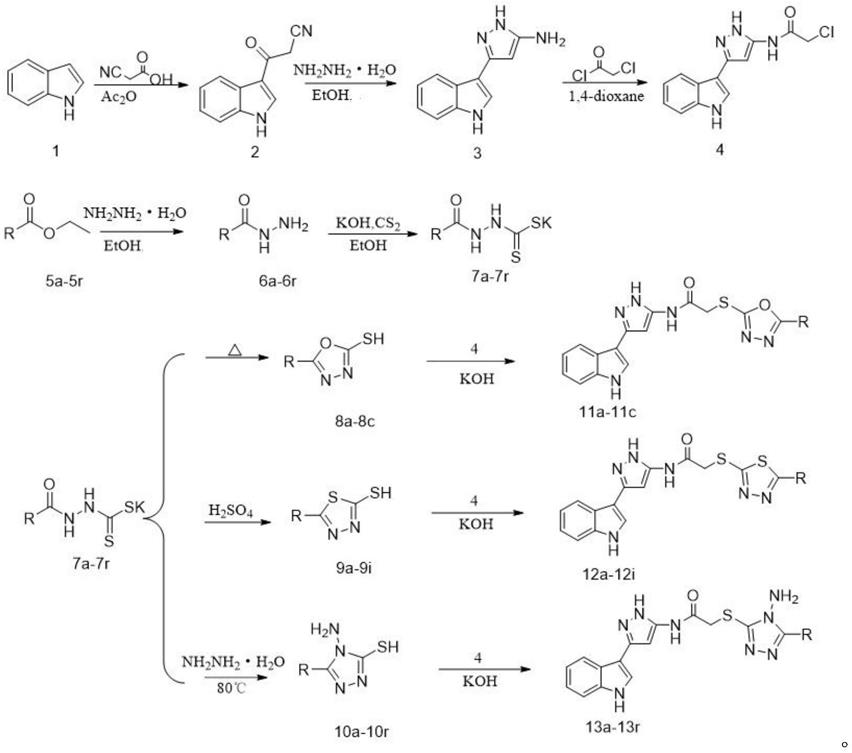
1.本发明涉及药物化学技术领域,尤其是一类含二唑、三唑及吡唑结构单元的吲哚衍生物结构及其应用。
背景技术:
2.由于环境等诸多因素的影响,恶性肿瘤的发病率逐年上升,癌症已成为人类健康的第二大杀手。癌症对人类健康的严重威胁尚未得到有效控制。因此,研究开发低毒高效的新型抗癌药物至关重要。
3.吲哚结构普遍存在于天然及合成的药物中,具有抗肿瘤、抗氧化、抗菌等生物活性。在吲哚3位引入不同的取代基,进行结构的修饰与改造一直是药物研究的热点。芦卡帕利(rucaparib)、甲磺酸奥希替尼(osimertinib)、帕比司他(panobinostat)、褪黑素(melatonin)等已上市的抗肿瘤药物都是以吲哚为骨架,治疗效果良好且愈后副作用小,应用前景十分广阔。
4.此外,吡唑及其衍生物也具有较好的生物活性,如抗痉挛、抗炎、抗菌、抗肿瘤、抗抑郁等,广泛用于医药领域。为了寻找高效低毒的抗肿瘤化合物,本发明以3
‑
吡唑吲哚为骨架,巯基为连接链,将可能提高目标化合物生物活性的二唑或三唑结构引入到此体系中,合成含二唑或三唑结构单元的3
‑
吡唑吲哚类化合物,考察其抗肿瘤活性,为新型抗肿瘤药物的研发和创制提供一定的参考价值。
5.吲哚类衍生物的生物活性研究进展如下:
6.2016年zhang等[zhang,d t.;wang,g t.;zhao,g l.;xu,w r.;huo,l y.synthesis and cytotoxic activity of novel 3
‑
(1h
‑
indol
‑3‑
yl)
‑
1h
‑
pyrazole
‑5‑
carbohydrazide derivatives[j].eur.j.med.chem.,2011,46(12):5868
‑
5877.]设计合成了一系列3
‑
吡唑吲哚类衍生物,并采用mtt法检测了所有化合物对4种人肿瘤细胞株的抗增殖活性。结果表明大部分化合物对hepg
‑
2(人肺癌细胞)、bgc823(人胃癌细胞)、及bt474(人乳腺癌细胞)的细胞毒性均高于阳性对照药5
‑
氟尿嘧啶。化合物对bt474细胞的抑制作用最高可达到对照药的52倍,ic
50
(半数最大抑制浓度)为1.39μmol/l。
[0007]
2017年kamath等[kamath,p r.;joseph,m m.;ajees,a a.;bisindole
‑
oxadiazole hybrids,t3p((r))
‑
mediated synthesis,and appraisal of their apoptotic,antimetastatic,and computational bcl
‑
2 binding potential[j].j.biochem.mol.toxic.,2017,31(11):e21962.]开发了一种绿色、高效的方法共合成了一系列含噁二唑的新型双吲哚衍生物。某些化合物对mcf
‑
7细胞具有选择性毒性,ic
50
最低为8.45μmol/l,表现出比长春新碱更强的抗增殖作用。进一步机制研究表明,这类化合物还可引发mcf
‑
7细胞的g1期细胞周期阻滞;并通过激活caspase
‑
2、caspase
‑
9(半胱氨酸蛋白),及抑制抗凋亡蛋白bcl
‑
2的活性诱发该细胞程序性死亡。
[0008]
2019年bhale等人[bhale,p s.;bandgar,b p.;dongare,s b.;shringare,s n.;sirsat,d m.;chavan,h v.ketene dithioacetal mediated synthesis of 1,3,4,5
‑
[0018]
r选自任意取代或未取代的芳基、任意取代或未取代的杂环。
[0019]
进一步优选地,r选自取代或未取代的芳基、取代或未取代的杂环;
[0020]
优选地,r选自取代或未取代的c6‑
c
15
芳基、取代或未取代的c6‑
c
10
杂环中的一个或一个以上,其中,所述取代的指的是被c1‑
c6烷氧基、氨基、羟基、卤素的一个或一个以上取代;
[0021]
更优选地,r选自取代或未取代的苯基,取代或未取代的吡啶及、取代或未取代的吡咯基、取代或未取代的吲哚基。其中,所述取代的指的是被c1‑
c6烷氧基、氨基、羟基、卤素中的一个或一个以上取代;
[0022]
最优选地,r选自苯基、2
‑
氟苯基、2
‑
氯苯基、2
‑
溴苯基、2
‑
羟基苯基、4
‑
氟苯基、4
‑
氯苯基、4
‑
溴苯基、4
‑
氨基苯基、4
‑
甲氧基苯基、3,5
‑
二氯苯基、3,4,5
‑
三甲氧基苯基、吡啶基、吲哚基、吡咯基。所述的含二唑、三唑及吡唑结构单元的吲哚化合物或其立体异构体、或其盐或其溶剂化物,选自下述化合物:
[0023]
[0024]
[0025][0026]
本发明还提供了一种制备所述的含二唑或三唑及吡唑结构单元的吲哚化合物或其立体异构体、或其盐或其溶剂化物的中间体化合物,如下所示:
[0027][0028]
本发明还提供了所述的含二唑或三唑及吡唑结构单元的吲哚化合物或其立体异构体、或其盐或其溶剂化物的制备方法,其包括下述步骤:
[0029][0030]
本发明还提供了一种组合物,其含有所述的化合物或其立体异构体、或其盐或其溶剂化物,以及肿瘤治疗上可用的助剂或抗肿瘤药物制剂;优选地,所述组合物的剂型选自丸剂、颗粒剂、片剂、口服液、注射剂、散剂等。
[0031]
所述的化合物或其立体异构体、或其盐或其溶剂化物,或所述的组合物可用于肿瘤的治疗,优选地,所述肿瘤为肺癌、肝癌、前列腺癌、结肠癌、宫颈癌或白血病;更优选地,所述肿瘤为肺癌、肝癌、前列腺癌及白血病。
[0032]
本发明还提供了一种制备抗肿瘤药物的方法。使所述的化合物或其立体异构体、或其盐或其溶剂化物,或所述的组合物作用于肿瘤细胞或其生活环境;优选地,所述肿瘤细胞为肝癌细胞、肺癌细胞、前列腺癌细胞、白血病细胞。
[0033]
本发明还提供了一种制备抗肿瘤药物的方法,包括使植物与所述的化合物或其立体异构体、或其盐或其溶剂化物,或所述的组合物接触的方法步骤。
[0034]
此处用到的术语“取代的”指的是在指定原子或基团上的任意一个或多个氢原子以选择的指定基团取代,前提是不超过指定原子的一般化合价。如果没有其它说明,取代基命名至中心结构。例如,可以理解的是当(环烷基)烷基是可能的取代基,该取代基至中心结构的连接点是在烷基部分中。此处使用的环双键是形成于两个临近环原子之间的双键(如c=c、c=n或n=n)。当提到取代时,特别是多取代时,指的是多个取代基在指定基团上的各个位置上取代,如二氯苯基指的是1,2
‑
二氯苯基、1,3
‑
二氯苯基和1,4
‑
二氯苯基。
[0035]
取代基和或变量的组合是允许的,仅当这些组合产生稳定的化合物或有用的合成
中间体。稳定的化合物或稳定结构暗示所述化合物以有用的纯度从反应混合物分离出来时是足够稳定的,随之配制形成有效的治疗试剂。优选地,目前所述化合物不包含n
‑
卤素、s(o)2h或s(o)h基。
[0036]
术语“芳基”指的是在环部分具有6到12个碳原子的单环或双环芳香烃基,如苯基,每个可被取代的。
[0037]
术语“卤素”或“卤素原子”指的是氯、溴、氟和碘。
[0038]
术语“杂芳基”指的是取代和非取代芳香5或6元单环基团,9
‑
或10
‑
元双环基团,和11到14元三环基团,在至少一个环中具有至少一个杂原子(o,s或n),所述含杂原子的环优选具有1、2或3个选自o、s和n中的杂原子。含杂原子的杂芳基的每个环可含一个或两个氧或硫原子和/或由1到4个氮原子,前提是每个环中杂原子的总数是4或更少,且每个环中具有至少一个碳原子。构成双环和三环基团的稠合环可仅含有碳原子,并可以是饱和、部分饱和或不饱和。氮和硫原子可被氧化,而且氮原子可被季铵化。双环或三环的杂芳基必须包括至少一个全芳香环,其它稠合环可为芳香性或非芳香性的。杂芳基可链接在任何环的任何可利用的氮或碳原子上。当化合价允许,如果所述其它环是环烷基或杂环,其另外任选以=o(氧)取代。
[0039]
示例性单环杂芳基包括吡咯基、吡啶基及其类似物。
[0040]
如果没有其它说明,本发明的化合物理解为包括游离态和其盐。术语“盐”表示以无机和/或有机酸和碱形成酸式和/或碱式盐。另外,术语“盐可包括两性离子(内盐),如当式i化合物含有碱性片段如胺或吡啶或咪唑环,和酸式片段如羧酸。药物上可接受的(即非毒性、生理学上可接受的)盐是优选的,如可接受的金属和胺盐,其中阳离子没有显著贡献毒性或盐的生物活性。然而,其它盐可是有用的,如在制备过程中采用分离或纯化步骤,因此也包含于本发明范围中。
[0041]
当提到取代基时,如卤素、芳基、杂芳基、羟基、烷氧基、氨基时,或这些取代基具体的为某个具体的卤素、芳基、杂芳基、烷氧基、羟基、氨基时,指的是一个到三个上述取代基。如甲基苯基指的是一个到三个甲基取代的苯基。
[0042]
通过采用上述技术方案,本发明以3
‑
吡唑吲哚结构为基础,合成一系列含二唑或三唑结构单元的吲哚类化合物,且发现该化合物对某些肿瘤细胞具有良好的抑制作用,针对抗肿瘤细胞[如a549(人非小细胞肺癌细胞)、hep
‑
g2(人肝癌细胞)、pc
‑
3(人前列腺癌细胞)、k562(人慢性髓原白血病细胞)]均具有特别好的抑制效果,ic
50
最低可达到10nm,为新型高活性抗肿瘤药物的研发和创制提供重要的科学基础。
实施例
[0043]
下面通过实施例对本发明作进一步说明。应该理解的是,本发明实施例所述方法仅仅是用于说明本发明,而不是对本发明的限制,在本发明的构思前提下对本发明制备方法的简单改进都属于本发明的范围。实施例中用到的所有原料和溶剂均为市售产品。
[0044]
实施例1:3
‑
(1h
‑
吲哚
‑3‑
基)
‑3‑
氧代
‑
丙腈的制备
[0045]
在250ml圆底烧瓶中依次投入吲哚(10.00g,85.36mmol)、氰基乙酸(10.17g,119.50mmol)、100ml醋酸酐,40℃回流4h。反应结束后,静止冷却至室温,抽滤,甲醇洗涤2~3次,干燥后得白色固体,收率77.3%.
[0046]
实施例2:3
‑
(1h
‑
吲哚
‑3‑
基)
‑
1h
‑
吡唑
‑5‑
胺的制备
[0047]
于500ml的圆底烧瓶中依次加入3
‑
(1h
‑
吲哚
‑3‑
基)
‑3‑
氧代
‑
丙腈(5.00g,27.14mmol)和80%的水合肼(6.79g,169.38mmol),再加入约250ml的无水乙醇作溶剂,80℃回流搅拌,tlc监测反应体系。反应结束后,静置,自然冷却,将反应混合物减压浓缩至稠状,冷却,抽滤,乙酸乙酯洗涤2~3次,得到棕色固体,产率54.1%。
[0048]
实施例3:n
‑
(3
‑
(1h
‑
吲哚
‑3‑
基)
‑
1h
‑
吡唑
‑5‑
基)
‑2‑
氯乙酰胺的制备
[0049]
在50ml的圆底烧瓶中分别加入3(2.00g,10.09mmol),无水碳酸钠(1.07g,10.69mmol),加入一部分1,4
‑
二氧六环,常温搅拌。再将氯乙酰氯(1.14g,10.09mmol)融入2ml 1,4
‑
二氧六环(共计20ml)中,再逐滴加入圆底烧瓶中,继续搅拌1h。tlc监测反应结束后,另取烧杯加入100ml水,加nacl,再倒入碎冰,倒入反应混合物,搅拌至出现沉淀,静置,减压过滤,水洗,得灰绿色固体。干燥后乙醇重结晶得4。白色固体,产率18%。
[0050]
实施例4:3,5
‑
二氯苯酰肼的制备
[0051]
在25ml的圆底烧瓶中加入3g 3,5
‑
二氯苯甲酸乙酯(1mmol
×
1.5ml水合肼),水合肼约21ml,80℃回流反应5h,放置冰箱下层过夜,过滤,得白色固体,收率81%。
[0052]
实施例5:4
‑
氨基
‑5‑
(3,5
‑
二氯苯基)
‑
4h
‑
1,2,4
‑
三唑
‑3‑
硫醇的制备
[0053]
将3,5
‑
二氯苯甲酰肼(13.69mmol),氢氧化钾(12.68mmol)投入100ml圆底烧瓶中,加入32ml乙醇常温搅拌,逐步滴加cs2(12.68mmol),继续搅拌5h,过滤,乙醇洗涤数次,干燥得黄白色固体(1mmol
×
3ml水合肼),80℃回流5h,反应混合物倒入冰水中,用盐酸调ph到2
‑
3,静置,过滤,水洗,干燥得白色固体,收率20%。
[0054]
实施例6:n
‑
(3
‑
(1h
‑
吲哚
‑3‑
基)
‑
1h
‑
吡唑
‑5‑
基)
‑2‑
((4
‑
氨基
‑5‑
(3,5
‑
二氯苯基)
‑
4h
‑
1,2,4
‑
三唑
‑3‑
基)硫)乙酰胺
[0055]
25ml圆底烧瓶中依次加入4
‑
氨基
‑5‑
(3,5
‑
二氯苯基)
‑
4h
‑
1,2,4
‑
三唑
‑3‑
硫醇(1.49mmol),koh(2.99mmol),再加入10ml无水乙醇,室温搅拌10min后加入n
‑
(3
‑
(1h
‑
吲哚
‑3‑
基)
‑
1h
‑
吡唑
‑5‑
基)
‑2‑
氯乙酰胺(1.49mmol),继续反应1h。反应结束后,另取一个烧杯中依次加入60ml冰水,边搅边倒入反应混合物,静置,抽滤,乙醇重结晶得白色固体,收率45.6%。
[0056]
其他目标化合物合物采用相应的原料或取代基,参照实施例4步及5步骤合成。
[0057]
合成的部分含二唑、三唑及吡唑结构单元的吲哚类化合物的结构及核磁共振氢谱、核磁共振碳谱相关表征数据如表1所示,物化性质如表2所示。
[0058]
表1部分化合物的核磁共振氢谱和碳谱数据
[0059]
[0060]
[0061]
[0062]
[0063]
[0064]
[0065][0066]
表2部分目标化合物的理化性质
[0067]
[0068][0069]
药理实施例1:
[0070]
通过mtt法测定化合物对a549(人非小细胞肺癌细胞)、pc
‑
3(人前列腺癌细胞)、hepg2(人肝癌细胞)、k562(人慢性髓系白血病细胞)及nrk
‑
52e(大鼠肾小管导管上皮细胞)的半数抑制浓度ic
50
值。
[0071]
化合物的体外抗肿瘤活性测定(初筛)
[0072]
接种细胞步骤(适用于贴壁细胞、悬浮细胞)
[0073]
1.选取细胞:选择根据细胞生长曲线,选取对数生长期的细胞;
[0074]
2.消化细胞:用0.25%胰蛋白酶消化单层培养细胞,用含10%fbs的dmem/1640培养液配成单个细胞悬液;
[0075]
3.细胞计数:先用无水乙醇浸泡细胞计数板及血盖片;取10μl细胞悬液,滴加在血盖片边缘,使悬液充满血盖片和计数板之间,不能溢出血盖片也不能溢入两侧的玻璃槽内;在显微镜下用l0
×
物镜观察计数板四角大方格中的细胞数。压线细胞只计左侧和上方的,不计右侧和下方的;按照公式计算:(4个大格细胞数之和/4)
×
104
×
细胞液体积=3
×
104
×
所需总的细胞混液(一般密度为3
×
104,也可根据细胞性质不同换成其他密度计算);
[0076]
4.呈色及测值(适用于贴壁细胞)
[0077]
加入mtt溶液(5mg/ml,10%mtt)20μl,孵育4h,终止培养;采用吸液的方式,将培养板内的溶液吸出;每孔加入150μl dmso溶解甲臜颗粒,摇床震荡10min(150rpm/min),混匀,使用酶标仪,在490、570nm测定od值,记录结果。使用软件分析结果。
[0078]
抑制率=1
‑
(加药组od值
‑
空白组od值)/(阴性组od值
‑
空白组od值)
×
100%;
[0079]
空白对照孔:200μl培养液 150μl dmso;
[0080]
阴性对照孔:180μl细胞混悬液 20μl dmso 20μlmtt 150μl dmso;
[0081]
加药孔:180μl细胞混悬液 不同浓度的药物20μl 20μlmtt 150μl dmso;
[0082]
选取抑制率接近50%的化合物进行复筛,待测化合物分别配制成10μm、5μm、2.5μ
m、1.25μm、0.625μm溶液,每孔加20μl,做5个复孔。其余处理步骤同化合物的初筛。本发明实施例辅以说明本发明的技术方案,但实施例的内容并不局限于此,目标化合物实验结果如表3所示。
[0083]
表3目标化合物对四种肿瘤细胞的抑制活性
[0084][0085][0086]
采用mtt法评估了所有化合物对人非小细胞肺癌细胞a549、人前列腺癌细胞pc
‑
3、人肝癌细胞hepg2及慢性骨髓性白血病细胞k562的细胞毒性,并测试了化合物对大鼠肾细胞nrk
‑
52e的细胞毒性以评估其安全性与选择性。结果表明,这类化合物普遍对k562细胞更敏感。大部分化合物对a549、pc
‑
3、k562、hepg2细胞表现出比阳性对照药5
‑
氟尿嘧啶更好的肿瘤抑制活性,抑制率最高可达到99.57%,ic
50
值最低为10nm。部分化合物的ic50值可达到纳摩尔级,其中化合物11a、11c、12a、12b、12e对a549细胞的ic50分别为0.47
±
0.07、0.31
±
0.06、0.43
±
0.10、0.12
±
0.05、0.83
±
0.09μm;化合物12a、12b、12e对pc
‑
3细胞的ic
50
分别0.14
±
0.01、0.85
±
0.72、0.85
±
0.43μm;化合物11a、11c、12b、12d、12e对hepg2细胞的ic
50
分别为0.57
±
0.15、0.63
±
0.25、0.21
±
0.07、0.73
±
0.15、0.80
±
0.09μm;对k562细胞而言,化合物11a、11b、11c、12a、12b、12c、12e、12f、13h、13l的活性较好,ic
50
分别为0.22
±
0.06、0.28
±
0.01、0.14
±
0.06、0.05
±
0.02、0.01
±
0.01、0.18
±
0.02、0.19
±
0.09、0.99
±
0.52、0.06
±
0.02、0.36
±
0.10μm。化合物12b、12e对这四株人肿瘤细胞的ic
50
值均在纳摩尔范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。