用于治疗癌症的方法
1.分案申请
2.本技术是2016年4月29日提交的名称为“用于治疗癌症的方法”,申请号为 201680038907.4的中国专利申请的分案申请。
3.相关申请的交叉引用
4.本技术要求2015年4月29日提交的美国临时申请第62/154,699号、2015年4月30日提交的美国临时申请第62/155,451号、2015年11月6日提交的美国临时申请第62/252,085 号、2015年12月10日提交的美国临时申请第62/265,696号、2015年5月7日提交的美国临时申请第62/158,469号、2015年11月9日提交的美国临时申请第62/252,916号、2015年12 月10日提交的美国临时申请第62/265,774号、2015年7月15日提交的美国临时申请第 62/192,940号、2015年12月10日提交的美国临时申请第62/265,658号、2016年4月15日提交的美国临时申请第62/323,572号、2015年7月15日提交的美国临时申请第62/192,944 号、2015年12月10日提交的美国临时申请第62/265,663号以及2016年4月15日提交的美国临时申请第号62/323,576号的权益;所有这些申请均通过引用以其全文并入本文。
背景技术:
5.基于三种受体:雌激素受体(er)、孕酮受体(pr)和人表皮生长因子受体
‑
2(her2) 的表达,将乳腺癌分为三种亚型。在许多乳腺癌患者体内发现了er的过度表达。er阳性 (er )乳腺癌占所有乳腺癌的三分之二。除乳腺癌之外,雌激素和er与例如卵巢癌、结肠癌、前列腺癌和子宫内膜癌有关。
6.er可以被雌激素激活并转移到细胞核中以结合dna,由此调节几种基因的活性。参见例如marino等人的“estrogen signaling multiple pathways to impact gene transcription”(curr genomics 7(8):497
‑
508(2006));和heldring等人的“estrogen receptors:how do they signal andwhat are their targets”(physiol.rev.87(3):905
‑
931(2007))。
7.在er阳性乳腺癌的治疗中,之前已经在使用或者正在开发抑制雌激素产生的药剂(例如芳香酶抑制剂(ai,例如来曲唑、阿那曲唑和阿诺新))、或者直接阻断er活性的那些药剂(例如选择性雌激素受体调节剂(serm,例如他莫昔芬、托瑞米芬、屈洛昔芬、艾多昔芬、雷洛昔芬、拉索昔芬、阿佐昔芬、米泼昔芬、左美洛昔芬、和em
‑
652(sch 57068)) 以及选择性雌激素受体降解剂(serd,例如氟维司群、tas
‑
108(sr16234)、zk191703、 ru58668、gdc
‑
0810(arn
‑
810)、gw5638/dpc974、srn
‑
927、ici182782和azd9496)。
8.经常使用serm(例如他莫昔芬)和ai作为er阳性乳腺癌的一线辅助系统治疗。他莫昔芬通常用于er阳性乳腺癌。ai通过阻断芳香酶的活性(其将体内的雄激素转化为雌激素) 来抑制外周组织中的雌激素产生。然而,ai不能阻止卵巢制造雌激素。因此,ai主要用于治疗绝经后的妇女。此外,由于ai比他莫昔芬有效得多且产生较少的严重副作用,所以ai也可以用于治疗其卵巢功能受抑制的绝经前妇女。参见例如francis等人的“adjuvant ovariansuppression in premenopausal breast cancer”(nengl.j.med.,372:436
‑
446(2015))。
9.虽然利用这些药剂进行的初始治疗可能是成功的,但许多患者最终复发耐药性乳腺癌。影响er的突变已经成为这种耐药性发展的潜在机制之一。参见例如robinson等人,“activating esr1 mutations in hormone
‑
resistant metastatic breast cancer,”nat.genet. 45:1446
‑
51(2013)。在21%的来自接受至少一线内分泌治疗患者的转移性er阳性乳房肿瘤样品中发现了er配体结合结构域(lbd)中的突变。jeselsohn等人,“esr1 mutations
‑
a mechanism for acquired endocrine resistance in breast cancer,”nat.rev.clin.oncol.,12:573
‑
83 (2015)。
10.氟维司群是目前唯一批准用于治疗抗雌激素治疗后具有疾病进展的er阳性转移性乳腺癌的serd。尽管其具有临床功效,但是氟维司群的效用已经受单次注射中可施用的药剂量和生物利用度降低限制。使用18f
‑
氟雌二醇正电子发射断层扫描(fes
‑
pet)的成像研究表明,甚至在500mg剂量水平,一些患者也可能不能完全抑制er,并且剂量不足可能是治疗失败的原因。
11.与雌激素指导的疗法相关的另一挑战在于它们可能对子宫、骨和其他组织具有不期望的影响。er指导几种组织和细胞类型中的雌激素应答基因的转录。随着绝经期间雌激素和其他卵巢激素的内源性水平减少,这些影响可能会特别显著。例如,由于对子宫内膜具有部分激动剂的作用,他莫昔芬可导致绝经前妇女骨质疏松,并增加罹患子宫内膜癌的风险。在绝经后妇女中,ai相比他莫昔芬可导致更多的骨质流失和更多的骨折。由于其作用机制,利用氟维司群治疗的患者也可能遭受骨质疏松症的风险。
12.磷脂酰肌醇3
‑
激酶(pi3k)/蛋白激酶b(akt)/雷帕霉素(mtor)途径的哺乳动物靶标是在调节细胞周期中重要的胞内信号传导途径。癌症中pi3k/akt/mtor途径的频繁激活及其在细胞生长和存活中的关键作用对寻找适宜的增殖
‑
分化量以在开发各种疗法时利用这种平衡带来了挑战。参见,例如gitto等,“recent insights into the pathophysiology of mtor pathwaydysregulation,”res.rep.bio.,2:1
‑
16(2015)。
13.与其他疗法结合施用时,pi3k途径的抑制剂显示出最大的希望。例如,2012年依维莫司 (一种变构型mtor抑制剂)被批准用于与ai依西美坦联用治疗患有晚期激素受体阳性 (hr )、her2
‑
乳腺癌的绝经后妇女(bolero
‑
2研究)。正在开发用于治疗hr 癌症的靶向pi3k途径其他组分的药物,例如pi3k和mtor的atp竞争性双重抑制剂(例如bez235, gdc
‑
0980)、抑制i类pi3k的全部四种同种型的泛pi3k抑制剂(例如bkm120、gdc
‑
0941)、各种pi3k同种型的异构体特异性抑制剂(例如byl719、gdc
‑
0032)、akt的变构和催化抑制剂(mk2206、gdc
‑
0068、gsk2110183、gsk2141795、azd5363)、和仅mtor的atp 竞争性抑制剂(azd2014、mln0128和cc
‑
223)。dienstmann等人,“picking the point ofinhibition:a comparative review of pi3k/akt/mtor pathway inhibitors,”mol.cancer ther, 13(5):1021
‑
31(2014)。
14.尽管它们具有巨大的潜力,但是与mtor抑制剂有关的不良副作用阻碍了它们作为有效癌症疗法的发展。kaplan等,“strategies for the management of adverse events associated withmtor inhibitors,”transplant rev(orlando),28(3):126
‑
133(2014);and pallet et al.,“adverseevents associated with mtor inhibitors,”expert opin.drug saf.12(2):177
‑
186(2013)。
15.仍然需要如下更为持久和有效的er靶向疗法:所述疗法可以克服与目前的内分泌
治疗相关的挑战,同时通过与第二治疗剂(例如,依维莫司和其它靶向pi3k/akt/mtor途径的药剂)结合对抗晚期癌症和/或对既往治疗具有耐药性的癌症来提供额外的益处。
技术实现要素:
16.本发明的一个方面涉及用于治疗受试者中的一种或多种癌症和/或肿瘤的方法,其包括向所述受试者施用治疗有效量的rad1901或其溶剂化物(例如水合物)或其盐的组合和如本文所述的一种或多种第二治疗剂(例如,依维莫司)。
17.在一些实施方案中,所述癌症是雌激素依赖性癌症,诸如乳腺癌、卵巢癌、结肠癌、子宫内膜癌或前列腺癌。在一些实施方案中,所述癌症是er阳性乳腺癌。
18.将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)组合施用至有需要的受试者。短语“组合”是指在施用所述第二治疗剂(例如依维莫司)之前、期间或之后施用rad1901或其溶剂化物(例如水合物)或其盐。例如,可以按以下时间施用 rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂:间隔约一周、间隔约6天、间隔约5天、间隔约4天、间隔约3天、间隔约2天、间隔约24小时、间隔约23小时、间隔约22小时、间隔约21小时、间隔约20小时、间隔约19小时、间隔约18小时、间约17小时、间隔约16小时、间隔约15小时、间隔约14小时、间隔约13小时、间隔约12小时、间隔约11小时、间隔约10小时、间隔约9小时、间隔约8小时、间约7小时、间隔约6小时、间隔约5小时、间隔约4小时、间隔约3小时、间隔约2小时、间隔约1小时、间隔约55分钟、间隔约50分钟、间隔约45分钟、间隔约40分钟、间隔约35分钟、间隔约30分钟、间隔约25分钟、间隔约20分钟、间隔约15分钟、间隔约10分钟或间隔约5分钟。在其它实施方案中,将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司) 同时或基本上同时施用至所述受试者。在这些实施方案的某些实施方案中,可以作为单一制剂的一部分来施用化合物。
19.在一些实施方案中,以分开的制剂施用rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂。在这些实施方案中的某些实施方案中,制剂可以是相同类型的制剂。例如,可以将两种制剂都设计用于口服施用(例如,通过两种分开的丸剂)或用于注射(例如,通过两种分开的可注射制剂)。在其他实施方案中,可以将rad1901或其溶剂化物(例如水合物) 或其盐和第二治疗剂(例如依维莫司)配制成不同类型的制剂。例如,一种化合物可以在设计用于口服给药的制剂中,而另一种化合物在设计用于注射的制剂中。
20.在其他实施方案中,作为单一制剂的一部分施用rad1901或其溶剂化物(例如水合物) 或其盐和第二治疗剂。例如,将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂配制成用于口服施用的单一丸剂或配置成单一注射剂。相应地,在某些实施方案中,本文提供了包含rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂的制剂。
21.rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂的施用途径包括但不限于局部施用、口服施用、皮内施用、肌内施用、腹膜内施用、静脉内施用、膀胱内灌注、皮下施用、透皮施用、和透粘膜施用。
附图说明
22.图1:rad1901在源自患者的几个异种移植(pdx)模型中抑制肿瘤生长(tgi),无论该肿瘤的esr1状态和既往内分泌治疗史。其显示了利用rad1901治疗的pdx模型中的肿瘤生
长抑制(tgi)的百分比。
23.图2a
‑
c:在野生型(wt)erα mcf
‑
7小鼠异种移植模型(pr ,her2
‑
)中,rad1901 和依维莫司的组合表现出肿瘤生长抑制和消退。(图2a):利用载体对照、依维莫司(2.5mg/kg,口服,每日一次)、氟维司群(3mg/剂,皮下注射,每周一次)、氟维司群(3mg/剂,皮下注射,每周一次)和依维莫司(2.5mg/kg,口服,每日一次)的组合、rad1901(60mg/kg,口服,每日一次)、和rad1901(60mg/kg,口服,每日一次)和依维莫司(2.5mg/kg,口服,每日一次)的组合治疗的mcf
‑
7异种移植模型中的肿瘤生长;单因素方差分析,“ns”为不显著,*p值<0.05,***p值<0.001;(图2b):用溶媒对照、依维莫司(2.5mg/kg,口服,每日一次)、氟维司群(3mg/剂,皮下注射,每周一次)、氟维司群(3mg/剂,皮下注射,每周一次)和依维莫司(2.5mg/kg,口服,每日一次)的组合、rad1901(60mg/kg,口服,每日一次)、和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5mg/kg,口服,每日一次)组合治疗的mcf
‑
7异种移植模型中从基线至研究结束时个体肿瘤尺寸的变化;(图 2c):利用溶媒对照、依维莫司(2.5mg/kg,口服,每日一次)、氟维司群(3mg/剂,皮下注射,每周一次)、氟维司群(3mg/剂,皮下注射,每周一次)和依维莫司(2.5mg/kg,口服,每日一次)的组合、rad1901(30或60mg/kg,口服,每日一次)、和rad1901(30或60mg/kg,口服,每日一次)和依维莫司(2.5mg/kg,口服,每日一次)的组合治疗的mcf
‑
7异种移植模型中的肿瘤生长。
24.图3a
‑
b:在wt erα pdx
‑
11模型(pr ,her2
‑
,之前曾用芳香酶抑制剂、氟维司群和化学疗法进行治疗)中,rad1901和依维莫司的组合表现出肿瘤生长抑制和消退。(图3a):利用溶媒对照、氟维司群(3mg/剂,皮下注射,每周一次)、依维莫司(2.5mg/kg,口服,每日一次)、rad1901(60mg/kg,口服,每日一次)、和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5mg/kg,口服,每日一次)的组合治疗的pdx
‑
11模型的肿瘤生长;(图 3b):在利用溶媒对照、氟维司群(3mg/剂,皮下注射,每周一次)、rad1901(60mg/kg,口服,每日一次)、和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5mg/kg,口服,每日一次)的组合治疗的pdx
‑
11模型中从基线到研究结束时个体肿瘤尺寸的变化。n= 8
‑
10/组。
25.图4a
‑
b:在wt er pdx
‑
2模型(pr ,her2
‑
,初次治疗)中,rad1901和依维莫司的组合表现出肿瘤生长抑制。(图4a):利用溶媒对照、rad1901(60mg/kg,口服,隔日一次)、氟维司群(3mg/剂,皮下注射,每周一次)、和rad1901(60mg/kg,口服,隔日一次) 与氟维司群(3mg/剂,皮下注射,每周一次)的组合治疗的pdx
‑
2模型的肿瘤生长;(图4b):利用溶媒对照、依维莫司(2.5mg/kg,口服,每日一次)、rad1901(60mg/kg,口服,隔日一次)、和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5mg/kg,口服,每日一次)的组合治疗的pdx
‑
2模型的肿瘤生长。n=8
‑
10/组。
26.图5:在rad1901治疗结束之后继续在wt erα pdx
‑
4模型(pr ,her2
‑
,初次治疗) 中进行雌二醇治疗,rad1901的功效持续至少两个月。
27.图6a
‑
b:在突变型(y537s)erα pdx
‑
5模型(pr ,her2 ,之前曾用芳香酶抑制剂治疗)中,rad1901与依维莫司的组合表现出肿瘤生长抑制。(图6a)利用溶媒对照、rad1901 (60mg/kg,口服,每日一次)、和氟维司群(3mg/剂,皮下注射,每周一次)治疗的pdx
‑
5 模型的肿瘤生长;(图6b)利用溶媒对照、rad1901(60mg/kg,口服,每日一次)、依维莫司(2.5mg,口服,每日一次)、和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5 mg,口服,每日一次)的组合治疗的pdx
‑
5模型的肿瘤生长。n=8
‑
10/组。
28.图7:裸鼠中的氟维司群药代动力学分析。其显示在1mg/剂(实心菱形)、3mg/剂(实心圆圈)和5mg/剂(实心三角形)下氟维司群的血浆浓度。在第1天给裸鼠皮下注射氟维司群,在第8天施用第二剂。在第二剂之后长达168小时的时间内,在指定的时间点监测氟维司群的血浆浓度。
29.图8:rad1901和氟维司群(faslodex)对颅内mcf
‑
7肿瘤模型中小鼠存活的影响。
30.图9a
‑
c:利用200和500mg rad1901(口服,每日一次)治疗的受试者的子宫的fes
‑
pet 扫描的代表性图像和rad1901治疗后er结合的变化。(图9a):在200mg rad1901治疗之前(a)和之后(c)的子宫ct扫描的横向视图以及在rad1901治疗之前(b)和之后(d) 的子宫fes
‑
pet扫描的横向视图;(图9b):在500mg rad1901治疗之前(上(a)图)和之后(下(a)图)的子宫ct扫描的矢状位图、在rad1901治疗之前(上(b)图)和之后 (下(b)图)的子宫fes
‑
pet扫描的矢状位图、在rad1901治疗之前(上(c)图)和之后(下(c)图)的子宫ct扫描的横向视图、在rad1901治疗之前(上(d)图)和之后(下 (d)图)的子宫fes
‑
pet扫描的横向视图;(图9c):与基线(rad1901治疗前)相比受试者1
‑
3(200mg)和受试者4
‑
7(500mg)进行rad1901治疗后的%er结合变化。
31.图10a
‑
b:rad1901治疗(500mg)之前(基线)和之后(治疗后)子宫(图10a)和垂体(图10b)的fes
‑
pet扫描的代表性图像。(a)侧向截面;(b)纵向截面;和(c)纵向截面。
32.图11:利用溶媒对照、rad1901、依维莫司、rad1901和依维莫司的组合、氟维司群、和氟维司群与依维莫司的组合治疗的mcf
‑
7异种移植模型中的pr和er表达。
33.图12a
‑
b:在体外,rad1901治疗导致mcf
‑
7细胞系(图12a)和t47d细胞系(图 12b)中er完全降解并抑制er信号传导。显示在以0.001 μm、0.01 μm、0.1μm和1μm的几种浓度下分别用rad1901和氟维司群治疗的两种细胞系中的er表达。通过测试的三种er靶基因显示er信号传导:pgr、greb1和tff1。
34.图13a
‑
c:在mcf
‑
7异种移植模型中,rad1901治疗导致er降解并消除er信号传导。 (图13a):蛋白质免疫印迹,其显示了在利用溶媒对照、30和60mg/kg rad1901和3mg/ 剂氟维司群治疗的mcf
‑
7异种移植模型中、在最后一剂后2小时或8小时时的pr和er表达;(图13b):在利用溶媒对照、30和60mg/kg rad1901和3mg/剂氟维司群治疗的mcf
‑
7 异种移植模型中、在最后一剂后2小时时的er蛋白表达;(图13c):在利用溶媒对照、30 和60mg/kg rad1901和3mg/剂氟维司群治疗的mcf
‑
7异种移植模型中、在最后一剂后8 小时时的pr蛋白表达。
35.图14a
‑
c:rad1901治疗导致mcf
‑
7异种移植模型中的pr快速降低。(图14a):蛋白质免疫印迹,其显示在利用溶媒对照和rad1901(30、60和90mg/kg)治疗的mcf
‑
7异种移植模型中、在单剂之后8小时或12小时时的pr表达;(图14b):蛋白质免疫印迹,其显示在利用溶媒对照和rad1901(30、60和90mg/kg)治疗的mcf
‑
7异种移植模型中、在第7剂之后4小时或24小时时的pr表达;(图14c):在利用30、60和90mg/kg rad1901 治疗的mcf
‑
7异种移植模型中pr表达的剂量依赖性降低。
36.图15a
‑
b:rad1901治疗导致mcf
‑
7异种移植模型中的增殖快速降低。(图15a):在单剂后8小时和第4剂后24小时时从利用溶媒对照和90mg/kg rad1901治疗的mcf
‑
7 异种移植模型采集的切片肿瘤的代表性照片,其中对增殖标志物ki
‑
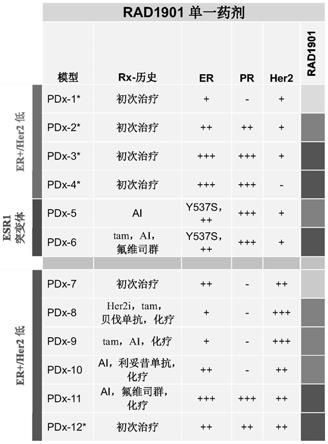
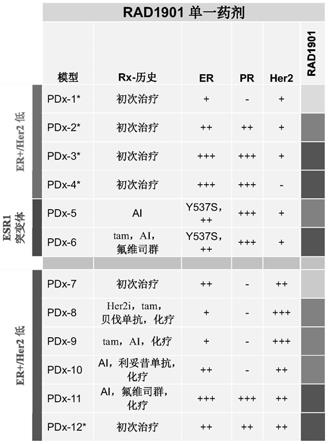
67进行染色;(图15b):直方图,其显示在单剂之后8小时和第4剂之后24小时时利用溶媒对照和90mg/kg rad1901 治疗的
mcf
‑
7异种移植模型中的增殖标志物ki
‑
67减少。
37.图16:在56天功效研究的最后一天,4小时时,30、60和120mg/kg rad1901治疗相比氟维司群(1mg/动物)更显著地降低pdx
‑
4模型的研究结束肿瘤中的ki67。
38.图17:60和120mg/kgrad1901治疗导致pdx
‑
5模型中的体内er信号传导减少,伴随 pr表达降低。
39.图18a
‑
d:rad1901对新断奶雌性sprague
‑
dawley大鼠子宫组织的影响。(图18a):在最后一剂后24小时时安乐死的大鼠的子宫湿重;(图18b):子宫组织切片中的上皮高度; (图18c):400倍放大的经甲苯胺蓝o
‑
染色的子宫组织的代表性切片,箭头指示子宫上皮; (图18d):从子宫组织提取并通过定量rt
‑
pcr分析其相对于18s核糖体rna看家基因的补体c3表达水平的总rna。
40.图19:在第7天给药200、500、750和1000mg/kg rad1901后的血浆药代动力学结果。
41.图20:3ert(i)。
42.图21:3ert(ii)。
43.图22:表11中汇总的erα lbd拮抗剂复合物的叠加。
44.图23a
‑
b:(图23a)rad1901
‑
1r5k;和(图23b)gw5
‑
1r5k的建模。
45.图24a
‑
b:(图24a)rad1901
‑
1sj0;和(图24b)e4d
‑
1sj0的建模。
46.图25a
‑
b:(图25a)rad1901
‑
2jfa;和(图25b)ral
‑
2jfa的建模。
47.图26a
‑
b:(图26a)rad1901
‑
2bj4;和(图26b)oht
‑
2bj4的建模。
48.图27a
‑
b:(图27a)rad1901
‑
2iok;和(图27b)iok
‑
2iok的建模。
49.图28:由利用1r5k和2ouz进行的ifd分析获得的rad1901构象的叠加。
50.图29:由利用2bj4和2jfa进行的ifd分析获得的rad1901构象的叠加。
51.图30a
‑
b:由利用2bj4、2jfa和1sj0进行的ifd分析获得的rad1901构象的叠加。
52.图31a
‑
c:利用2bj4进行的rad1901的ifd。
53.图32a
‑
c:通过ifd对接在2bj4中的rad1901的蛋白质表面相互作用。
54.图33a
‑
c:利用2bj4进行的氟维司群的ifd。
55.图34a
‑
b:利用2bj4进行的氟维司群和rad1901的ifd。
56.图35a
‑
b:利用2bj4进行的氟维司群和rad1901的ifd的叠加。
57.图36:利用wt和lbd突变体的erα构建体进行的rad1901体外结合测定。
58.表1.治疗40天后在植入mcf7细胞的小鼠的血浆、肿瘤和大脑中的rad1901水平。 blq:低于定量极限。
59.表2.用200mg剂量(口服,每日一次)治疗6天的人受试者的子宫、肌肉和骨的suv。
60.表3.用500mg剂量(口服,每日一次)治疗6天的人受试者(n=4)的子宫、肌肉和骨的suv。
61.表4.rad1901对卵巢切除大鼠中的bmd的影响。对成年雌性大鼠进行假手术或卵巢切除手术,然后利用载体、e2(0.01mg/kg)或rad1901(3mg/kg)每天一次(n=20/每个治疗组)开始治疗。在基线和治疗4周后通过双发射x射线吸收测量法测量bmd。数据表示为平均值 sd。相对于对应的ovx veh对照,*p<0.05。bmd为骨矿物质密度;e2为β雌二醇;ovx为卵巢切除;veh为溶媒。
62.表5.rad1901对卵巢切除大鼠的股骨微构造的影响。对成年雌性大鼠进行假手术
或卵巢切除手术,然后利用载体、e2(0.01mg/kg)或rad1901(3mg/kg)每天一次(n=20/每个治疗组)开始治疗。在4周后,使用微计算机断层扫描技术评估骨的微结构。数据表示为平均值
±
sd。相对于对应的ovx veh对照,*p<0.05。abd为表观骨密度;bv/tv为骨体积密度;connd为连接密度;e2为β雌二醇;ovx为卵巢切除;tbn为骨小梁数目;tbth为骨小梁厚度;tbsp为骨小梁间距;veh为溶媒。
63.表6.rad1901的1期剂量递增研究的关键基线人口统计学。
64.表7.在rad1901的1期剂量递增研究中最频繁(>10%)的治疗相关ae。ae根据ctcaev4.0进行分级。经历同一优先项目的多种情形的任何患者都只以最严重的等级计算一次。 *>10%的总活跃组中具有任何相关teae的患者。n=给定类别中具有至少一个治疗相关ae 的受试者数目。
65.表8.rad1901的1期剂量递增研究中的药物动力学参数(第7天)。
66.表9.lbd突变的频率。
67.表10.er
‑
α lbd
‑
拮抗剂复合物与3ert的残基构型差异。
68.表11.通过rmsd计算评估er
‑
α lbd
‑
拮抗剂复合物的结构重叠。
69.表12.er
‑
αlbd
‑
拮抗剂复合物中配体结合的分析。
70.表13.rad1901对接的模型评估。
71.表14.rad1901与1r5k、1sj0、2ifa、2bj4和2ouz的诱导契合对接评分。
具体实施方式
72.如以下实施例部分阐述的,在几种乳腺癌异种移植模型中,rad1901和依维莫司的组合 (rad1901
‑
依维莫司组合)(结构见下)比单独使用rad1901表现出更大的肿瘤生长抑制,所述几种乳腺癌异种移植模型包括:野生型(wt)erα mcf
‑
7异种移植物模型(图2a
‑
c)、 wt erα pdx
‑
2(图4a
‑
b)和pdx
‑
11模型(图3a
‑
b)、和突变型(例如y537s)erα pdx
‑
5 模型(图6a
‑
b),无论该肿瘤的esr1状态以及实施例i中描述的既往内分泌治疗史。pdx
‑
2、 pdx
‑
5和pdx
‑
11模型具有表达wt或突变型(例如y537s)erα、具有pr表达,具有高或低her2表达、以及具有或没有既往内分泌治疗(例如,ai、氟维司群)、和/或化疗(chemo) (图1)的肿瘤。单独使用rad1901也抑制图1中列出的所有其他pdx模型中的肿瘤生长,即具有表达wt或突变型(例如,y537s)erα、具有pr表达、具有高或低her2表达、以及有或没有既往内分泌疗法(例如,他莫昔芬(tam)、ai、氟维司群)、化疗(chemo)、her2 抑制剂(her2i,例如曲妥珠单抗、拉帕替尼)、贝伐单抗和/或利妥昔单抗的肿瘤。
73.er wt pdx模型和er突变型pdx模型对于单独使用氟维司群、单独使用依维莫司、和 /或氟维司群和依维莫司的组合(氟维司群
‑
依维莫司组合)进行的治疗可以具有不同的反应水平。然而,与单独使用rad1901或单独使用依维莫司进行的治疗相比,rad1901
‑
依维莫司组合表现出改善的肿瘤生长抑制和/或肿瘤消退,无论该pdx模型是否对氟维司群治疗和/或氟维司群
‑
依维莫司组合治疗有反应。换言之,rad1901
‑
依维莫司组合可以在氟维司群耐药性癌症中抑制肿瘤生长和/或产生肿瘤消退。
74.与单独使用氟维司群或利用氟维司群
‑
依维莫司组合进行的治疗相比,rad1901
‑
依维莫司组合治疗表现出改善的肿瘤生长抑制和/或肿瘤消退。例如,与单独使用氟维司群、单独使用rad1901、或单独使用依维莫司进行的治疗相比,rad1901
‑
依维莫司组合在更
多的wt er 异种移植模型中产生更显著的肿瘤消退,即使这些异种移植模型对氟维司群治疗具有不同的反应性(例如,对氟维司群治疗具有反应的mcf7细胞系异种移植模型(图2);对于氟维司群治疗有反应的pdx
‑
11模型(图3);以及对氟维司群治疗反应最少的pdx
‑
2模型(图 4))也是如此。与利用氟维司群
‑
依维莫司组合进行的治疗相比,rad1901
‑
依维莫司组合还在更多的wt er mcf7细胞系异种移植模型和pdx
‑
11模型中产生更为显著的肿瘤消退(图 2和3)。rad1901
‑
依维莫司组合提供了与30mg/kg或60mg/kg剂量rad1901相似的效果,但是以30mg/kg单独使用rad1901在抑制肿瘤生长方面不如以60mg/kg单独使用rad1901 有效(图2c)。所述结果表明,具有较低剂量的rad1901(例如30mg/kg)的rad1901
‑
依维莫司组合足以使所述异种移模型中的肿瘤生长抑制/肿瘤消退效果最大化。
75.rad1901
‑
依维莫司组合在对氟维司群治疗几乎没有反应的突变型er (例如y537s)pdx 模型中表现出肿瘤消退或改善的肿瘤生长抑制(图6a)。例如,pdx
‑
5是在对氟维司群治疗几乎没有反应的er y537s突变型pdx模型(pr ,her2 ,具有ai既往治疗)。rad1901
‑ꢀ
依维莫司组合在pdx
‑
5模型中表现出肿瘤消退,而单独使用依维莫司或单独使用rad1901 仅抑制肿瘤生长而不引起肿瘤消退(图6b)。与单独使用rad1901、单独使用依维莫司、单独使用氟维司群、相比,rad1901
‑
依维莫司组合在突变型pdx
‑
5模型中产生更显著的肿瘤生长抑制(图6b)。因此,当与rad1901组合施用时,添加依维莫司使pdx
‑
5模型受益。因此, rad1901
‑
依维莫司组合为表达wt或突变型er、具有pr表达、具有高或低her2表达、以及对氟维司群具有或没有耐药性的er 乳腺癌提供了强力的抗肿瘤疗法。
[0076][0077]
本文提供的结果还显示,rad1901可以被递送至大脑(实施例ii),并且所述递送改善了表达野生型erα的颅内肿瘤模型(mcf
‑
7异种移植模型,实施例i(b))中的小鼠存活。依
维莫司已被批准用于治疗室管膜下巨细胞星形细胞瘤(sega),这是一种见于结节性硬化症 (ts)的脑肿瘤。因而,rad1901
‑
依维莫司组合的两种组分都可能能够穿过脑
‑
血屏障并治疗大脑中的er 肿瘤。这代表了与氟维司群
‑
依维莫司组合相比在治疗大脑内er 肿瘤方面的额外优势,原因是氟维司群不能穿过血
‑
脑屏障(vergote1等,“fulvestrant,a new treatment optionfor advanced breast cancer:tolerability versus existing agents,”ann.oncol.,17(2):200
‑
204 (2006))。能够穿过血
‑
脑屏障的rad1901与其它第二治疗(例如,mtor抑制剂,例如雷帕霉素类似物(geoerger等人,“antitumor activity of the rapamycin analog cci
‑
779in human primitiveneuroectodermal tumor/medu1loblastoma models as single agent and in combinationchemotherapy,”cancer res.61:1527
‑
1532(2001))的组合也可以对大脑中的er 肿瘤具有相似的治疗效果。
[0078]
在rad1901治疗结束后继续进行雌二醇治疗(例如pdx
‑
4模型)的情况下rad1901显示出持续的抑制肿瘤生长的功效。因此,rad1901
‑
依维莫司组合可能通过在治疗结束后抑制肿瘤生长而使患者受益,特别是当由于不良反应而可能中断(例如,对依维莫司而言29%) 或减少或延迟(对于依维莫司治疗的患者而言70%)第二治疗剂时如此。 http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm488028.htm.
[0079]
与单独使用依维莫司或使用依维莫司与其他激素疗法(例如,ai(例如来曲唑)和serd (利如氟维司群)的组合进行的治疗相比,rad1901
‑
依维莫司组合可能具有较低的和/或较不严重的副作用。例如,ai和氟维司群两者都可能导致治疗患者的骨质流失。rad1901不太可能有类似的副作用。已发现,rad1901优先积累在肿瘤中,其中肿瘤rad1901水平:血浆rad1901水平(t/p比)高达约35(实施例ii)。计算以约200mg至高达约500mg(每天一次)的剂量利用rad1901治疗的人受试者的子宫、肌肉和骨的标准摄取值(suv)(实施例iii(a))。给药后的子宫信号接近来自“非标靶组织”(不表达雌激素受体的组织)的水平,其表明rad1901治疗后的fes
‑
pet摄取完全减弱。在不显著表达雌激素受体(例如肌肉、骨)的组织中,在治疗前和治疗后的pet扫描中几乎没有观察到变化(实施例iii(a))。最后, rad1901治疗拮抗卵巢切除(ovx)大鼠中雌二醇对子宫组织的刺激(实施例iv(a)),并且大幅保持接受治疗的受试者的骨质量。例如,利用rad1901治疗的ovx大鼠显示出维持的bmd和股骨微构造(实施例iv(a))。因此,因此,rad1901
‑
依维莫司组合对于患有骨质疏松症或具有较高骨质疏松症风险的患者可能尤其有用。
[0080]
此外,已经报道基因表达分析对于鉴别对依维莫司治疗有反应的患者是有效的。yoon等人,“gene expression profiling identifies responsive patients with cancer of unknown primarytreated with carboplatin,paclitaxel,and everolimus:ncctg n0871(alliance),”ann.oncol., 27(2):339
‑
44(2016)。研究nct00805129发现依维莫司在tsc1中存在体细胞突变的患者中更有效,原因是所述突变导致复发增加并且对依维莫司的反应时间增加。因此,本文公开的方法可以进一步包括待治疗受试者进行基因分析,以识别具有更大反应和/或更长反应时间的受试者。
[0081]
此外,已经发现,rad1901在mcf
‑
7细胞系异种移植模型中降解野生型erα并消除体内er信号传导,并且在这些mcf
‑
7细胞系异种移植模型中显示出pr的剂量依赖性降低(实施例iii(b))。rad1901降低mcf
‑
7细胞系异种移植模型和pdx
‑
4模型中的增殖,如从接受治疗
的受试者采集的肿瘤中的增殖标志物ki67减少所证实的。在对氟维司群治疗几乎没有反应的突变型er pdx模型中,rad1901还降低了体内er信号传导(实施例iii(b))。
[0082]
rad1901
‑
依维莫司组合对氟维司群治疗几乎没有反应的肿瘤和表达突变型erα的肿瘤中的出乎意料的功效可能是由于rad1901和erα之间的独特相互作用引起的。对结合至 rad1901和其他erα
‑
结合化合物的erα的结构模型进行分析,以获得关于特异性结合相互作用的信息(实施例v)。计算机建模显示,rad1901
‑
erα相互作用不太可能受erα的lbd 突变体影响,所述lbd突变体例如为y537x突变体,其中x是s、n或c;d538g;和s463p,其占接受至少一线内分泌治疗的患者的转移性er阳性乳房肿瘤样品的最近研究中发现的 lbd突变的约81.7%(表9,实施例v)。因此,一种或多种第二治疗剂(例如依维莫司)和 rad1901或其盐或其溶剂化物(例如水合物)的组合可能具有与本文所公开的rad1901
‑
依维莫司类似的治疗效果,同时具有相对低的副作用。计算机建模使得能够识别出对于结合至关重要的erα c
‑
末端配体结合结构域中的特定残基,该信息可用于开发不仅结合和拮抗野生型erα而且还结合并拮抗其某些突变体和其变体的化合物,当与第二治疗剂(例如依维莫司) 组合时所述化合物可提供与本文公开的rad1901
‑
依维莫司组合类似的强抗肿瘤疗法,同时具有相对低的副作用。
[0083]
基于这些结果,本文提供了用于通过向受试者施用治疗有效量的rad1901或其溶剂化物 (例如水合物)或其盐与本文所述一种或多种第二治疗剂(例如依维莫司)的组合而在有需要的受试者中抑制erα阳性肿瘤生长或使其消退的方法。
[0084]
在某些实施方案中,除了抑制肿瘤生长(包括例如抑制癌细胞增殖或抑制erα活性(例如,通过抑制雌二醇结合或通过下调erα))之外,施用rad1901或其盐或其溶剂化物(例如水合物)还具有另外的治疗益处。在某些实施方案中,该方法对肌肉、骨、乳房和子宫不产生负面影响。
[0085]
在某些实施方案中,rad1901或其溶剂化物(例如水合物)或其盐调节和/或降解erα和突变型erα。
[0086]
在本文提供的肿瘤生长抑制或肿瘤消退方法的某些实施方案中,提供了通过向受试者施用治疗有效量的rad1901或其溶剂化物(例如水合物)或其盐与本文所述一种或多种第二治疗剂的组合而在有需要的受试者中抑制erα阳性肿瘤生长或使其消退的方法。在这些实施方案的某些实施方案中,它的盐是具有以下结构的rad1901二盐酸盐:
[0087][0088]
第二治疗剂
[0089]
用于本文提供方法中的第二种治疗剂可以是化疗剂或akt抑制剂、雄激素受体、血管生成抑制剂、芳香化酶、极光激酶a、bcl2、egfr、雌激素途径、雌激素信号途径、雌激素受体、her2、her3、热休克蛋白90(hsp90)、hedgehog(hh)信号传导途径、组蛋白脱乙酰酶(hdac)、kit途径、mtor(例如torc1和/或torc2)、微管、myc、核苷代谢、 parp、泛pi3k、pi3k、
蛋白激酶ck2、ras途径、类固醇硫酸酯酶(sts)、tk、top2a、酪氨酸激酶、vegf受体酪氨酸激酶或其任意组合。第二治疗剂也可以是抗体,例如抗tgf β抗体、抗1型胰岛素样生长因子受体抗体、抗trop
‑
2抗原抗体、抗her3抗体、抗pd1 抗体或其药物缀合物。
[0090]
第二治疗剂的进一步实例包括但不限于乙酸阿比特龙、adi
‑
peg 20、ado
‑
trastuzumabemtansine、阿法替尼、alisertib、阿那曲唑、紫杉醇和紫杉醇衍生物(例如ang1005、紫杉醇聚合物胶束)、arn
‑
810、阿扎胞苷、azd2014、azd5363、贝伐单抗、bp
‑
c1、buparlisib (bkm120)、byl719、卡培他滨、卡铂、马来酸西地替尼、西妥昔单抗、顺铂/ac4
‑
cddp4、 cr1447、cx
‑
4945、达沙替尼、denosumab、多西他赛、多柔比星、恩尿嘧啶、entinostat、恩杂鲁胺、表柔比星、艾日布林、依西美坦、依维莫司、氟尿嘧啶、氟维司群、fresolimumab、 ganetespib、ganitumab、gdc
‑
0032、gdc
‑
0941、吉西他滨、glembatumumab vedotin、gnrh 激动剂(例如醋酸戈舍瑞林)、grn1005、gsk 2141795、伊班膦酸盐、immu
‑
132、依立替康、irosustat、埃博霉素(例如伊沙匹隆)、拉帕替尼、sonidegib(lde225)、来曲唑、lgk974、 ljm716、lucitanib、甲氨蝶呤、mk
‑
2206、mk
‑
3475、mln0128、mm
‑
302、来那替尼、niraparib、奥拉帕尼、抗雄激素药(例如orteronel)、奥沙利铂、帕唑帕尼、帕妥珠单抗、pf
‑
05280014、 pm01183、孕酮、pyrotinib、罗米地辛、鲁索替尼、索拉非尼、舒尼替尼、talazoparib、他莫昔芬、紫杉烷、t
‑
dm1、telapristone(cdb
‑
4124)、替莫唑胺、替西罗莫司、四硫钼酸盐、tesetaxel、 tlr7激动剂、tpi 287、曲美替尼、曲妥珠单抗、trc105、trebananib(amg 386)、曲普瑞林、veliparib、长春氟宁、长春瑞滨、vorinostat、诺雷得和唑来膦酸,包括其溶剂化物(例如水合物)和其盐。
[0091]
在某些实施方案中,第二治疗剂选自ado
‑
trastuzumab emtansine、极光激酶a抑制剂(例如alisertib)、ai(例如阿那曲唑、依西美坦、来曲唑)、arn
‑
810,mtor抑制剂(例如,依维莫司、azd2014、bez235、gdc
‑
0980、cc
‑
223、mln0128)、akt抑制剂(例如azd5363、 gdc
‑
0068、gsk2110183、gsk2141795、gsk690693、mk2206)、pi3k抑制剂(例如bkm120、 byl719、gdc
‑
0032、gdc
‑
0941)、选择性组蛋白脱乙酰酶(hdac)抑制剂(例如entinostat)、 gnrh激动剂(例如醋酸戈舍瑞林)、grn1005及其与曲妥珠单抗、拉帕替尼、酪氨酸激酶抑制剂(例如,lucitanib、来那替尼)、抗雄激素药(例如,orteronel)、帕妥珠单抗、替莫唑胺和抗体(例如,keytruda和bym338)。
[0092]
在某些实施方案中,第二治疗药剂可以为ai(例如阿那曲唑、aromasin和来曲唑)、另一种serm(例如阿佐昔芬、屈洛昔芬、em
‑
652(sch 57068)、吲哚昔酚、拉索昔芬、左美洛昔芬、米泼昔芬、雷洛昔芬、他莫昔芬和托瑞米芬)、或另一种serd(例如氟维司群、 gdc
‑
0810(arn
‑
810)、gw5638/dpc974、ici182782、ru58668、srn
‑
927、tas
‑
108(sr16234) 和zk191703),包括其溶剂化物(例如水合物)和其盐。
[0093]
第二治疗剂的其他实例包括但不限于:abraxane、amg 386、卡巴他赛、楷莱、卡培他滨、多西他赛、艾日布林、吉西他滨、赫赛汀、来那替尼、帕唑帕尼(gw786034)、rapalog (雷帕霉素及其类似物)、紫杉醇(包括类似物/替代制剂)、tdm1、temozolamide、泰立沙、 veliparib(abt
‑
888)和长春瑞滨,包括其溶剂化物(例如水合物)和其盐。
[0094]
第二治疗剂靶向pi3k/akt/mtor途径
[0095]
在一些实施方案中,第二治疗剂靶向pi3k/akt/mtor途径,并且可以是mtor抑制剂、 mtor双重抑制剂、pi3k/mtor抑制剂。在一些实施方案中,第二治疗剂是雷帕霉素衍生
物 (aka rapalog),例如雷帕霉素(西罗莫司或雷帕鸣,pfizer)、依维莫司(afinitor或rad001, novartis)、ridaforolimus(ap23573或mk
‑
8669,merck和ariad pharmaceuticals)、替西罗莫司(torisel或cci779,pfizer),包括其溶剂化物(例如水合物)和其盐。在一些实施方案中,第二治疗剂是抑制mtorc1和mtorc2两者的mtor双重抑制剂,例如mln0128(去势抵抗性前列腺癌(crpc),斯隆
‑
凯特林纪念癌症中心)、cc115和cc223(celgene)、osi
‑
027 (osi pharmaceuticals)、以及azd8055和azd2014(astrazeneca),包括其溶剂化物(例如水合物)和其盐。在一些实施方案中,第二治疗剂是pi3k/mtor抑制剂,例如gdc
‑
0980、 sar245409(xl765)、ly3023414(eli lilly)、nvp
‑
bez235(novartis)、nvp
‑
bgt226(novartis)、 sf1126、和pki
‑
587(pfizer),包括其溶剂化物(例如水合物)和其盐。
[0096]
在某些实施方案中,上文公开的多于一种的第二治疗剂可以与rad1901或其溶剂化物 (例如水合物)或其盐一起使用。例如,mtor抑制剂可以与另一种mtor抑制剂或与 pi3k/mtor抑制剂一起使用。而且,本领域已知,可以将包括mtor抑制剂、mtor双重抑制剂和pi3k/mtor抑制剂在内的上述第二治疗剂与其它活性剂一起施用,以增强治疗效果。例如,可以将mtor抑制剂与jak2抑制剂(bogani等,plos one,8(1):e54826(2013))、化学治疗剂(yardley,breast cancer(auckl)7:7
‑
22(2013))、或内分泌疗法(例如他莫昔芬或依西美坦(vinayak等,“mtor inhibitors in the treatment of breast cancer,”oncology,published january 15,2013(http://www.cancernetwork.com/breast
‑
cancer/mtor
‑
inhibitors
‑
treatment
‑ꢀ
breast
‑
cancer))组合使用。相应地,第二治疗剂也包括这些辅助活性剂。
[0097]
组合疗法
[0098]
rad1901或其溶剂化物(例如水合物)或其盐与一种或多种第二治疗剂的组合
[0099]
当单独施用于受试者时,rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)对一种或多种癌症或肿瘤具有治疗效果(实施例i(a)和i(b))。出乎意料地,已经发现,当组合施用于受试者时,rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂 (例如依维莫司)对癌症/肿瘤具有显著的改善效果(实施例i(a)和i(b))。
[0100]
如本文所用的erα阳性肿瘤的“抑制生长”可以指减慢肿瘤生长的速度、或完全停止肿瘤生长。
[0101]
如本文所用的,erα阳性肿瘤“肿瘤消退”或“消退”可指减小肿瘤的最大尺寸。在某些实施方案中,施用本文所述一种或多种第二治疗剂(例如依维莫司)(例如,ribociclib、 abemaciclib和依维莫司)与rad1901或其溶剂化物(例如水合物)或其盐的组合可导致肿瘤尺寸相对于基准(即开始治疗之前的尺寸)减小或甚至根除或部分根除肿瘤。相应地,在某些实施方案中,本文提供的肿瘤消退方法可以替代地表征为相对于基准减小肿瘤尺寸的方法。
[0102]
如本文所用的,“肿瘤”是恶性肿瘤,并且可与“癌症”互换使用。
[0103]
肿瘤生长抑制或消退可以定位于具体组织或器官内的单个肿瘤或一组肿瘤,或者可以是全身性的(即,影响所有组织或器官中的肿瘤)。
[0104]
由于已知rad1901优先结合erα而不是雌激素受体β(erβ),所以除非另有说明,雌激素受体、雌激素受体α、erα、er、野生型erα和esr1在本文中可互换使用。如本文所用的“雌激素受体α”或“erα”是指包含由基因esr1编码的野生型erα氨基酸序列的多肽、由通过基因
esr1编码的野生型erα氨基酸序列组成的多肽或基本上由通过基因esr1编码的野生型erα氨基酸序列组成的多肽。如本文所用的“雌激素受体α阳性”、“erα阳性”、“er ”或“erα ”的肿瘤是指其中一种或多种细胞表达至少一种erα同种型的肿瘤。在某些实施方案中,这些细胞过度表达erα。在某些实施方案中,患者具有在肿瘤内的表达一种或多种形式雌激素受体β的一种或多种细胞。在某些实施方案中,erα阳性肿瘤和/或癌症与乳腺癌、子宫癌、卵巢癌或垂体癌相关。在这些实施方案中的某些实施方案中,患者具有位于乳房、子宫、卵巢或垂体组织中的肿瘤。在患者具有位于乳房中的肿瘤的那些实施方案中,肿瘤可能与对her2可能呈阳性或可能不呈阳性的管腔乳腺癌相关,并且对于her2 肿瘤,肿瘤可能表达高her2或低her2(例如图1)。在其他实施方案中,患者具有位于另一组织或器官 (例如,骨、肌肉、大脑)中的肿瘤,但仍然与乳腺癌、子宫癌、卵巢癌或垂体癌相关(例如源自乳腺癌、子宫癌、卵巢癌或垂体癌的迁移或转移的肿瘤)。相应地,在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,被靶向肿瘤是转移性肿瘤和/或肿瘤在其他器官(例如骨和/或肌肉)中具有er过度表达。在某些实施方案中,被靶向肿瘤是大脑肿瘤和/或癌症。在某些实施方案中,被靶向肿瘤对rad1901和本文公开的第二治疗剂的治疗比用以下进行的治疗更为敏感:另一种serd(例如氟维司群、tas
‑
108(sr16234)、zk191703、ru58668、 gdc
‑
0810(arn
‑
810)、gw5638/dpc974、srn
‑
927、ici182782和azd9496)、her2抑制剂 (例如曲妥珠单抗、拉帕替尼、曲妥珠单抗艾坦素和/或培妥珠单抗)、化疗(例如abraxane、阿霉素、卡铂、环磷酰胺、柔红霉素、doxil、ellence、氟尿嘧啶、健择、helaven、伊沙匹隆、甲氨蝶呤、丝裂霉素、micoxantrone、去甲长春碱、紫杉酚、泰索帝、塞替派、长春新碱和希罗达)、芳香酶抑制剂(例如阿那曲唑、依西美坦和来曲唑)、选择性雌激素受体调节剂(例如他莫昔芬、雷洛昔芬、拉索昔芬、和/或托瑞米芬)、血管生成抑制剂(例如贝伐单抗)、和 /或利妥昔单抗。
[0105]
在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,所述方法还包括在施用rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)的组合之前确定患者是否具有表达erα的肿瘤的步骤。在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,所述方法还包括在施用rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)的组合之前确定患者是否具有表达突变型erα的肿瘤的步骤。在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,所述方法还包括在施用rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司) 的组合之前确定患者是否具有对氟维司群治疗具有反应或没有反应的、表达erα的肿瘤的步骤。这些确定可以利用本领域已知的任何表达检测方法来进行,并且可以利用从受试者中取出的肿瘤或组织样品在体外进行。
[0106]
除了证明rad1901在表达野生型erα的肿瘤中抑制肿瘤生长的能力之外,本文提供的结果还显示rad1901表现出出乎意料的、抑制表达突变形式erα(即y537serα)的肿瘤生长的能力(实施例i(a))。erα突变实施例的计算机模拟评估显示,预计这些突变中没有一个会影响lbd,也不会特异性阻碍rad1901结合(实施例v(a)),例如选自以下的具有一个或多个突变体的erα:具有y537x突变体的erα,其中x是s、n或c;具有d538g突变体的erα;和具有s463p突变体的erα。基于这些结果,本文提供了通过对受试者施用治疗有效量的一种或多种第二治疗剂(例如依维莫司)和rad1901或其溶剂化物(例如水合物) 或其盐的组合来抑制患癌受试者中的肿瘤生长或导致其消退的方法,所述肿瘤对在配体结合结构域(lbd)内
具有一个或多个突变体的erα呈阳性,所述突变体选自y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或q;p535h;v534e;s463p;v392i;e380q;特别是y537serα。本文所用的“突变型erα”是指包含一个或多个替换或缺失的erα和其变体,其包含与erα的氨基酸序列具有至少80%、至少85%、至少90%、至少95%、至少 97%、至少98%、至少99%、或至少99.5%同一性的氨基酸序列,或由所述氨基酸序列构成,或基本上由所述氨基酸序列构成。
[0107]
除了在动物异种移植模型中抑制乳腺癌肿瘤生长之外,本文公开的结果还显示rad1901 在肿瘤细胞内表现出明显的积累,并且能够穿透血脑屏障(实施例ii)。通过显示rad1901 施用明显延长大脑转移异种移植模型中的存活证实了穿透血脑屏障的能力(实施例i(b))。相应地,在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,被靶向erα阳性肿瘤位于大脑中或中枢神经系统的其他地方。在这些实施方案中的某些实施方案中,erα阳性肿瘤主要与脑癌相关。在其他实施方案中,erα阳性肿瘤是主要与另一类型的癌症(例如乳腺癌、子宫癌、卵巢癌或垂体癌)或已从另一组织或器官转移的肿瘤相关的转移性肿瘤。在这些实施方案中的某些实施方案中,肿瘤是脑转移,例如乳腺癌脑转移(bcbm)。在本文公开方法的某些实施方案中,rad1901或其盐或其溶剂化物累积在标靶肿瘤内的一个或多个细胞中。
[0108]
在本文公开方法的某些实施方案中,rad1901或其溶剂化物(例如水合物)或其盐优选以约15或更高、约18或更高、约19或更高、约20或更高、约25或更高、约28或更高、约30或更高、约33或更高、约35或更高、或约40或更高的t/p(肿瘤中的rad1901浓度 /血浆中的rad1901浓度)比累积在肿瘤中。
[0109]
本文提供的结果显示,rad1901施用防止了卵巢切除大鼠中的骨质流失(实施例iv(a))。相应地,在本文提供的肿瘤生长抑制或消退方法的某些实施方案中,施用一种或多种第二治疗剂(例如依维莫司)和rad1901或其溶剂化物(例如水合物)或其盐的组合对骨没有不良作用,包括例如对经治疗受试者的骨体积密度、骨表面密度、骨矿物质密度、骨小梁数目、骨小梁厚度、骨小梁间距、连接密度和/或表观骨密度的不良影响。他莫昔芬可能与绝经前妇女的骨质流失有关,氟维司群可能由于其作用机制而损害骨结构。一种或多种第二治疗剂(例如依维莫司)和rad1901或其溶剂化物(例如水合物)或其盐的组合可特别用于绝经前女性、对他莫昔芬或抗雌激素疗法具有耐药性的肿瘤、和具有骨质疏松症和/或具有高骨质疏松症风险的患者。
[0110]
本文提供的结果显示,rad1901拮抗卵巢切除大鼠中雌二醇对子宫组织的刺激(实施例 iv(a))。此外,以200mg或高达500mg(每日一次)的剂量利用rad1901治疗的人受试者中,未显著表达er的子宫、肌肉和骨组织的标准摄取值(suv)显示在治疗前和治疗后几乎没有任何信号改变(实施例iii(a))。相应地,在某些实施方案中,这样的施用也不会对其他组织(包括例如子宫、肌肉或乳房组织)造成不良影响。
[0111]
将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)组合施用至有需要的受试者。短语“组合”是指在施用第二治疗剂(例如依维莫司)之前、期间或之后施用rad1901或其溶剂化物(例如水合物)或其盐。例如,可以按以下时间施用rad1901 或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司):间隔约一周、间隔约6 天、间隔约5天、间隔约4天、间隔约3天、间隔约2天、间隔约24小时、间隔约23小时、间隔约22小时、间隔约21小时、间隔约20小时、间隔约19小时、间隔约18小时、间约 17小时、间隔约16
小时、间隔约15小时、间隔约14小时、间隔约13小时、间隔约12小时、间隔约11小时、间隔约10小时、间隔约9小时、间隔约8小时、间约7小时、间隔约 6小时、间隔约5小时、间隔约4小时、间隔约3小时、间隔约2小时、间隔约1小时、间隔约55分钟、间隔约50分钟、间隔约45分钟、间隔约40分钟、间隔约35分钟、间隔约 30分钟、间隔约25分钟、间隔约20分钟、间隔约15分钟、间隔约10分钟或间隔约5分钟。在其它实施方案中,将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)同时或基本上同时施用至受试者。在这些实施方案的某些实施方案中,可以作为单一制剂的一部分来施用rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)。
[0112]
在一些实施方案中,将rad1901或其溶剂化物(例如水合物)或其盐和单一第二治疗剂 (例如依维莫司)的组合施用至受试者。在其他实施方案中,将rad1901或其溶剂化物(例如水合物)或其盐和多于一种的第二治疗剂(例如依维莫司)的组合施用至受试者。例如,可以将rad1901或其溶剂化物(例如水合物)或其盐与两种或多种第二治疗剂(例如依维莫司)组合用于治疗癌症/肿瘤。
[0113]
(2)剂量
[0114]
用于本文公开方法的rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)的组合的治疗有效量为如下量:当在特定时间间隔内施用时,所述量导致实现一个或多个治疗基准(例如,减缓或停止肿瘤生长、导致肿瘤消退、症状停止等)。可以将用于本文公开的方法的组合按一次或多次施用至对象。在其中按多次施用化合物的那些实施方案中,可以以设定的间隔(例如每天、每隔一天、每周或每月)来施用化合物。或者,可以以不规则的间隔来施用化合物,例如基于症状、患者健康等根据需要施用。可以按每天一次施用治疗有效量的组合1天、至少2天、至少3天、至少4天、至少5天、至少6 天、至少7天、至少10天、或至少15天。任选地,在治疗期间或之后,监测癌症的状态或肿瘤的消退,例如通过受试者的fes
‑
pet扫描来监测。可以根据所检测的癌症状态或肿瘤消退来增加或减少施用至受试者的组合的剂量。
[0115]
理想地,治疗有效量不超过最大耐受剂量,即50%或更多的接受治疗的受试者经历恶心或其它毒性反应(其阻止进一步的药物施用)的剂量。受试者的治疗有效量可以根据多种因素变化,所述多种因素包括受试者的症状种类和程度、性别、年龄、体重或一般健康、给药方式和盐或溶剂化物类型、对药物的敏感性变化、具体的疾病类型等。
[0116]
用于本文公开方法中的治疗有效量的rad1901或其溶剂化物(例如水合物)或其盐的实例包括但不限于:对于具有耐药性er引起的肿瘤或癌症的受试者而言为约150至约1500mg、约200至约1500mg、约250至约1500mg或约300至约1500mg日剂量;对于同时具有野生型er引起的肿瘤和/或癌症和耐药性肿瘤和/或癌症的受试者而言为约150至约1500mg、约200至约1000mg或约250至约1000mg或约300至约1000mg日剂量;以及对于具有主要野生型er引起的肿瘤和/或癌症的受试者而言为约300至约500mg、约300至约550mg、约300至约600mg、约250至约500mg、约250至约550mg、约250至约600mg、约200 至约500mg、约200至约550mg、约200至约600mg、约150至约500mg、约150至约550 mg或约150至约600mg日剂量。
[0117]
第二治疗剂(例如依维莫司)的治疗有效量或剂量取决于其具体类型。通常,第二治疗剂(例如依维莫司)的日剂量的范围为约1mg至约1500mg、约1mg至约1200mg、约1mg 至约1000mg、约1mg至约800mg、约1mg至约600mg、约1mg至约500mg、约1mg 至约200mg、约1mg至
约100mg、约1mg至约50mg、约1mg至约30mg、约1mg至约 20mg、约1mg至约10mg、约1mg至约5mg、约50mg至约1500mg、约100mg至约1200 mg、约150mg至约1000mg、约200mg至约800mg、约300mg至约600mg、约350mg 至约500mg。第二治疗剂(例如依维莫司)的日剂量可以为约1至约100mg/kg、约1至约 75mg/kg、约l至约50mg/kg、约1至约45mg/kg、约1至约40mg/kg、约l至约30mg/kg、约1至约20mg/kg、约1至约10mg/kg、约2至约100mg/kg、约2至约75mg/kg、约2至约50mg/kg、约2至约45mg/kg、约2至约40mg/kg、约2至约30mg/kg、约2至约20mg/kg、约2至约10mg/kg、约2.5至约100mg/kg、约2.5至约75mg/kg、约2.5至约50mg/kg、约 2.5至约45mg/kg、约2.5至约40mg/kg、约2.5至约30mg/kg、约2.5至约20mg/kg、或约 2.5至约10mg/kg。
[0118]
在某些实施方案中,治疗有效量的组合可以利用治疗有效量的单独施用的任一种化合物。在其他实施方案中,由于组合实现了显著改善的协同治疗效果,所以在组合中施用的 rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)的治疗有效量可以小于单独施用时所需的rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)的治疗有效量;并且可以以低于分开给药时通常施用的剂量低的剂量施用一种或两种化合物。不受任何特定理论限制,通过降低rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)中的至少一种或全部的剂量,组合疗法显著改善了效果,从而消除或减轻不期望的毒性副作用。
[0119]
在一些实施方案中,当作为组合的一部分施用时,rad1901或其溶剂化物(例如水合物) 或其盐的治疗有效量为单独施用时rad1901或其溶剂化物(例如水合物)或其盐的治疗有效量的约30%至约200%、约40%至约200%、约50%至约200%、约60%至约200%、约70%至约200%、约80%至约200%、约90%至约200%、约100%至约200%、30%至约150%、约 40%至约150%、约50%至约150%、约60%至约150%、约70%至约150%、约80%至约150%、约90%至约150%、约100%至约150%、约30%至约120%、约40%至约120%、约50%至约 120%、约60%至约120%、约70%至约120%、约80约120%、约90%至约120%、约100%至约120%、30%至约110%、约40%至约110%、约50%至约110%、约60%至约110%、约 70%至约110%、约80%至约110%、约90%至约110%、或约100%至约110%。在一些实施方案中,当作为组合的一部分施用时,第二治疗剂(例如依维莫司)的治疗有效量为单独施用时第二治疗剂(例如依维莫司)的治疗有效量的约30%至约200%、约40%至约200%、约 50%至约200%、约60%至约200%、约70%至约200%、约80%至约200%、约90%至约200%、约100%至约200%、约30%至约150%、约40%至约150%、约50%至约150%、约60%至约 150%、约70%至约150%、约80%至约150%、约90%至约150%、约100%至约150%、约 30%至约120%、约40%至约120%、约50%至约120%、约60%至约120%、约70%至约120%、约80%至约120%、约90%至约120%、约100%至约120%、30%至约110%、约40%至约110%、约50%至约110%、约60%至约110%、约70%至约110%、约80%至约110%、约90%至约 110%、或约100%至约110%。
[0120]
在某些实施方案中,癌症或肿瘤是耐药性er
‑
引起的癌症或肿瘤(例如具有突变型er 结合结构域(例如包含一个或多个突变的erα,所述一个或多个突变包括但不限于y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或q;p535h;v534e;s463p;v392i; e380q及其组合),er或肿瘤和/或癌症增殖的过表达体变成配体非依赖性的,或者在利用以下治疗的情况下出现进展的肿瘤和/癌症:serd(例如氟维司群,tas
‑
108(sr16234)、 zk191703、
ru58668、gdc
‑
0810(arn
‑
810)、gw5638/dpc974、srn
‑
927、ici182782和 azd9496)、her2抑制剂(例如曲妥珠单抗、拉帕替尼、曲妥珠单抗艾坦素、和/或培妥珠单抗)、化疗(例如abraxane、阿霉素、卡铂、环磷酰胺、柔红霉素、doxil、ellence、氟尿嘧啶、健择、helaven、伊沙匹隆、甲氨蝶呤、丝裂霉素、micoxantrone、去甲长春碱、紫杉酚、泰索帝、塞替派、长春新碱和希罗达)、芳香酶抑制剂(例如阿那曲唑、依西美坦和来曲唑)、选择性雌激素受体调节剂(例如他莫昔芬、雷洛昔芬、拉索昔芬、和/或托瑞米芬)、血管生成抑制剂(例如贝伐单抗)、和/或利妥昔单抗。
[0121]
在某些实施方案中,通常用于成人受试者的本公开方法的、与本文所述第二治疗剂(例如依维莫司)(例如,ribociclib、abemaciclib和依维莫司)组合的rad1901或其溶剂化物(例如水合物)或其盐的剂量可以为约30μg至2000mg、100μg至1500mg、或150mg至1500 mg(口服,每日一次)。可以通过单次施用或多次施用来实现该日剂量。
[0122]
可以将一种或多种第二治疗剂(例如依维莫司)和rad1901或其溶剂化物(例如水合物) 或其盐的组合按一次或多次施用于受试者。在其中按多次施用化合物的那些实施方案中,可以以设定的间隔(例如每天、每隔一天、每周或每月)施用所述化合物。或者,可以以不规则的间隔施用化合物,例如基于症状、患者健康等根据需要施用。
[0123]
(3)制剂
[0124]
在一些实施方案中,以分开的制剂施用rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)。在这些实施方案中的某些实施方案中,制剂可以是相同类型的制剂。例如,可以将两种制剂都设计用于口服施用(例如,通过两种分开的丸剂)或用于注射(例如,通过两种分开的可注射制剂)。在其他实施方案中,可以将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)配制成不同类型的制剂。例如,一种化合物可以在设计用于口服给药的制剂中,而另一种化合物在设计用于注射的制剂中。
[0125]
在其他实施方案中,作为单一制剂的一部分施用rad1901或其溶剂化物(例如水合物) 或其盐和第二治疗剂(例如依维莫司)。例如,将rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)配制成用于口服施用的单一丸剂或单一注射剂。在某些实施方案中,本文提供了包含rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)的组合制剂。在某些实施方案中,以单一制剂施用化合物改善了患者的依从性。
[0126]
当以组合形式施用时,每种化合物的治疗有效量可以低于单独施用每种化合物的治疗有效量。
[0127]
在一些实施方案中,包含rad1901或其溶剂化物(例如水合物)或其盐、一种或多种第二治疗剂(例如依维莫司)、或rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)两者的制剂可以进一步包含一种或多种药物赋形剂、载体、佐剂和/或防腐剂。
[0128]
可以将用于本公开方法中的rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂 (例如依维莫司)配制成单位剂型,即适合作为经受治疗的受试者的单位剂量的物理分离单位,其中每个单位包含经计算可产生期望治疗效果的预定量活性物质,其任选地与合适的药物载体结合。单位剂型可以用于单个日剂、或多个日剂中的一剂(例如,每天约1至4次或更多次)。当使用多个日剂时,对于每一剂,单位剂型可以相同或不同。在某些实施方
案中,可以将化合物配制成受控释放。
[0129]
可以根据任何可用的常规方法配制用于本公开方法的rad1901或其溶剂化物(例如水合物)或其盐和第二治疗剂(例如依维莫司)。优选剂型的实例包括片剂、粉剂、微粒剂、颗粒剂、包衣片剂、胶囊剂、糖浆剂、锭剂、吸入剂、栓剂、注射剂、软膏剂、眼用软膏剂、眼滴剂、滴鼻剂、滴耳剂、巴布膏剂、洗剂等。在制剂中,可以施用通常使用的添加剂,例如稀释剂、粘合剂、崩解剂、润滑剂、着色剂、调味剂,以及必要情况下的稳定剂、乳化剂、吸收促进剂、表面活性剂、ph调节剂、防腐剂、抗氧化剂等。另外,根据常规方法,还通过组合通常用作药物制剂原料的组合物来进行制剂。这些组合物的实例包括例如:(1)油,例如大豆油、牛油和合成甘油酯;(2)烃,例如液体石蜡、角鲨烷和固体石蜡;(3)酯油,例如辛基十二烷基肉豆蔻酸和异丙基肉豆蔻酸;(4)高级醇,例如鲸蜡硬脂醇和山嵛醇;(5) 硅树脂;(6)硅油;(7)表面活性剂,例如聚氧乙烯脂肪酸酯、山梨糖醇酐脂肪酸酯、甘油脂肪酸酯、聚氧乙烯山梨糖醇酐脂肪酸酯、固体聚氧乙烯蓖麻油和聚氧乙烯聚氧丙烯嵌段共聚物;(8)水溶性高分子,例如羟乙基纤维素、聚丙烯酸、羧基乙烯基聚合物、聚乙二醇、聚乙烯吡咯烷酮和甲基纤维素;(9)低级醇,例如乙醇和异丙醇;(10)多元醇,例如甘油、丙二醇、二丙二醇和山梨糖醇;(11)糖,例如葡萄糖和蔗糖;(12)无机粉末,例如无水硅酸、硅酸铝镁和硅酸铝;(13)纯化水等。用于上述制剂的添加剂可包括例如:1)作为稀释剂的乳糖、玉米淀粉、蔗糖、葡萄糖、甘露糖醇、山梨糖醇、结晶纤维素和二氧化硅;2)作为粘合剂的聚乙烯基醇、聚乙烯基醚、甲基纤维素、乙基纤维素、阿拉伯胶、黄蓍胶、明胶、虫胶、羟丙基纤维素、羟丙基甲基纤维素、聚乙烯吡咯烷酮、聚丙二醇
‑
聚氧乙烯嵌段共聚物、葡甲胺、柠檬酸钙、糊精、果胶等;3)作为崩解剂的淀粉、琼脂、明胶粉、结晶纤维素、碳酸钙、碳酸氢钠、柠檬酸钙、糊精、果胶、羧甲基纤维素/钙等;4)作为润滑剂的硬脂酸镁、滑石、聚乙二醇、二氧化硅、浓缩植物油等;5)可接受其添加在药学上可接受的任何着色剂作为着色剂;6)作为调味剂的可可粉、薄荷醇、芳香剂、薄荷油、肉桂粉;7)其添加在药学上可接受的抗氧化剂,例如抗坏血酸或α
‑
苯酚。
[0130]
可以将用于本公开方法的rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依维莫司)作为本文所述的任一种或多种活性化合物和生理上可接受的载体 (也称为药学上可接受的载体或溶液或稀释剂)配制成药物组合物。这样的载体和溶液包括在本发明方法中使用的化合物的药学上可接受的盐和溶剂化物,以及包含两种或更多种这样的化合物、所述化合物的药学上可接受的盐和所述化合物的药学上可接受的溶剂化物的混合物。根据可接受的药学方法制备这样的组合物,例如在remington’s pharmaceutical sciences(第 18版.alfonso r.gennaro,mack publishing company,eaton,pa.(1990))中描述的,其通过引用并入本文。
[0131]
术语“药学上可接受的载体”是指在其施用的患者中不引起过敏反应或其它不良反应并且与制剂中的其他成分相容的载体。药学上可接受的载体包括例如根据预期给药形式合适地选择并符合常规药学实践的药学稀释剂、赋形剂或载体。例如,固体载体/稀释剂包括但不限于树胶、淀粉(例如玉米淀粉、预胶化淀粉)、糖(例如乳糖、甘露糖醇、蔗糖、右旋糖)、纤维素材料(例如,微晶纤维素)、丙烯酸酯(例如聚丙烯酸甲酯)、碳酸钙、氧化镁、滑石或其混合物。药学上可接受的载体可进一步包含少量的辅助物质,例如润湿剂或乳化剂、防腐剂或缓冲剂,其提高治疗剂的保质期或有效性。
[0132]
可以通过常规方法将游离形式的一种或多种第二治疗剂(例如依维莫司)和rad1901 或其溶剂化物(例如水合物)或其盐转化成盐。本文使用的术语“盐”不受限制,只要该盐是利用rad1901或其溶剂化物(例如水合物)或其盐形成并且在药学上可接受即可;盐的优选实例包括氢卤酸盐(例如盐酸盐、氢溴酸盐、氢碘酸盐等)、无机酸盐(例如硫酸盐、硝酸盐、高氯酸盐、磷酸盐、碳酸盐、碳酸氢盐等)、有机羧酸盐(例如乙酸盐、马来酸盐、酒石酸盐、富马酸盐、柠檬酸盐等)、有机磺酸盐(例如甲磺酸盐、乙磺酸盐、苯磺酸盐、甲苯磺酸盐、樟脑磺酸盐等)、氨基酸盐(例如天冬氨酸盐、谷氨酸盐等)、季铵盐、碱金属盐(例如钠盐、钾盐等)、碱土金属盐(镁盐、钙盐等)等。另外,优选盐酸盐、硫酸盐、甲磺酸盐、乙酸盐等作为根据本发明的化合物的“药学上可接受的盐”。
[0133]
可以使用通用分离手段将rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂(例如依维莫司)的异构体(例如几何异构体、旋光异构体、旋转异构体、互变异构体等) 纯化成单一异构体,所述分离手段包括例如重结晶、旋光拆分如非对映异构盐法、酶分级分离法、各种色谱法(例如,薄层色谱法、柱色谱法、玻璃色谱法等)。本文的术语“单一异构体”不仅包括纯度为100%的异构体,而且还包括包含除目标之外的、甚至通过常规纯化操作也仍然存在的异构体。对于rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂 (例如依维莫司),有时存在晶体多晶型物,并且其所有的晶体多晶型物都包括在本发明中。晶体多晶型物有时是单一的,有时是混合物,两者均包括在本文中。
[0134]
在某些实施方案中,rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂(例如依维莫司)可以为前药形式,这意味着其必须经历一些改变(例如氧化或水解)来实现其活性形式。或者,rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂(例如依维莫司)可以是通过将亲本前药改变为其活性形式而产生的化合物。
[0135]
(4)施用途径
[0136]
本文公开的rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂(例如依维莫司)的施用途径包括但不限于局部施用、口服施用、皮内施用、肌内施用、腹膜内施用、静脉内施用、膀胱内灌注、皮下施用、透皮施用和透粘膜施用。
[0137]
(5)基因分析
[0138]
在某些实施方案中,本文提供的肿瘤生长抑制方法还包括对受试者进行基因分析,其中待分析的基因是选自以下的一种或多种基因:abl1、akt1、akt2、alk、apc、ar、arid1a、 asxl1、atm、aurka、bap、bap1、bcl2l11、bcr、braf、brca1、brca2、ccnd1、 ccnd2、ccnd3、ccne1、cdh1、cdk4、cdk6、cdk8、cdkn1a、cdkn1b、cdkn2a、 cdkn2b、cebpa、ctnnb1、ddr2、dnmt3a、e2f3、egfr、eml4、ephb2、erbb2、 erbb3、esr1、ewsr1、fbxw7、fgf4、fgfr1、fgfr2、fgfr3、flt3、frs2、hif1a、 hras、idh1、idh2、igf1r、jak2、kdm6a、kdr、kif5b、kit、kras、lrp1b、map2k1、 map2k4、mcl1、mdm2、mdm4、met、mgmt、mll、mpl、msh6、mtor、myc、 nf1、nf2、nkx2
‑
1、notch1、npm、nras、pdgfra、pik3ca、pik3r1、pml、pten、 ptprd、rara、rb1、ret、rictor、ros1、rptor、runx1、smad4、smarca4、 sox2、stk11、tet2、tp53、tsc1、tsc2、和vhl。
[0139]
在一些实施方案中,第二剂是依维莫司,并且受试者存在tsc1中的体细胞突变。
[0140]
在一些实施方案中,本发明提供了如下方法:治疗乳腺癌患者亚群,其中所述亚群具有增加的一个或多个以下基因的表达;并且根据本公开内容中描述的给药实施方案利用有效剂量的rad1901或其溶剂化物(例如水合物)或其盐和一种或多种第二治疗剂(例如依
维莫司) 的组合治疗所述亚群。
[0141]
(6)剂量调整
[0142]
除了确定rad1901抑制肿瘤生长的能力之外,本文提供的结果显示,rad1901抑制雌二醇与子宫和垂体中的er结合(实施例iii(a))。在这些实验中,通过fes
‑
pet成像来评估雌二醇与子宫和垂体组织中的er结合。在利用rad1901治疗后,所观察到的er结合水平处于背景水平或低于背景水平。这些结果证实,可以通过实时扫描来评估rad1901对er活性的拮抗作用。基于这些结果,本文提供了用于通过测量一种或多种标靶组织中的雌二醇
‑
er 结合来监测本文公开的组合疗法中的rad1901或其溶剂化物或其盐治疗功效的方法,其中结合减少或消失表明产生了功效。
[0143]
还提供了基于雌二醇
‑
er结合来调整rad1901或其溶剂化物或其盐在组合中的剂量的方法。在这些方法的某些实施方案中,在一次或多次施用第一剂量的化合物之后的某个时间点时对结合进行测量。如果雌二醇
‑
er结合不受影响或表现出低于预定阈值的降低(例如,相比基线水平,结合降低小于5%、小于10%、小于20%、小于30%或小于50%),则认为第一剂量过低。在某些实施方案中,这些方法包括施用增加的第二剂量化合物的额外步骤。可以重复这些步骤,反复增加剂量,直到实现期望的雌二醇
‑
er结合降低。在某些实施方案中,可以将这些步骤并入本文提供的抑制肿瘤生长的方法中。在这些方法中,可以将雌二醇
‑
er 结合作为肿瘤生长抑制的替代指标,或评估生长抑制的补充手段。在其他实施方案中,可以将这些方法与rad1901或其溶剂化物(例如水合物)或其盐的施用结合使用,以用于除了抑制肿瘤生长之外的目的,包括例如抑制癌细胞增殖。
[0144]
在某些实施方案中,本文提供的用于调整组合疗法中rad1901或其盐或其溶剂化物(例如,水合物)的剂量的方法包括:
[0145]
(1)施用第一剂量的rad1901或其溶剂化物(例如水合物)或其盐(例如,约350至约500mg,或约200至约600mg/天)3、4、5、6或7天;
[0146]
(2)例如使用本文公开的fes
‑
pet成像来检测雌二醇
‑
er结合活性;其中:
[0147]
(i)如果er结合活性不可检测或低于预定阈值水平,则继续施用第一剂量(即维持剂量水平);或者
[0148]
(ii)如果er结合活性是可检测的或高于预定阈值水平,则施用比第一剂量更大的第二剂量(例如,第一剂量加上约50至约200mg)3、4、5、6或 7天,然后进行步骤(3);
[0149]
(3)例如使用本文公开的fes
‑
pet成像检测雌二醇
‑
er结合活性;其中,
[0150]
(i)如果er结合活性不可检测或低于预定的阈值水平,则继续施用第二剂量(即维持剂量水平);或者
[0151]
(ii)如果er结合活性是可检测的或高于预定阈值水平,则施用大于第二剂量的第三剂量(例如,第二剂量加上约50至约200mg)3、4、5、6或7天,然后进行步骤(4);
[0152]
(4)通过第四剂量、第五剂量等重复上述步骤,直到检测不到er结合活性。
[0153]
在某些实施方案中,本发明包括使用pet成像来检测er敏感性或er耐药性癌症和/或对其给药。
[0154]
(7)用于本文公开方法的组合
[0155]
本发明的另一个方面涉及用于本文所阐述的组合方法的药物组合物,其包含如本文公开的治疗有效量的rad1901或其溶剂化物(例如水合物)或其盐和/或第二治疗剂(例如
依维莫司)。
[0156]
rad1901
‑
erα相互作用
[0157]
(1)来自接受至少一线内分泌治疗的患者的er阳性乳房肿瘤样品中的突变型 erα
[0158]
在过去两年中报道的五项研究中,对来自接受至少一线内分泌治疗的患者的总计187个转移性er阳性乳房肿瘤样品进行测序,并且在39名患者(21%)中鉴别出er lbd突变 (jeselsohn)。在39名患者中,最频繁的6种lbd突变在方案1(改编自jeselsohn)中示出。
[0159][0160]
所有lbd突变的频率汇总于表9中。
[0161]
表9
[0162]
lbd突变频率
[0163] 频率(%)d538g29.5y537s25.0y537n13.6y537c9.1e380q6.8s463p4.5l536r2.3l536q2.3p535h2.3v392i2.3v534e2.3
[0164]
计算机建模显示,rad1901
‑
erα相互作用不太可能受erα的lbd突变体影响,所述突变体例如为y537x突变体,其中x是s、n或c;d538g;和s463p,其占对接受至少一线内分泌治疗的患者的转移性er阳性乳房肿瘤样品的最新研究中发现的lbd突变的约81.7% (表10,实施例v)。
[0165]
本文提供了结合erα和/或突变型erα的rad1901的复合物和晶体,所述突变型erα包括一个或多个突变,其包括但不限于y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或q;p535h;v534e;s463p;v392i;e380q及其组合。
[0166]
在本文提供的方法的某些实施方案中,erα和突变型erα的lbd包含af
‑
2。在其他实施方案中,lbd包含erα的氨基酸299
‑
554,由erα的氨基酸299
‑
554组成,或基本上由erα的氨基酸299
‑
554组成。在某些实施方案中,突变型erα的lbd包括一个或多个突变,其包括但不
限于y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或q;p535h; v534e;s463p;v392i;e380q及其组合。本文使用的术语“和/或”包括“和”情形以及“或”情形。
[0167]
在某些实施方案中,本文提供了在有需要的受试者中治疗与erα和/或突变型erα活性或表达相关的病症的方法,其包括向受试者施用一种或多种第二治疗剂(例如依维莫司)和能够经由lbd与erα和/或突变型erα结合的一种或多种化合物。在某些实施方案中,受试者是哺乳动物,并且在这些实施方案中的某些实施方案中,受试者是人。在某些实施方案中,所述病症是肿瘤和/或癌症,包括但不限于如本文所公开的er阳性肿瘤和/或癌症。
[0168]
在本文提供的化合物和方法的某些实施方案中,erα和突变型erα的lbd包含af
‑
2。在其他实施方案中,lbd包含erα的氨基酸299
‑
554,由erα的氨基酸299
‑
554组成,或基本上由erα的氨基酸299
‑
554组成。在某些实施方案中,突变型erα的lbd包括一个或多个突变,其包括但不限于y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或 q;p535h;v534e;s463p;v392i;e380q及其组合。
[0169]
在本文提供的化合物和方法的某些实施方案中,能够经由lbd与erα和/或突变型erα结合的化合物是选择性雌激素受体降解剂(serd)或选择性雌激素受体调节剂(serm)。在某些实施方案中,能够经由lbd与erα和/或突变型erα结合的化合物经由选自以下的一种或多种相互作用来完成结合:与残基e353、d351、r349和/或l536的氢键相互作用和与 erα和/或突变型erα的残基f404的π
‑
相互作用。这种化合物的一个实例是rad1901。
[0170]
在某些实施方案中,本文提供了治疗与突变型erα的活性或表达相关的病症的方法,所述突变型erα包含一个或多个突变,所述一个或多个突变包括但不限于y537x1,其中x1是s、n或c;d538g;l536x2,其中x2是r或q;p535h;v534e;s463p;v392i;e380q 及其组合,其中所述方法包括向所述受试者施用一种或多种第二治疗剂(例如,依维莫司) 和能够经由lbd结合erα的一种或多种化合物。在某些实施方案中,所述病症是癌症,包括但不限于er阳性癌症、乳腺癌、er阳性乳腺癌和转移性乳腺癌,并且在某些实施方案中,所述化合物是rad1901或其药学上可接受的溶剂化物(例如水合物)或其药学上可接受的盐。
[0171]
提供以下实施例以更好地举例说明所要求保护的发明,而不应被解释为限制本发明的范围。就所提及的具体材料的范围而言,这仅仅是为了说明的目的,而无意于限制本发明。本领域的技术人员可以在不发挥创造能力且不偏离本发明的范围的情况下开发等同手段或反应物。将会理解,可以对本文描述的程序进行许多变化而仍然在本发明的范围内。本发明人意在将这样的变化包括在本发明的范围内。
[0172]
实施例
[0173]
材料和方法
[0174]
测试化合物
[0175]
在以下实施例中使用的rad1901是由irix pharmaceuticals公司(florence,sc)制造的(6r)
‑6‑
(2
‑
(n
‑
(4
‑
(2
‑
(乙基氨基)乙基)苄基)
‑
n
‑
乙基氨基)
‑4‑
甲氧基苯基)
‑
5,6,7,8
‑
四氢萘
‑2‑
醇二盐酸盐。rad1901以干粉形式储存,在去离子水中配制成0.5%(w/v)甲基纤维素的均匀悬浮液待用,并口服施用至动物模型。他莫昔芬、雷洛昔芬和雌二醇(e2)得自sigma
‑
aldrich (密苏里州圣路易斯市),并通过皮下注射施用。氟维司群得自tocris biosciences(明尼苏达州明尼阿波利斯市)并通过皮下注射施用。除非另有说明,其他实验室试剂购自sigma
‑
aldrich。
[0176]
细胞系
[0177]
从美国典型培养物保藏中心(马里兰州罗克维尔市)购买mcf
‑
7细胞(人类乳房转移性腺癌)并以5%co2常规保持在包含2mm l
‑
谷氨酰胺和earle’s bss、0.1mm非必需氨基酸和补充有0.01mg/ml牛胰岛素和10%胎牛血清(invitrogen,加利福尼亚州卡尔斯巴德)的1mm 丙酮酸钠的无酚红最低基本培养基(mem)中。
[0178]
在补充有10%fbs和5μg/ml人胰岛素的rpmi生长基质中,将t47d细胞在5%co2培养箱中的10cm培养皿中培养至约75%汇合。
[0179]
体内异种移植模型
[0180]
将所有小鼠置于单独通风的鼠笼中的无病原体房舍(具有无菌和无尘用品)中,可随意获得消毒的食物和水,处于暗光周期(12
‑
14小时的人造光昼夜节律周期)和受控室温和湿度。每周用游标卡尺测量肿瘤两次,并利用下式计算体积:(l
×
w2)
×
0.52。
[0181]
pdx模型
[0182]
来自患者的异种移植模型(pdx模型)的一些实例显示在图1中。由已经在动物(无胸腺裸鼠(nu(ncf)
‑
foxn1nu))中连续传代有限次数以维持肿瘤异质性的可存活人肿瘤组织或流体来建立源自患者的乳腺癌肿瘤的pdx模型。对于每个实验,在其估计开始日期之前大约一周开始记录研究前的肿瘤体积。当肿瘤达到适当的肿瘤体积引发(tvi)范围(150
‑
250mm3) 时,将动物随机分成治疗组和对照组并开始给药(第0天,每组8
‑
10个受试者);所有研究中的动物逐个进行每个实验。初始剂量从第0天开始;所有组的动物按重量给药(0.01ml/g; 10ml/kg)。利用溶媒(对照,口服,每日一次至终点)、他莫昔芬(1mg/受试者,皮下注射,隔日一次至终点)、氟维司群(1mg/受试者或3mg/受试者,皮下注射,每周
×
5,如有必要的话延长)、或rad1901(30、60或120mg/kg受试者,口服,每日一次至终点) 按照说明从第0天开始对每个组进行治疗。根据模型,治疗期持续56
‑
60天。在这些pdx模型的饮用水中补充17β
‑
雌二醇。
[0183]
药剂功效
[0184]
对于所有研究,从第0天开始,通过数字卡尺测量肿瘤尺寸,并且针对各组记录数据,所述数据包括个体和平均估计肿瘤体积(平均tv
±
sem);利用公式(yasui等,invasion metastasis 17:259
‑
269(1997),其通过引用并入本文)来计算肿瘤体积:tv=(宽度)2×
(长度)
×
0.52。一旦评估的组平均肿瘤体积达到肿瘤体积(tv)终点(时间终点为60天;体积终点为组平均2cm3),就结束每个组或研究;从研究中取出肿瘤体积达到2em3或更多的个体小鼠,并且将最终测量包括在组平均中,直到组平均达到体积终点或研究达到时间终点。
[0185]
功效计算和统计分析
[0186]
在单个时间点(当对照组达到肿瘤体积或时间终点时)计算%肿瘤生长抑制(%tgi)值,并且通过下式(corbett th等人,in vivo methods for screening and preclinical testing.in:teicherb,ed.,anticancer drug development guide.totowa,nj:humana.2004:99
‑
123.)利用初始(i) 和最终(f)肿瘤测量值对每个治疗组(t)
‑
对照(c)报道该%肿瘤生长抑制(%tgi)值:%tgi =1
‑
tf
‑
ti/cf
‑
ci。
[0187]
统计
[0188]
tgi研究
‑
单因素方差分析 dunnett多重比较检验(corbett th等人)。
[0189]
样品采集
[0190]
在终点时移除肿瘤。将一个片段快速冷冻,而将另一个片段置于10%nbf中至少24小时,并用福尔马林固定石蜡包埋(ffpe)。将快速冷冻样品储存在
‑
80℃;将ffpe块在室温下储存。
[0191]
蛋白质免疫印迹
[0192]
采集细胞并利用标准做法来分析蛋白质表达。在给药最后一天之后的指定时间点收集肿瘤,使用tissuelyser(qiagen)在具有蛋白酶和磷酸酶抑制剂的ripa缓冲液中匀浆。通过 mw分离等量的蛋白质,将其转移至硝酸纤维素膜并使用标准做法利用以下抗体进行印迹分析:
[0193]
·
雌激素受体(santacruz(hc
‑
20);sc
‑
543)
[0194]
·
孕酮受体(cell signaling technologies;3153)
[0195]
·
纽蛋白(sigma
‑
aldrich,v9131)
[0196]
按以下进行qpcr分析:采集细胞,提取mrna,将其等量用于cdna分析以及利用孕酮受体、greb1和tff1(lifetech)特异性引物进行qpcr。使用1d quant软件(ge)量化条带。
[0197]
免疫组化
[0198]
收集肿瘤,利用福尔马林固定并包埋入石蜡中。将包埋的肿瘤切片(6μm)并利用er、 pr和her2特异性抗体染色。按如下进行定量:对阳性细胞(0
‑
100%)和染色强度(0
‑
3 ) 计数五个区域。利用以下公式计算h评分(0
‑
300):%阳性
×
强度。
[0199]
实施例i.在具有不同既往内分泌治疗的情况下rad1901
‑
依维莫司组合增强表达wt er或突变型er(例如y537s)的肿瘤和/或癌症中的肿瘤生长抑制。
[0200]
i(a).rad1901对动物异种移植模型的有效性
[0201]
i(a)(i)rad1901在pdx模型(pdx
‑
1至pdx
‑
12)中抑制肿瘤生长,无论该肿瘤的er状态租既往内分泌疗法史。
[0202]
图1显示出对于单独利用rad1901治疗的小鼠在几种pdx模型中的肿瘤生长抑制作用。筛选12个源自患者的异种移植模型以测试具有不同er、pr和her2水平的几种遗传背景下的rad1901反应。对标记有“*”的pdx模型(pdx
‑
1至pdx
‑
4和pdx
‑
12)进行全功效研究,其中n=8
‑
10。对于其他pdx模型(pdx
‑
5至pdx
‑
11)进行筛选研究(其中n=3),利用溶媒(阴性对照)或rad1901以60mg/kg剂量(口服,每日一次)治疗60天。如图1所示出的,其中由er和另外的驱动因子(例如pr 和/或her2 )驱动生长的pdx模型获得了 rad1901治疗的益处。rad1901有效地抑制具有er突变和/或her2高表达水平(pdx)的模型中的肿瘤生长,无论该肿瘤的既往治疗史,即初次治疗或利用芳香化酶抑制剂、他莫昔芬(tam)、化疗(chemo)、her2抑制剂(her2i,例如曲妥珠单抗、拉帕替尼)、贝伐单抗、氟维司群和/或利妥昔单抗进行的治疗。
[0203]
i(a)(ii)rad1901
‑
依维莫司组合在表达wt er的异种移植模型中相比单独使用rad1901 引起更多的消退
[0204]
i(a)(ii)(1)rad1901
‑
依维莫司在对氟维司群治疗有反应的mcf
‑
7异种移植物中相比单独使用rad1901引起更多的消退。
[0205]
mcf
‑
7异种移植模型
[0206]
在细胞植入前两天,用0.18/90天释放的17β
‑
雌二醇丸粒接种balb/c
‑
nude小鼠。采集 mcf
‑
7细胞(pr ,her2
‑
),并将1
×
107个细胞皮下植入balb/c
‑
nude小鼠的右侧腋下。
当肿瘤平均达到200mm3时,按肿瘤体积将小鼠随机分成治疗组,并用测试化合物进行治疗。利用溶媒(对照,口服,每日一次,至终点)、氟维司群(3mg/受试者,皮下注射,每周一次,
×
5,如果必要的话延长)、rad1901(30mg/kg或60mg/kg受试者,口服,每日一次,至终点)、依维莫司(2.5mg/kg,口服,每日一次,至终点)或规定剂量的rad1901
‑ꢀ
依维莫司组合从第0天开始对每个组进行治疗。治疗期持续28天。
[0207]
利用溶媒(阴性对照)、rad1901(60mg/kg,口服,每日一次)、依维莫司(2.5mg/kg,口服)、rad1901(30mg/kg或60mg/kg,口服,每日一次)和依维莫司(2.5mg/kg,口服) 的组合、氟维司群(3mg/剂,皮下注射,每周一次)、或氟维司群(3mg/剂,皮下注射,每周一次)和依维莫司(2.5mg/kg,口服)的组合治疗mcf7异种移植小鼠。在几个时间点测量肿瘤尺寸,测量27天。
[0208]
结果显示在图2a
‑
b中。利用rad1901(60mg/kg)和依维莫司(2.5mg/kg)的组合治疗再次导致显著的肿瘤消退,该结果优于单独使用rad1901、依维莫司或氟维司群、或使用氟维司群和依维莫司组合进行的治疗(图2b)。
[0209]
图2c显示,具有30mg/kg或60mg/kg rad1901的rad1901
‑
依维莫司组合提供了相似的效果,但是以30mg/kg单独使用rad1901在抑制肿瘤生长方面不及以60mg/kg单独使用 rad1901有效。所述结果表明,具有较低剂量rad1901(例如30mg/kg)的rad1901
‑
依维莫司组合足以使所述异种移植模型中的肿瘤生长抑制/肿瘤消退效果最大化。
[0210]
与单独使用rad1901、依维莫司或氟维司群、或使用氟维司群和依维莫司组合进行的治疗相比,利用rad1901和依维莫司组合进行的治疗在降低mcf7异种移植模型体内er和 pr表达方面也更有效(图11);在最后一次给药后两个小时时采集肿瘤)。
[0211]
i(a(ii)(2)在制氟维司群治疗有反应的pdx
‑
11和pdx
‑
2模型中,rad1901
‑
依维莫司比单独使用rad1901引起更多的肿瘤消退。
[0212]
er wt pdx模型pdx
‑
2(pr ,her2 ,初次治疗)和pdx
‑
11(pr ,her2 ,利用ai、氟维司群和化疗进行治疗)对氟维司群(3mg/剂,皮下注射,每周一次)表现出不同的敏感性。使用rad1901(60mg/kg,口服,每日一次)和依维莫司(2.5mg/kg,口服)的组合、单独使用rad1901(60mg/kg,口服,每日一次)、单独使用依维莫司(2.5mg/kg,口服)、或单独使用氟维司群(3mg/剂,皮下注射,每周一次)治疗pdx
‑
2和pdx
‑
11模型。
[0213]
在pdx
‑
11模型中,单独施用氟维司群或依维莫司显著抑制肿瘤生长,而利用氟维司群治疗的小鼠在肿瘤生长抑制方面表现出更好的效果。氟维司群治疗表现出轻微的肿瘤消退(图 3b)。出乎意料地,单独施用rad1901或与依维莫司组合施用导致显著的肿瘤消退,且在野生型esr1 pdx模型中该组合实现甚至更显著的肿瘤消退效果(图3b)。
[0214]
在pdx
‑
2模型中,与单独注射氟维司群相比,单独口服施用rad1901获得了更好的抑制肿瘤生长的效果(图4a)。此外,单独施用rad1901或依维莫司显著抑制肿瘤生长。出乎意料地,rad1901与依维莫司组合施用导致甚至更强的肿瘤生长抑制效果(图4b)。
[0215]
此外,在对氟维司群治疗(1mg/剂,皮下注射,每周一次)有反应的pdx
‑
4模型中,在 rad1901治疗(30mg/kg,口服,每日一次)期结束之后继续进行雌二醇治疗的情况下, rad1901介导的肿瘤生长抑制在不进行治疗的情况下维持至少两个月(图5)。
[0216]
因此,一种或多种第二治疗剂与rad1901的组合可能有益于患者在治疗结束后抑制肿瘤生长,特别是当一种或多种第二治疗剂(例如依维莫司)由于不良反应而可能被减少
或延迟时如此。
[0217]
i(a)(iii)在表达突变型er(erαy537s)的异种移植模型中,rad1901
‑
依维莫司比单独使用rad1901引起更多的消退
[0218]
i(a)(iii)(1)在对氟维司群治疗几乎没有反应的pdx
‑
5模型中,rad1901
‑
依维莫司比单独使用rad1901引起更多的消退。
[0219]
按照上文针对pdx模型所述的类似方案制备pdx
‑
5模型。用游标卡尺每周测量两次给药组的肿瘤尺寸,并使用公式(l
×
w2)
×
0.52计算体积。
[0220]
利用本文所述方法评估rad1901(60mg/kg,口服,每日一次)、依维莫司(2.5mg/kg,口服)和rad1901(60mg/kg,口服,每日一次)与依维莫司(2.5mg/kg)的组合对突变型 er pdx
‑
5模型(患有来源于具有y537s雌激素受体突变、pr 、her2 的乳腺癌肿瘤患者的 pdx模型,具有芳香酶抑制剂的既往治疗)的肿瘤生长抑制。对于表达某些erα突变(例如 y537s)的肿瘤,rad1901和依维莫司的组合治疗在抑制肿瘤生长方面比单独使用任一种药剂进行的治疗更有效(图6b)。这些pdx模型对氟维司群(3mg/剂)治疗不敏感。rad1901 和依维莫司的组合治疗在rdx
‑
5模型中引起肿瘤消退方面比单独使用任一种药剂进行的治疗更为有效(图6b)。
[0221]
因此,结果显示rad1901是增强靶向药物肿瘤生长抑制的有效内分泌骨架。此外, rad1901在来源于已经接受多次既往内分泌治疗(包括对氟维司群不敏感的那些)的患者的 pdx模型中显示出有效的抗肿瘤活性。
[0222]
i(a)(iv)氟维司群治疗对非荷瘤小鼠的药代动力学评估。
[0223]
向小鼠施用几种剂量的氟维司群,其对受试者表现出显著的剂量暴露(图7)。
[0224]
在第1天(d1 rx)和第8天(d8 rx,n=4/剂水平)时将氟维司群以1、3或5mg/剂经皮下施用至裸鼠。在第二剂之后长达168小时的时间内在指定的时间点采集血液,离心分离,并通过液相色谱
‑
质谱分析血浆。
[0225]
i(b)rad1901在脑转移的小鼠异种移植模型(mcf
‑
7颅内模型)中促进存活。
[0226]
使用mcf
‑
7颅内肿瘤异种移植模型进一步评估rad1901穿过血脑屏障并抑制肿瘤生长的潜在能力。
[0227]
使用雌性无胸腺裸鼠(cr1:nu(ncr)
‑
foxn1nu)用于肿瘤异种移植研究。在肿瘤细胞植入前三天,使用灭菌的套管针将雌激素丸粒(0.36mg e2,60天释放(innovative research ofamerica,弗罗里达州萨拉索塔市))经皮下植入所有测试动物的肩胛骨之间。在包含10%胎牛血清、100单位/ml青霉素g、100μg/ml硫酸链霉素、2mm谷氨酰胺、10mm hepes、 0.075%碳酸氢钠和25g/ml庆大霉素的rpmi
‑
1640培养基中将mcf
‑
7人乳腺癌细胞培养至对数生长中期。在肿瘤细胞植入当天,用胰蛋白酶消化细胞,沉淀,并以5
×
10
7个
细胞/ml 的浓度重新悬浮于磷酸盐缓冲盐水中。对每只测试小鼠经颅内植入1
×
106个mcf
‑
7细胞。
[0228]
在肿瘤细胞植入(指定为研究的第1天)后第5天,将小鼠随机分成三组,每组12只动物,并用载体、氟维司群(0.5mg/动物,每天一次)或rad1901(120mg/kg,每天一次) 进行治疗,如上文所述的。
[0229]
终点定义为对照组的死亡率或3
×
存活率,以先到者为准。通过体重测量和频繁观察治疗相关不良反应的临床征兆来评估治疗耐受性。将一次测量的体重减轻超过30%的动
物或三次测量的体重减轻超过25%的动物人道安乐死并归类为治疗相关死t。可接受的毒性被定义为在研究期间组平均体重减轻小于20%,并且在10只接受治疗的动物中具有不多于一个的治疗相关死亡(或10%)。在研究结束时,在异氟烷麻醉下,通过终末心脏穿刺将动物安乐死。利用lc
‑
ms/ms测定血浆和肿瘤中的rad1901和氟维司群浓度。
[0230]
kaplan meier存活分析表明,rad1901相比氟维司群显著延长存活(p<0.0001;图8)。对照组或氟维司群组中没有动物分别存活超过第20天和第34天,而rad1901治疗的动物中 41%(5/12)存活至第54天研究结束时。
[0231]
rad1901在血浆中的浓度为738
±
471ng/ml,在颅内肿瘤中的浓度为462
±
105ng/g,其支持rad1901能够有效穿过血脑屏障的假设。与此相比,血浆(21
±
10ng/ml)和颅内肿瘤(8.3 0.8ng/g)内的氟维司群浓度明显较低。
[0232]
i(c).rad1901治疗er 晚期乳腺癌的1期研究。
[0233]
在1期研究中,在44位健康的绝经后女性中评估安全性、耐受性和药代动力学。没有观察到剂量限制毒性,未确定最大耐受剂量(mtd)。在所测试的剂量范围内,血浆暴露量随剂量增加而成比例增加。
[0234]
受试者
[0235]
招募8名患有晚期乳腺癌(具有不少于1%ihc染色的er 肿瘤、具有0或1的ecog 表现状态的her2
‑
阴性肿瘤)的绝经后女性作为该1期研究的受试者。受试者必须接受以下既往治疗:
[0236]
·
在晚期/转移性情形中不超过2个既往化疗方案
[0237]
·
6个月的既往内分泌治疗,并且在既往内分泌治疗中取得进展
[0238]
·
排除未经治疗或有症状的中枢神经系统转移或在以下时间窗内接受既往抗癌治疗的患者:
[0239]
·
在第一剂量研究治疗之前他莫西芬<14天
[0240]
·
在第一剂量研究治疗之前氟维司群<90天
[0241]
·
在第一剂量研究治疗之前化疗<28天
[0242]
·
在第一剂量研究治疗之前lhrh类似物<12个月
[0243]
dlt标准
[0244]
·
不低于3非血液学毒性(排除尚未用最佳药物治疗的脱发和恶心、呕吐或腹泻)的任何等级
[0245]
·
不低于3血液学毒性的任何等级
[0246]
·
导致研究药物中断>7天的任何等级毒性
[0247]
·
剂量限制毒性观察期是第1周期的第1
‑
28天
[0248]
治疗紧急不良事件(teae)
[0249]
在整个研究中记录teae。初步数据汇总在表12中。“n”是在给定类别中具有至少一个治疗相关ae的受试者的数目(ae根据不良事件通用标准(ctcae)v4.0进行分级),并且经历同一优先项目的多种情形的任何患者都只以最严重的等级计算一次。没有观察到死亡或剂量限制性毒性,未确定最大耐受剂量(mtd)。大多数ae为1或2级。最常见的治疗相关 ae为消化不良(5/8的患者)和恶心(3/8的患者)。观察到两例严重ae(sae),一例是3 级治疗相关便秘,另一例是呼吸短促(胸腔积液),与治疗无关。
[0250]
该1期研究中经过严重预治疗的受试者包括之前曾用多种内分泌和靶向药剂(例如 cdk4/6、pi3k和mtor抑制剂)治疗的受试者。在以200mg剂量(口服,每日一次)治疗长达6个月和400mg剂量(口服,每日一次)治疗长达2个月的rad1901治疗之后,没有观察到剂量限制性毒性。因此,rad1901显示出治疗er 晚期乳腺癌的潜力,尤其是在之前曾用内分泌和/或靶向药剂如cdk4/6、pi3k和mtor抑制剂治疗的受试者中如此。
[0251]
实施例ⅱ.rad1901优先在肿瘤中累积并且可以递送至脑。
[0252]
使用lc
‑
ms/ms进一步评价实施例i(a)(i)中所述的mcf
‑
7异种移植物的血浆和肿瘤中的 rad1901浓度。在研究结束时,在60mg/kg剂量水平下,血浆中rad1901的浓度为344
±
117 ng/ml,肿瘤中的浓度为11,118
±
3801ng/ml。在其中肿瘤浓度为血浆浓度的约20
‑
30倍的较低剂量水平下也观察到类似的肿瘤与血浆比。接受治疗40天的小鼠的血浆、肿瘤和大脑中的 rad1901水平汇总在表1中。将显著量的rad1901递送到接受治疗的小鼠的大脑(例如,参见b/p比(大脑中的rad1901浓度/血浆中的rad1901浓度)),其表明rad1901能够穿过血脑屏障(bbb)。出乎意料地,rad1901优先积累在肿瘤中。参见例如表1中所示的t/p (肿瘤中的rad1901浓度/血浆中的rad1901浓度)比。
[0253]
表1
[0254]
治疗40天后植入mcf7细胞的小鼠血浆、肿瘤和大脑中的rad1901水平。
[0255][0256]
*blq:低于定量极限
[0257]
实施例iii.rad1901抑制er途径和降解er。
[0258]
iii(a).在健康的绝经后女性认类受试者中,rad1901降低了在子宫和垂体中的er结合。
[0259]
受试者具有至少12个月的闭经持续时间和与更年期一致的血清fsh。受试者为40
‑
75岁,其中bmi为18.0
‑
30kg/m2。受试者有完整的子宫。排除如下受试者:具有临床相关病理证据、中风风险增加或具有静脉血栓事件史,或在进入临床研究中心之前不到14天内使用伴随用药 (允许至多3天之前使用扑热息痛)。
[0260]
在基线和暴露于rad1901 6天之后进行fes
‑
pet以评价子宫中的er结合。在200mg (7名受试者)和500mg(6名受试者)剂量水平下,rad1901分别占据子宫中83%和92%的er。
[0261]
在用200mg或500mg(口服,每日一次,6天)进行rad1901治疗后,fes
‑
pet成像显示经标记的雌二醇与子宫和垂体的结合显著降低。
[0262]
由于er表达高,所以子宫在rad1901治疗之前的基线处显示出强fes
‑
pet信号(图 9a,利用200mg剂量水平治疗的受试者3的子宫fes
‑
pet扫描的基线横向视图;图9b,分别为用500mg剂量水平治疗的受试者7的子宫fes
‑
pet扫描的基线矢状位图和横向视图)。然而,
当在研究的第6天给药后4小时进行扫描时,几乎看不见子宫(处于背景fes
‑
pet信号水平或接近背景fes
‑
pet信号(图9a,第6天时受试者3子宫扫描的横向视图;以及图 9b,分别为第6天受试者7子宫扫描的矢状位图和横向视图)。这些数据与er降解和/或与受体结合的竞争一致。图9a和9b还包括由fes
‑
pet扫描的子宫ct扫描,其显示在rad1901 治疗前和治疗后存在子宫。
[0263]
进一步量化fes
‑
pet子宫扫描结果以显示7名受试者中给药后的er
‑
结合相对于基线的变化(图9c),其分别显示作为200mg剂量组和500mg剂量组实施例的受试者1
‑
3和受试者4
‑
7。rad1901在较低剂量水平(200mg)下显示出稳定的er结合。
[0264]
图10a
‑
b显示在6天之后,以500mg(口服,每日一次)进行rad1901治疗之前(基线)和之后(治疗后)的子宫(a)和垂体(b)的fes
‑
pet扫描的代表性图像。图10a按如下显示子宫的fes
‑
pet扫描:(a)侧向截面;(b)纵向截面;和(c)纵向截面。
[0265]
受试者的子宫和垂体的给药后fes
‑
pet剂量扫描分别显示,在子宫(图10a,治疗后) 和垂体(图10b,治疗后)处没有明显的er结合信号。
[0266]
因此,结果显示,在6天之后,在200mg和500mg的剂量(口服,每日一次)下rad1901 有效地结合人体内的er。
[0267]
计算子宫、肌肉和骨的标准摄取值(suv),并且将针对200mg和500mg(口服,每日一次)的rad1901治疗分别汇总在表2和3中。给药后子宫信号非常接近来自“非标靶组织”的水平,其表明rad1901治疗后的fes
‑
pet摄取完全减弱。在不显著表达雌激素受体的组织中,在治疗前和治疗后的pet扫描中几乎没有观察到变化。
[0268]
表2
[0269]
用200mg剂量(口服,1次天)治疗6天的人受试者的子宫、肌肉和骨suv
[0270][0271]
表3
[0272]
用500mg剂量(口服,1次/天)治疗6天的人受试者(n=4)的子宫、肌肉和骨的suv。
[0273][0274]
因此,rad1901或其盐或其溶剂化物(例如其水合物)可用于治疗具有er过度表达的癌症和/或肿瘤细胞(例如,乳腺癌、子宫癌和卵巢癌),而对其他器官(例如,骨、肌肉) 没有负效应。rad1901或其盐或其溶剂化物(例如水合物)可能特别适用于治疗转移性癌症和/或在其他器官中具有er过度表达的肿瘤,例如转移至其他器官(例如骨、肌肉)的原发性乳
腺癌、子宫癌和/或卵巢癌,以治疗其他器官(例如,骨、肌肉)中的乳腺癌、子宫癌和 /或卵巢癌病变,而对所述器官没有负面影响。
[0275]
iii(b).rad1901降低er表达并抑制er途径。
[0276]
iiiiii(b)(i)(1)rad1901
‑
依维莫司组合在降低mcf7异种移植模型中的er和pr表达方面相比单独使用rad1901、依维莫司或氟维司群或氟维司群依维莫司组合进行的治疗更为有效。
[0277]
与单独使用rad1901、依维莫司或氟维司群进行的治疗、或利用氟维司群和依维莫司组合进行的治疗相比,利用rad1901和依维莫司组合进行的治疗在降低mcf7异种移植模型 (如实施例i(a)(ii)中描述的)中的体内er和pr表达降低方面也更有效(图11);在最后一次给药后两个小时时采集肿瘤)。
[0278]
iii(b)(i)(2)rad1901和氟维司群在mcf7和t47d细胞系中的比较。
[0279]
使用mcf
‑
7和t47d细胞系比较不同浓度的rad1901和氟维司群的效果,所述细胞系均为人乳腺癌细胞系,所述不同浓度为0.01μm、0.1μm和1μm(图12a为mcf
‑
7细胞系分析;图12b为t47d细胞系)。使用以下三种er标靶基因作为标志物:孕酮受体(pgr)、通过乳腺癌1的雌激素进行的生长调节(greb1)和三叶因子1(tff1)。rad1901引起几乎完全的er降解并抑制er信号传导(图12a
‑
b)。特别是对于mcf
‑
7细胞系,当以相同浓度施用时,氟维司群在抑制er信号传导方面表现出相当的或甚至略高的功效。出乎意料地,与氟维司群相比,rad1901在抑制肿瘤生长和引起肿瘤消退方面与其相当或更为有效,如在实施例i(a)和i(b)中公开的。
[0280]
iii(b)(i)(3)在上文i(a)(ii)(1)实施例中喵述的mcf
‑
7异种移植模型ii中rad1901治疗导致er降解并消除er信号传导。
[0281]
rad1901治疗导致体内er降解(图13a和13b,学生t检验:*p值<0.05,**p值<0.01) 并且抑制体内er信号传导(图13a和13c,学生t检验:*p值<0.05,**p值<0.01)。
[0282]
在rad1901的最终剂(30mg/kg,60mg/kg,口服,每日一次)或氟维司群(3mg/剂,皮下注射,每周一次)之后2小时从mcf
‑
7异种移植物采集的肿瘤显示er和pr表达显著降低(图13a
‑
b)。在氟维司群治疗的最终剂之后8小时从mcf
‑
7异种移植物采集的肿瘤显示出不同的pr和er表达。然而,在rad1901治疗的最终剂之后8小时从mcf
‑
7异种移植物采集的肿瘤显示pr和er表达降低(图13a和13c)。
[0283]
在单剂rad1901(30mg/kg、60mg/kg或90mg/kg,口服,每日一次)之后8小时或12 小时从mcf
‑
7异种移植物采集的肿瘤显示pr表达迅速降低(图14a
‑
c)。在第7剂rad1901 (30mg/kg、60mg/kg或90mg/kg,口服,每日一次)之后4小时或24小时从mcf
‑
7异种移植物采集的肿瘤显示出稳定且一致的er信号传导抑制作用(图14b)。在rad1901(30 mg/kg、60mg/kg或90mg/kg,口服,每日一次)治疗期间的几个时间点对从mcf
‑
7异种移植物采集的肿瘤进行蛋白质免疫印迹分析的定量分析显示出剂量依赖性的pr降低(图14c)。
[0284]
rad1901治疗导致mcf
‑
7异种移植模型中的增殖快速降低。例如,将单剂rad1901(90 mg/kg,口服,每日一次)之后8小时和第4剂rad1901(90mg/kg,口服,每日一次)之后24小时时从mcf
‑
7异种移植模型中采集的肿瘤切片并染色,其显示增殖标志物ki67快速降低(图15a和15b)。
[0285]
这些结果表明rad1901治疗导致er降解并抑制wt er异种移植物中的体内er信号
传导。
[0286]
iii(b)(i)(4)rad1901治疗在实施例i(a)(ii)中描述的pdx
‑
4模型中导致er降解并消除er 信号传导。
[0287]
rad1901治疗导致pdx
‑
4模型中的增殖快速降低。例如,将在56天功效研究最后一天的最后一剂之后四小时时从利用rad1901(30、60或120mg/kg,口服,每日一次)或氟维司群(1mg/动物,每周一次)治疗的pdx
‑
4模型采集的肿瘤切片;与利用氟维司群治疗的 pdx
‑
4模型相比,其显示增殖标志物ki67快速降低(图16)。
[0288]
这些结果表明,rad1901治疗导致er降解并抑制wt er异种移植物中的体内er信号传导。
[0289]
iii(b)(ii)rad1901治疗导致上文实施例i(a)(iii)(1)中描述的突变型er异种移植模型 (pdx
‑
5)中的er信号传导降低。
[0290]
在给药最后一天(除非另有说明)后的指定时间点采集肿瘤,并使用tissuelyser(qiagen) 在具有蛋白酶和磷酸酶抑制剂的ripa缓冲液中匀浆。通过mw分离等量的蛋白质,将其转移至硝酸纤维素膜,并利用按照材料和方法部分所述的以下抗体进行印迹:孕酮受体(pr, cell signaling technologies;3153)。
[0291]
使用1d quant软件(ge)量化条带,并且从如实施例i(a)(iii)(1)中所述的pdx
‑
5模型获得的pr(allred评分)示于图17中。氟维司群对pr表达几乎没有影响,而rad1901在60 mg/kg和120mg/kg(口服,每日一次,图17)剂量下均显示出功效。
[0292]
这些结果表明,对于表达某些erα突变(例如y537s)的肿瘤,rad1901在抑制肿瘤生长方面比氟维司群更为有效,并且在抑制对氟维司群治疗几乎没有反应的肿瘤生长方面尤其有效(例如,剂量为3mg/剂,皮下注射,每周一次,图6a显示pdx
‑
5)。此外,对于氟维司群治疗没有良好反应的肿瘤(例如pdx
‑
5),rad1901在降低体内pr表达方面有效,而氟维司群无效(图17)。
[0293]
实施例iv rad1901治疗对子宫组织和/或bmd的影响
[0294]
iv(a(1)):rad1901拮抗雌二醇对子宫组织的刺激。
[0295]
通过评估未成熟大鼠的子宫重量、组织学和c3基因表达的变化来研究rad1901对子宫的影响。得自代表性研究的结果在图18a
‑
d中示出。
[0296]
评估子宫活性
[0297]
将sprague
‑
dawley大鼠幼仔在19日龄断奶,随机分组(n=4),连续3天施用溶媒(含水甲基纤维素)、e2(0.01mg/kg)、雷洛昔芬(3mg/kg)、他莫昔芬(1mg/kg)、单独的rad1901 (0.3至100mg/kg)或rad1901(0.01至10mg/kg)与e2(0.01mg/kg)的组合,视情况采用皮下注射或口服(见上文的试剂)施用,每日一次。最终剂之后24小时,通过吸入二氧化碳使所有的动物安乐死。记录每只动物的体重和子宫湿重。在大鼠和小鼠中还利用rad1901 (0.03至100mg/kg)进行类似的测定(charles river laboratories,魁北克省蒙特利尔市)。
[0298]
将来自每只大鼠的新鲜子宫组织固定在4%的多聚甲醛中,用乙醇脱水并且包埋在jb4 塑料树脂中。将切片切成8μm,用0.1%甲苯胺蓝o染色。使用spot advanced程序,利用zeissaxioskop 40显微镜测量子宫内膜上皮的厚度;计算每个样本9次测量的平均值。
[0299]
子宫补体成分3(c3)基因表达
[0300]
为了测定接受治疗的子宫组织中c3的相对表达水平,使用微至中量总rna纯化试剂盒 (invitrogen,加利福尼亚州卡尔斯巴德市)根据制造商的说明从剩余组织中提取rna。对 rna进行定量,并使用高容量cdna存档试剂盒(applied biosystems,加利福尼亚州福斯特城)对等量rna进行逆转录。
[0301]
使用abi prism 7300系统(applied biosystems)进行定量pcr。使用taqman通用母液混合器进行pcr,该混合器具有用于c3和用于作为参照基因的18s核糖体rna的探针组。热循环条件包括95℃下10分钟的初始变性步骤和随后在95℃下15秒和在60℃下1分钟的 40个循环。
[0302]
通过将每个样品归一化至内源性对照(18s)并与校准物(溶媒)比较来测定相对基因表达。使用下式确定相对基因表达:2
‑
δδct(其中ct=循环阈值或首次检测pcr产物的循环次数,δct=归一化样品值,δδct=给药受试者与溶媒之间的归一化差异)。在每个研究中对每个剂量进行5次重复的基因表达测定。
[0303]
与单独的溶媒相比,用e2(0.01mg/kg)、雷洛昔芬(ral,3mg/kg)或他莫昔芬(tam, 1mg/kg)的治疗导致子宫湿重显著增加,而0.3
‑
100mg/kg剂量范围的rad1901治疗不显著影响子宫湿重(图18a)。所示数据(图18a)是平均值(
±
sem);每组n=4只大鼠;p
‑
溶媒:*<0.05;p
‑
e2:此外,当与e2(0.01mg/kg)联合施用时,rad1901以剂量依赖性方式拮抗e2介导的子宫刺激,其在0.1mg/kg和更高的剂量下对子宫活性表现出显著抑制,并且在3mg/kg剂量下完全抑制子宫活性。rad1901的ec
50
约为0.3mg/kg。在其中 rad1901剂量0.03至100mg/kg对子宫湿重或上皮厚度没有影响的小鼠中也获得了类似的结果(数据未显示)。
[0304]
通过定量显微组织学进一步研究子宫组织中的治疗依赖性变化。在以0.01和0.3mg/kg 利用e2治疗之后,子宫内膜上皮厚度存在统计学显著性增加(图18b)。在利用他莫昔芬(1 mg/kg)或雷洛昔芬(3mg/kg)治疗后,也观察到上皮厚度显著增加。与此相比,rad1901 治疗在高达100mg/kg的最高评价剂量下也没有使子宫内膜上皮厚度增加。图18c中显示子宫内膜上皮的代表性图像。
[0305]
与子宫重量和子宫内膜上皮厚度两者的变化一致,e2、他莫昔芬和雷洛昔芬均显著增加雌激素调节的补体基因c3的表达(图18d)。与此相比,在所测试的任何剂量(0.3至100mg/kg) 下,rad1901均未增加c3基因表达。而且,1、3和10mg/kg的rad1901显著抑制e2刺激的c3基因表达。
[0306]
rad1901不刺激未成熟雌性大鼠的子宫
[0307]
连续3天给未成熟雌性大鼠(口服,每日一次)施用溶媒(veh)、雌二醇(e2)、雷洛昔芬(ral)、他莫昔芬(tam)、rad1901或rad1901 e2。测量子宫湿重。所示数据(图18)是平均值(
±
sem);每组n=4只大鼠;p
‑
溶媒:*<0.05;p
‑
e2:
[0308]
实施例ii(a)(2).rad1901治疗防止卵巢切除大鼠中的骨质流失
[0309]
在卵巢切除大鼠中检查rad1901的骨特异性效应。
[0310]
作为绝经后骨质流失的模型,对麻醉的成年雌性sprague
‑
dawley大鼠进行卵巢切除术,以假手术作为对照。手术后,利用溶媒、e2(0.01mg/kg)或rad1901(0.1、0.3、1、3mg/kg) 治疗卵巢切除的大鼠4周(每日一次,如上所述施用),每组20只动物。用溶媒治疗假手术组中的动物。在最终剂后24小时,通过吸入二氧化碳使所有动物安乐死。使用piximus双
发射x射线吸收测定法在基线处评估骨矿物质密度,并且治疗4周后再次评估。
[0311]
在尸体剖检时,取出每只动物的左股骨,解剖掉软组织,并在分析之前保存在70%乙醇中。使用micro
‑
ct40系统(scanco systems,宾夕法尼亚州韦恩市)进行详细的定性和定量 3
‑
d评价。对于每个样本,获取远端股骨干骺端的250个图像切片。在预先选择的分析区域中使用直接3d方法确定形态测定参数。在小梁骨中测定的参数包括骨体积密度、骨表面密度、骨小梁数量、骨小梁厚度、骨小梁间距、连接密度和表观骨密度。
[0312]
在卵巢切除术后,与基线相比,未治疗(溶媒对照)大鼠在整个全股骨和腰椎骨中都出现了骨矿物质密度降低(表4)。e2治疗与预防股骨和脊柱两者中的骨质流失相关。rad1901 治疗对卵巢切除术诱发的骨质流失产生了剂量依赖性的和统计学显著的抑制(数据显示3 mg/kg治疗组)。在0.1mg/kg至3mg/kg的剂量下,接受rad1901治疗的大鼠中的骨矿物质密度是完整的,与e2治疗组无统计学显著差异。
[0313]
表4
[0314]
rad1901对卵巢切除大鼠中的bmd的影响。
a
[0315][0316]
a
在利用溶媒、e2(0.01mg/kg)或rad1901(3mg/kg)每天一次(n=20/处理组)开始治疗前,对成年雌性大鼠进行假手术或卵巢切除手术。在基线和治疗4周后通过双发射x射线吸收测量法测量bmd。数据表示为平均值
±
sd。相对于相应的ovx 溶媒对照,*p<0.05。bmd为骨矿物质密度;e2为β雌二醇:ovx为去卵巢切除:veh为溶媒。
[0317]
远端股骨的micro
‑
ct分析(表5)证实,与假手术动物相比,卵巢切除术诱发多种关键微观结构参数的显著变化。这些变化与骨质减少相一致,包括骨体积减少、骨小梁数、厚度和密度减小、以及骨小梁分离增加。与用rad1901治疗后观察到的保持骨矿物质密度的结果一致,在关键微观结构参数中观察到了明显的骨小梁结构保存(表5)
[0318]
表5
[0319]
rad1901对卵巢切除大鼠的股骨微结构的影响
a
[0320][0321]
a
在利用溶媒、e2(0.01mg/kg)或rad1901(3mgkg)每天一次(n=20处理组)开始治疗前,对成年雌性大鼠进行假手术或卵巢切除手术。在4周后,使用微计算机断层扫描技术
评价骨微结构。数据表示为平均值
±
sd。相对于相应的ovx 溶媒对照,*p<0.05。abd为表观骨密度;bvtv为骨体积密度; connd为连接密度;e2为β雌二醇;ovx为卵巢切除;tbn为骨小梁数;tbth 为骨小梁厚度;tbsp为骨小梁间距;veh为溶媒。
[0322]
实施例iv(b):在健康的绝经后妇女中的rad1011期剂量递增研究
[0323]
在1期研究中,在44名健康的绝经后女性中评价安全性、耐受性和药代动力学。没有观察到剂量限制性毒性(dlt),未确定耐受剂量(mtd)。在所测试的剂量范围内,血浆暴露量随剂量增加而按比例增加。
[0324]
受试者
[0325]
招募44名健康的绝经后女性作为1期研究的受试者。受试者的闭经时间为至少12个月,血清fsh与绝经一致。受试者为40
‑
75岁,bmi为18.0
‑
30kg/m2。排除如下受试者:具有临床相关病理证据、中风风险增加或有静脉血栓事件史,或在进入临床研究中心前不到14天内使用伴随用药(允许在至多3天前使用扑热息痛)。
[0326]
给药
[0327]
在少量早餐之后利用安慰剂或至少一剂(口服,每日一次)治疗受试者7天,剂量分别为200mg、500mg、750mg和1000mg。在1期研究中招募的44名健康的绝经后女性的关键基线人口统计学汇总在表6中。
[0328]
表6
[0329]
rad1901 1期剂量递增研究的关键基线人口统计学
[0330][0331]
治疗紧急不良事件(teae)
[0332]
记录teae,并将最频繁的(>10%的具有任何相关teae的总活性组中的患者)不良事件(ae)汇总在表7中,“n”是在给定类别中具有至少一个治疗相关ae的受试者的数目(ae 根据不良事件通用标准(ctcae)v4.0进行分级),并且经历同一优先项目的多种情形的任何患者都只以最严重的等级计算一次。没有观察到剂量限制毒性,未确定最大耐受剂量 (mtd)。
[0333]
表7
[0334]
在rad1901 1期剂量递增研究中最频繁(>10%)的治疗相关ae
[0335][0336]
药代动力学评估
[0337]
在研究期间采集一系列血液样品用于分析血浆中的rad1901。通过留置iv导管或直接静脉穿刺采集每个5ml的血液样品至包含k3‑
edta作为抗凝血剂的试管中。在治疗第5天达到稳定状态。评价rad1901的几何平均(geo
‑
mean)血浆浓度
‑
时间曲线。作为实例,将研究第7天(n=35)利用rad1901(200、500、750或1000mg)治疗的组的血浆药代动力学结果提供在表8和图19中。中位t
1/2
在37.5
‑
42.3小时之间(表8)。在施用多剂rad1901 后,中值t
max
在给药后3
‑
4小时之间。
[0338]
表8
[0339]
rad1901 1期剂量递增研究中的药物动力学参数(第7天)
[0340][0341]
实施例v(a)
‑
1.使用选用的公开er结构对rad1901
‑
erα结合进行建模。
[0342]
除非另外指定,当通过其杆状模型显示结构时,将键的每个末端着色为与其所连接的原子相同的颜色,其中灰色是碳,红色是氧,蓝色是氮,白色是氢。
[0343]
通过仔细评价,从96个公开的模型中选择14个与几种er配体复合的erα配体结合结构域(lbd)的公开结构(即模型)。14个模型中的一个模型是3ert(与4
‑
羟基他莫昔芬(oht) 结合的人erα lbd)。oht是他莫昔芬的活性代谢物和第一代serm,其在乳房组织中起拮抗剂的作用。
[0344]
在3ert(图20和21)中,erα结合位点采用形成包括螺旋3(h3)、螺旋5(h5)和螺旋11(h11)的疏水口袋的三层“螺旋三明治”(图20)。图21中的虚线框表示结合位点和结合位点内重要或受oht结合影响的残基。oht通过将h12置换到lxxll共激活剂结合的位点而起拮抗
剂的作用。oht占据l540正常填充的空间并修饰螺旋11的c端子上的四个残基(g521、h524、l525和m528)的构象。oht也与d351形成盐桥,导致电荷中和。
[0345]
将另外十三个erα lbd
‑
er配体模型与3ert进行比较。表10中汇总了它们残基构型中的差异。十四个模型(图22)的erα结构叠加显示这些结构在残基e380、m421、g521、 m522、h524、y526、s527、m528、p535、y537、l540及其各种组合处明显不同。
[0346]
表10
[0347]
er
‑
α lbd
‑
拮抗剂复合物与3ert的残基构型差异
[0348][0349]
在表11中汇总了十四种模型中的任一对模型的均方根偏差(rmsd)计算。当在表11中汇总了十四种模型中的任一对模型的均方根偏差(rmsd)计算。当时认为结构是重叠的。表11显示,所有十四个模型的rmsd均使用条件格式分析表明,1r5k和3uuc与其他模型最不相似(分析未显示)。因此,将1r5k和3uuc视为独特的、独立的结构类群来考查。
[0350]
表11
[0351]
通过rmsd计算评价er
‑
α lbd
‑
拮抗剂复合物的结构重叠:
[0352][0353]
在表12汇总了十四种模型中的由配体结合的erα残基。表12还显示了erαlbd
‑
拮抗剂复合物中的ec
50
。在十四个模型中,十三个模型显示配体和e353之间的氢键相互作用;十二个模型显示配体与f404之间的π相互作用;五个模型显示配体和d351之间的氢键相互作用;六个模型显示配体和h524之间的氢键相互作用;四个模型显示配体和r394之间的氢键相互作用;以及一个模型(3uuc)显示配体和t347之间的相互作用。
[0354]
表12
[0355]
在er
‑
α lbd
‑
拮抗剂复合物中的配体结合的分析
[0356][0357]
使用十四个模型中的每个模型对接1000个化合物加配体的随机文库,(所述模型
已公开为具有已知的拮抗剂),以确定模型是否能够识别和优先考虑已知的拮抗剂。如果模型能够识别已知的拮抗剂,则确定该模型能够预测其自己公开的配体的构型。然后计算ef
50
来量化模型强度,以查看它如何比随机选择要好多少。rad1901对接在选定的模型中(例如,图 23a&b
‑
27a&b)。确定所公开配体和rad1901在模型中的对接评分。还确定ec
50
。目视检查rad1901显示它“服从”所显示的与1r5k、1sj0、2jfa、2bj4和2ouz中的公开配体的相互作用。没有发现空间冲突。在某些实施方案中,例如在1r5k和2bj4中,rad1901具有比公开的配体更高的对接评分。
[0358]
表13汇总了9个模型(1err、3ert、3ucc、2iok、1r5k、1jj0、2jfa、2bj4和2ouz) 的评估结果。
[0359]
表13
[0360]
rad1901对接的模型评估
[0361][0362]
1err和3ert不能预测其结晶配体的正确构型。rad1901没有对接在3ucc中。 2iok
‑
rad1901中的四氢萘胺以非传统方式结合。
[0363]
模型1r5k、1sj0、2jfa、2bj4和2ouz之间的主要差别是螺旋11的c
‑
末端中的残基 (g521
‑
m528)。
[0364]
图23a&b显示了rad1901
‑
1r5k(a)和gw5
‑
1r5k(b)的建模。rad1901利用氢键相互作用结合至e353、r394和l536;并利用p
‑
相互作用结合至f404。
[0365]
图24a&b显示了rad1901
‑
1sj0(a)和e4d
‑
1sj0(a)的建模。rad1901利用氢键结合至e353和d351;并利用p
‑
相互作用与f404结合。
[0366]
图25a&b显示了rad1901
‑
2jfa(a)和ral
‑
2jfa(b)的建模。rad1901利用p
‑ꢀ
相互作用与f404结合。
[0367]
图26a&b显示了rad1901
‑
2bj4(a)和oht
‑
2bj4(b)的建模。rad1901利用氢键相互作用结合至e353和r394;并且利用p
‑
相互作用与f404结合。
[0368]
图27a&b显示rad1901
‑
2iok(a)和iok
‑
2iok(b)的建模。rad1901利用氢键相互作用与e353、r394和d351结合;并且利用p
‑
相互作用与f404结合。
[0369]
模型中的公开配体具有以下结构:
[0370][0371]
gw5,(2e)
‑3‑
{4
‑
[(1e)
‑
1,2
‑
二苯基丁
‑1‑
烯基]苯基}丙烯酸
[0372][0373]
e4d,(2s,3r)
‑2‑
(4
‑
(2
‑
(哌啶
‑1‑
基)乙氧基)苯基)
‑
2,3
‑
二氢
‑3‑
(4
‑
羟基苯基)苯并[b][1,4]氧硫杂环己二烯
‑6‑
醇
[0374][0375]
oht,4
‑
羟基他莫昔芬
[0376][0377]
iok,n
‑
[(1r)
‑3‑
(4
‑
羟基苯基)
‑1‑
甲基丙基]
‑2‑
(2
‑
苯基
‑
1h
‑
吲哚
‑3‑
基)乙酰胺
[0378]
实施例v(a)
‑
2.erα与rad1901和氟维司群的诱导契合对接(ifd)
[0379]
通过对5种erα晶体结构1r5k、1sj0、2jfa、2bj4和2ouz的ifd分析来进一步优化 erα中rad1901的结合构象。ifd分析考虑了适应其正确结合构象的受体灵活性(在配体结合后)。
[0380]
通过寻找作为围绕可旋转键的旋转的函数的局部最小值来产生每个配体(例如rad1901 和氟维司群)的不同构象文库。rad1901的文库有25种不同的构象。
[0381]
制备五种erα晶体结构并使其最小化。使用公开的x射线结构中的对应配体来定义erα结合口袋。
[0382]
将rad1901构象对接到所制备的erα结构中,其中使它们诱导侧链或主链移动到位于结合口袋中的残基。这些移动使得erα能够改变其结合位点,使得其更紧密地顺应rad1901 构象的形状和结合模式。在一些实施例中,在ifd分析中允许受体结构中的主链少量松弛和显著的侧链构象变化。
[0383]
使用经验评分函数来近似获得配体结合自由能以提供对接评分或g评分。g评分也被称为glidescore,在该实施例中其可以与对接得分互换使用。对接评分是结合亲和力的
估计值。因此,对接评分的值越低,则配体与其受体结合“越好”。对接评分为
‑
13至
‑
14对应于非常良好的结合相互作用。
[0384]
将分别用1r5k、1sj0、2jfa、2bj4和2ouz进行的ifd分析得到的rad1901构象进行叠加以显示它们的差异(图28
‑
30a&b,显示为杆状模型)。每个rad1901构象中的所有键在图28、29和30a中均用相同的颜色显示。
[0385]
由1r5k(蓝色)和2ouz(黄色)的ifd分析得到的rad1901构象在正面具有rad1901 的n
‑
苄基
‑
n
‑
乙基苯胺基团(图28)。利用2bj4(绿色)和2jfa(粉色)进行ifd分析得到的rad1901构象在背面具有rad1901的n
‑
苄基
‑
n
‑
乙基苯胺基团(图29)。利用2bj4(绿色)、2jfa(粉色)和1sj0(棕色)进行ifd分析得到的rad1901构象与通过其叠加所示的构象(图30a和30b)非常相似。rad1901 ifd对接评分汇总在表14中。
[0386]
表14
[0387]
rad1901与1r5k、1sj0、2ifa、2bj4和2ouz的诱导契合对接评分
[0388]
er
‑
α晶体结构rad1901ifd对接评分1r5k
‑
14.11sjo
‑
13.12jfa
‑
13.92bj4
‑
13.82ouz
‑
13.4
[0389]
利用2bj4进行的rad1901 ifd显示与e353和d351的氢键相互作用和与f404的π
‑
相互作用(图31a
‑
31c)。图31a显示了结合位点内适合于氢键受体基团(红色)、氢键供体基团(蓝色)和疏水性基团(黄色)的区域。在图31a和31b中,浅蓝色是rad1901的碳。图32a
‑
32c显示了利用2bj4进行的rad1901的ifd的蛋白质
‑
表面相互作用。图32a和32b 是正视图,图32c是侧视图。rad1901的分子表面在图32a中为蓝色,而在图32c中为绿色。图32b和32c是erα的溶剂可及表面的静电图示,其中红色代表电负性,蓝色代表电正性。
[0390]
如上所述利用2bj4对氟维司群进行类似的ifd分析。氟维司群
‑
2bj4 ifd得到
‑
14.945的 g评分,并显示与e353、y526和h524的氢键相互作用以及与f404的π
‑
相互作用(图 33a
‑
33c)。图33a显示了结合位点内适合氢键受体基团(红色)、氢键供体基团(蓝色)和疏水性基团(黄色)的区域。在图33a中,浅蓝色是rad1901的碳。
[0391]
图34a和34b显示了通过ifd对接在2bj4中的rad1901和氟维司群,两者与f404具有π
‑
相互作用并且与e353具有氢键相互作用。此外,rad1901与d351具有氢键相互作用 (蓝色表示rad1901分子表面,图34b),而氟维司群与y526和h524具有氢键相互作用。在图35a和35b中显示了与rad1901和氟维司群对接的2bj4的叠加。在图35a中,绿色代表氟维司群分子表面,蓝色代表rad1901分子表面。在图35b中,棕色结构是氟维司群,蓝色结构是rad1901。
[0392]
实施例example v(a)
‑
3.选用的erα突变的建模评估。
[0393]
评估几种erα突变对c末端配体结合结构域的影响。所评估的具体erα突变是y537x 突变体,其中x是s、n或c;d538g;和s463p。
[0394]
y537位于螺旋12中。它可以在磷酸化后调节配体结合、同源二聚化和dna结合,并且可以允许erα脱离磷酸化介导的控制并提供具有潜在选择性致瘤优势的细胞。此外,它可能会引起构象变化,从而使受体具有组成活性。
[0395]
y537s突变有利于转录活性闭合的口袋构象,而不管是否被配体占据。闭合但未被占据的口袋可以解释erα的组成活性(carlson等人,biochemistry 36:14897
‑
14905(1997))。ser537 与asp351形成氢键相互作用,导致螺旋11
‑
12环的构象改变,并且导致leu536包埋在溶剂不可到达的位置处。这可能促进y537s突变蛋白的组成性活性。y537s表面突变对lbd口袋的结构没有影响。
[0396]
y537n常见于erα阴性转移性乳腺癌。在该位点的突变可能使erα脱离磷酸化介导的控制,并提供具有潜在的选择性致瘤优势的细胞。具体而言,y537n的替换诱发erα中的构象变化,其可模拟激素结合,不影响受体二聚化的能力,但为受体提供组成转活功能(zhang 等人,cancer res 57:1244
‑
1249(1997))。
[0397]
y537c具有与y537n相似的作用。
[0398]
d538g可通过稳定活性构象和非活性构象来改变整个能量全景,但是更优选活性构象。这可能在不存在激素的情况下导致这种突变体具有组成活性,如在激素耐药性乳腺癌中观察到的(huang等人,“a newfound cancer activating mutation reshapes the energy landscape of estrogen
‑
binding domain,”j.chem.theory comput.10:2897
‑
2900(2014))。
[0399]
预期这些突变都不会影响配体结合结构域,也不会特异性阻碍rad1901结合。y537和 d538可引起构象变化,其导致与配体结合无关的组成受体活化。
[0400]
实施例v(b).野生型和lbd突变型erα构建体与rad1901和其他化合物的体外结合测定
[0401]
野生型(wt)和lbd突变型erα构建体与rad1901的体外结合测定显示,rad1901 结合至突变型erα的亲和力与结合至wt erα的亲和力相似。
[0402]
通过表达和纯化相应的lbd残基302
‑
552来制备wt和lbd突变型erα的构建体,所述lbd残基302
‑
552具有n
‑
末端硫氧还蛋白和通过tev蛋白酶切割的6xhis标签。
[0403]
根据制造商(polar screen,invitrogen)的说明,利用2nm荧光酮、100nm wt或lbd 突变型erα的构建体,使用荧光偏振(fp)来测定受试化合物(rad1901、氟维司群、巴多昔芬、雷洛昔芬、他莫昔芬和azd9496)与erα的结合。每组进行两次测试,并测试一种测试化合物来测定不同erα构建体的ic
50
(图36显示rad1901结合测定)。
[0404]
如上所述,前文仅意在举例说明本发明的几种实施方案。上文讨论的具体修改方案不应被解释为限制本发明的范围。本领域技术人员明显可知,在不脱离本发明范围的情况下可以做出各种等同方案、变化方案和修改方案,并且应当理解,这样的等同实施方案也被包括在本文中。本文引用的所有参考文献均通过引用并入本文,如同在本文中完全记载一样。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。