n2,50l/h;数据采集模式为hdms
e
,hdms
e
设定每次扫描时间0.3s,扫描范围m/z为50
‑
1500;数据采集模式为hddda,hddda设定每次扫描时间0.15s,低碰撞能量设定为6ev;高能裂解能量:ms
e
为esi :20
‑
70ev,esi
‑
:30
‑
70ev;dda为esi :20
‑
40ev/30
‑
50ev,esi
‑
:20
‑
50ev/30
‑
80ev;dda采集一级响应前3强离子的二级信息;
[0091]
采用unifi软件进行数据处理,包括:使用unifi软件实现数据的自动峰注解,未校正的hdms
e
数据首先由m/z 554.2620(esi
‑
)和m/z 556.2766(esi )处的lockmass进行校
正,添加数据库,有效时间设定为26分钟,低能量阈值(low energy intensity)和高能量阈值(high energy intensity)分别设置为200和100,目标匹配容差(target match tolerance)和片段匹配容差(fragment match tolerance)分别设置为10.0ppm和10.0mda,加合离子设置包括:esi :[m h]
、[m na]
;esi
‑
:[m
–
h]
‑
、[m hcoo]
‑
。
[0092]
(3)获得5种药材中鉴定的化学成分的数据:分别取红花、赤芍、川芎、丹参、当归5种药材粉碎,分别过五号筛,过筛率>80%,得到各药材粉末,每种药材称取500mg粉末,分别溶于10ml 20%甲醇中,涡旋2分钟,40℃水浴超声提取30分钟,提取液14000r/min离心10min,取上清液,过0.22μm微孔滤膜过滤,按步骤(2)的色谱条件和质谱条件进行检测,得到各药材的总离子流图和质谱图,按步骤(2)的数据处理方法进行数据处理,获得5种药材中已知化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值。
[0093]
向步骤(1)的数据库中添加步骤(2)获得的数据和步骤(3)获得的数据,获得含有1089个成分的数据库,其中包括红花化学成分293个(正离子模式96个、负离子模式197个),赤芍化学成分182个(正离子模式62个、负离子模式120个),川芎化学成分216个(正离子模式100个、负离子模式116个),丹参化学成分257个(正离子模式111个、负离子模式146个),当归化学成分141个(正离子模式68个、负离子模式73个)。
[0094]
实施例1待测样品鉴定
[0095]
(1)取待测血必净注射液,经0.22μm微孔滤膜过滤,得到待测样品溶液;
[0096]
(2)通过超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱,获得待测样品溶液中各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值;
[0097]
其中,超高效液相色谱的色谱条件包括:
[0098]
色谱柱:zorbax eclipse plus c18色谱柱(2.1
×
100mm,1.8μm);
[0099]
流动相:a相为体积分数为0.1%的甲酸水溶液,b相为乙腈;柱温:40℃;流速:0.3ml/分钟;进样量v1:3μl;梯度洗脱:0
‑
3分钟,1
‑
5%b相;3
‑
4分钟,5
‑
15%b相;4
‑
7分钟,15
‑
15%b相;7
‑
12分钟,15
‑
20%b相;12
‑
16分钟,20
‑
21%b相;16
‑
17分钟,21
‑
55%b相;17
‑
20分钟,55
‑
70%b相;20
‑
22分钟,70
‑
80%b相;22
‑
24分钟,80
‑
95%b相;24
‑
26分钟,95
‑
95%b相;
[0100]
离子淌度
‑
四级杆飞行时间质谱的质谱条件包括:
[0101]
采用电喷雾离子源,正、负离子全扫描检测模式,质谱参数为:
[0102]
毛细管电压:esi : 1.0kv,esi
‑
:
‑
1.0kv;锥孔电压:esi :60v,esi
‑
:80v;源偏移:80v;源温度:120℃;去溶剂气体温度:氮气,500℃;去溶剂气体流量:n2,800l/h;锥形气体流量:n2,50l/h;数据采集模式为hdms
e
,hdms
e
设定每次扫描时间0.3s,扫描范围m/z为50
‑
1500;数据采集模式为hddda,hddda设定每次扫描时间0.15s,低碰撞能量设定为6ev;高能裂解能量:ms
e
为esi :20
‑
70ev,esi
‑
:30
‑
70ev;dda为esi :20
‑
40ev/30
‑
50ev,esi
‑
:20
‑
50ev/30
‑
80ev;dda采集一级响应前3强离子的二级信息;
[0103]
(3)将获得的待测样品中各化学成分的保留时间、母离子质荷比、二级碎片离子质荷比和母离子碰撞截面值,与上述包含1089个成分的数据库比对,对待测样品的化学成分进行鉴定,共鉴定出382种化学成分,包括102种黄酮类化合物、74种萜类化合物、55种苯酞类化合物、47种有机酸类化合物、23种苯丙素类化合物、21种生物碱类化合物、4种菲醌类化合物和56种其他化合物。
[0104]
以下为根据已获得的化合物相关信息,采用比对的方法,对血必净注射液中主要类别的化合物黄酮类、萜类、有机酸类及菲醌类成分分类鉴定的举例:
[0105]
黄酮类成分的鉴定标准:以[m
‑
h]
‑
为分子离子峰。
[0106]
在全扫描质谱中,化合物68#的ms2质谱图(二级质谱图)和裂解示意图如图5的a图所示,其给出了m/z 611.1611(c
27
h
32
o
16
)处的[m
‑
h]
‑
离子,在ms2质谱图中,观察到所获得的离子在m/z 491.1186(c
23
h
23
o
12
)产生特征片段[m
–
h
–
c4h8o4]
–
,对应于标准品羟基红花黄色素a不仅色谱行为一致,还观察到m/z 491.1186碎片的进一步损失在m/z325.0705(c
18
h
13
o6)的[m
–
h
–
c4h8o4–
c5h
10
o6]
–
和m/z 119.0494(c8h7o5)的[m
–
h
–
c4h8o4–
c5h
10
o6–
c
10
h6o5]
–
产生的相同的特征碎片,因此,化合物68#被鉴定为羟基红花黄色素a(hsya)。
[0107]
化合物264#的ms2质谱图和裂解示意图如图5的b图所示,在m/z 613.1556(c
30
h
30
o
14
)处显示出[m
‑
h]
‑
离子,在ms2质谱图中,m/z 361.1073(c
22
h
17
o5)处的[m
–
h
–
c8h
12
o9]
–
离子和m/z 119.0494(c8h7o5)处的[m
–
h
–
c8h
12
o9‑
c
14
h
10
o4]
–
查尔酮特征碎片离子是由c8h
12
o9单位的损失和c
14
h
10
o4的进一步损失产生的,这些碎片断裂特征和羟基红花黄色素c(safflomin c)相同,化合物264#被推测认定为safflomin c或其异构体。
[0108]
萜类化合物的鉴定标准:以[m
‑
h]
‑
为分子离子峰,特征碎片常见为脱去苯甲酰和己糖部分。
[0109]
化合物288#(t
r
=15.73min)的ms2质谱图和裂解示意图如图6的a图所示,在m/z599.1762处具有较强的母离子丰度,根据高分辨数据推断其分子式为c
30
h
32
o
13
,它的[m
–
h]
–
母离子裂解产生碎片m/z 447.1283((c
23
h
25
o
11
),[m
–
h
–
c8h8o3]
–
)和二次裂解产生的碎片m/z 281.0657((c
13
h
13
o7),[m
–
h
–
c
17
h
18
o
36
]
–
),m/z 137.0237((c7h5o3),[m
–
h
–
c8h8o3–
o
–
c
15
h
18
o6]
–
),表明其存在o
‑
苯甲酰单元、苯甲酰单元和己糖部分。通过与同条件下采集的标准品比较分析进行鉴定,将其表征苯甲酰氧化芍药苷(benzoyloxypaeoniflorin)。
[0110]
化合物148#(t
r
=6.93min)的ms2质谱图和裂解示意图如图6的b图所示,根据高分辨数据推断其分子式为c
23
h
28
o
11
(m/z 479.1555),它的[m
–
h]
–
母离子裂解失去一分子ch4o产生特征诊断离子m/z 449.1448([m
–
h
–
ch4o]
–
)和脱去苯甲酰和己糖部分的典型碎片m/z121.0287([m
–
h
–
ch4o
–
c
15
h
20
o8]
–
),另外,低丰度碎片m/z 327.1078和m/z 165.0548来自于m/z 449.1448([m
–
h
–
ch4o]
–
)脱去苯甲酰单元(c7h6o2)和薄荷烷骨架(c9h9o3),所以将其鉴定为芍药苷(paeoniflotin)或其同分异构体。
[0111]
有机酸类化合物的鉴定标准:以[m
‑
h]
‑
为分子离子峰。
[0112]
在负离子全扫描模式下,化合物283#(mass error:
‑
0.9ppm;t
r
=15.34min)在m/z 717.1500(c
36
h
30
o
16
)处显示出[m
–
h]
–
离子,其ms2质谱图和裂解示意图如图7的a图所示,它的两个质谱数据显示在m/z 519.0926((c
27
h
19
o
11
),[m
–
h
–
c9h
10
o5]
–
),m/z 339.0500((c
18
h
11
o7),[m
–
h
–
c9h
10
o5–
c9h8o4]
–
),和m/z 339.0500丢失一份子h2o产生的高丰度特征碎片m/z321.0393((c
18
h9o6),[m
–
h
–
c9h
10
o5–
c9h8o4–
h2o]
–
)碎片以及丢失co2产生的碎片离子m/z 295.0600([m
–
h
–
c9h
10
o5–
c9h8o4–
co2]
–
),表明化合物283#是丹酚酸b或其异构体,通过与标准品的比较,确定化合物283#是丹酚酸b(salvianolic acid b)。
[0113]
类似地,化合物44#被鉴定为绿原酸或其异构体,因为存在与绿原酸相关的诊断碎片离子。化合物44#的ms2质谱图和裂解示意图如图7的b图所示,在它的全扫描负离子模式质谱中在m/z 353.0873(c
16
h
18
o9)处显示[m
–
h]
–
离子,在它的质谱中发现了前体离子失去
ch2o2基团产生的m/z 307.1760((c
15
h
16
o7),[m
–
h
–
ch2o2]
–
)和失去羧基六元烷环产生的m/z 191.0555((c
10
h6o4),[m
–
h
–
c6h
11
o5]
–
)离子,在m/z 191.0555的碎片结构种由于碰撞能量继续断裂在m/z 135.0446[m
–
h
–
c6h
11
o5–
c2o2]
–
处检测到的离子,这些断裂特征与绿原酸的特征信息相关,推测化合物44#为绿原酸(chlorogenic acid)或其异构体。
[0114]
菲醌类化合物的鉴定标准:以[m h]
‑
为分子离子峰,特征碎片为co、ch3、co2损失,包括几种特征碎片同时出现的情况。
[0115]
菲醌类化合物主要来自于血必净注射液中的丹参药材。化合物370#的ms2质谱图和裂解示意图如图8的a图所示,它的[m h]
离子在m/z 277.0947处显示,表明其分子式为c
18
h
12
o3,保留时间为20.93min,误差在负全扫描模式下为
‑
3.2ppm,m/z 249.0886(c
17
h
13
o2)、221.0938(c
16
h
13
o)、178.0763(c
14
h
10
)处的碎片离子分别由一分子co损失、一氧化碳进一步损失和在m/z 221.0938进一步损失c2h3o产生的[m h
–
co]
、[m h
–
co
–
co]
、[m h
–
co
–
co
–
c2h3o]
离子,在m/z 204.1368(c
15
h8o)处的离子是在m/z 221.0938处碎片损失
‑
·
ch3后生成的离子,这些离子是与丹参酮i相关的特征中性损失,经过标准品比对发现,化合物370#保留时间与色谱行为与标准品丹参酮i一致,最后确定化合物370#为丹参酮i(tanshinone i)。
[0116]
化合物319#(t
r
=17.74min)的ms2质谱图和裂解示意图如图8的b图所示,根据高分辨数据推断其分子式为c
19
h
18
o4(m/z 309.1104),它的准分子母离子m/z 309.1104([m h]
)裂解产生脱去一分子co2的碎片m/z 265.1212([m h
–
co2]
)和脱去c2h3o后在m/z223.0745处产生的较高丰度离子[m h
–
c2h3o]
,该裂解特征能够在丹参醛中发生,所以推测化合物319#为丹参醛(tanshinaldehyde)或其同分异构体。
[0117]
本技术建立超高效液相色谱
‑
离子淌度
‑
四级杆飞行时间质谱联用方法,通过合理选择色谱条件和质谱条件,实现血必净注射液中多类别化学成分的鉴定,此方法简便、灵敏度高、分析速度快、专属性强。
[0118]
本说明书中的各个实施例均采用相关的方式描述,各个实施例之间相同相似的部分互相参见即可,每个实施例重点说明的都是与其他实施例的不同之处。尤其,对于系统实施例而言,由于其基本相似于方法实施例,所以描述的比较简单,相关之处参见方法实施例的部分说明即可。
[0119]
以上所述仅为本技术的较佳实施例,并非用于限定本技术的保护范围。凡在本技术的精神和原则之内所作的任何修改、等同替换、改进等,均包含在本技术的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。