1.本发明属于催化剂技术领域,具体涉及一种铋基催化剂的制备方法和应用。
背景技术:
2.随着现代工业的迅速发展,传统化石能源的消耗量持续上升,化石能源的储量日益减少,而其燃烧所带来的环境污染问题也日益严峻。开发可以替代传统化石燃料的新型绿色能源显得愈加重要。二氧化碳是化石燃料燃烧的主要产物之一,作为一种温室效应极强的气体,其过量排放会对生态环境产生显著的负面影响。因此,充分利用二氧化碳,将其转化为可再利用的燃料资源或具有工业生产意义的化学原料,能够在遏制气候变暖的同时,缓解当下的能源危机,具有重大意义。
3.二氧化碳稳定的化学性质使得其转化条件相对苛刻,现有的二氧化碳转化技术包括化学重整法、生物转化法,以及电化学还原法等。其中电化学还原法以电能作为直接能量来源,与其他的绿色能源体系(风能、太阳能等)具备协同发展的潜力,因此是最具备实际应用价值的二氧化碳转化技术。二氧化碳电化学还原过程具有将其转化为高附加值的化学品的潜力,然而二氧化碳较高的反应能垒和低选择性,使其转化过程需要高活性和高选择性的二氧化碳还原催化剂。
4.甲酸是二氧化碳电化学还原的产物之一,作为最基本的有机化工原料之一,其广泛应用于医药、农药、橡胶等工业领域。目前已经有大量针对于银、汞、铂、钯等金属作为催化材料应用于二氧化碳电催化还原制甲酸过程的相关研究。由于材料毒性和经济成本的限制,上述催化材料应用在大规模工业生产中的潜力十分有限。铋基催化剂具备相当的催化二氧化碳电还原制甲酸活性,与上述材料相比,其成本低、无毒性。在研究中发现,多种优化方法,如构筑纳米结构、开发异质结构等,对铋基催化材料的催化活性,尤其是其在二氧化碳电催化还原过程中所能达到的产甲酸法拉第效率,具有良好的提升作用。但是,目前的优化方法对甲酸法拉第效率的提升主要局限在某些较小的电压窗口内,实际工业应用价值较小。
5.因此,亟需提供一种co2还原制甲酸的催化剂,能在大电压窗口下表现出优良催化活性和法拉第效率。
技术实现要素:
6.本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明提出一种铋基催化剂的制备方法,制备的铋基复合催化剂能在
‑
0.7到
‑
1.15v的大电压窗口下表现出优良催化活性和法拉第效率。
7.本发明第一方面提供了一种铋基催化剂的制备方法。
8.具体的,一种铋基催化剂的制备方法,包括以下步骤:
9.将醇与乙酸混合得混合液,然后向所述混合液中加入铋盐进行溶剂热反应,反应完毕后过滤,将滤渣干燥,再煅烧,制得所述铋基催化剂。
10.优选的,所述醇为乙醇。
11.优选的,所述铋盐为硝酸铋。如五水硝酸铋。
12.优选的,所述铋盐的质量与所述混合液的体积的比为1mg:(10
‑
30)ml;进一步优选的,所述铋盐的质量与所述混合液的体积的比为1mg:(15
‑
25)ml。
13.优选的,所述醇与所述乙酸的体积比为(1
‑
4):1;进一步优选的,所述醇与所述乙酸的体积比为(1
‑
3):1。
14.优选的,所述溶剂热反应的温度为120
‑
180℃,所述溶剂热反应的时间为200
‑
600min;进一步优选的,所述溶剂热反应的温度为150
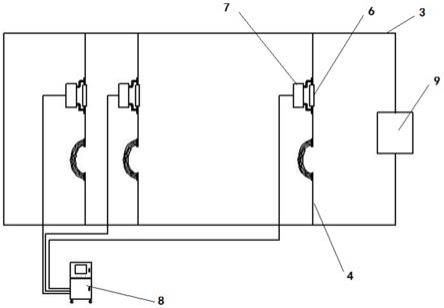
‑
180℃,所述溶剂热反应的时间为300
‑
500min。
15.优选的,所述煅烧的过程为:在氮氢混合气体下,以3
‑
10℃/min的升温速度升温至150
‑
300℃,然后保持10
‑
120min;进一步优选的,所述煅烧的过程为:在氮氢混合气体下,以3
‑
8℃/min的升温速度升温至180
‑
250℃,然后保持20
‑
100min。
16.优选的,所述氮氢混合气体包括氮气和氢气,所述氢气的体积占所述氮氢混合气体的体积的5%
‑
30%;进一步优选的,所述氢气的体积占所述氮氢混合气体的体积的5%
‑
20%。
17.优选的,在所述煅烧前,先通入氮氢混合气体置换出空气,以确保反应体系中无氧气的存在。
18.本发明第二方面提供了一种铋基催化剂,所述铋基催化剂由上述制备方法制得,所述铋基催化剂具有金属铋和三氧化二铋组成的异质结构。
19.本发明第三方面提供了一种工作电极。
20.具体的,所述工作电极,包括电极材料和所述铋基催化剂,所述铋基催化剂附着于所述电极材料表面。
21.优选的,所述电极材料选自碳布、碳纸、玻碳或泡沫镍中的一种。
22.本发明第四方面提供了所述工作电极的制备方法。
23.具体的,所述工作电极的制备方法,包括以下步骤:
24.将所述铋基催化剂分散于溶剂中,加入黏连剂,混合,得到催化剂分散液;然后将所述催化剂分散液滴加或涂覆于所述电极材料上,干燥,制得所述工作电极。
25.本发明第五方面提供了一种二氧化碳还原催化反应系统。
26.一种二氧化碳还原催化反应系统,包括所述工作电极、对电极和参比电极。
27.优选的,所述对电极为铂片对电极。
28.优选的,所述参比电极为ag/agcl参比电极。
29.优选的,所述二氧化碳还原催化反应系统还包括电解液、质子交换膜和电解池。
30.优选的,所述电解池为h型电解池。
31.本发明第六方面提供了一种二氧化碳还原催化反应系统的催化剂原位测试系统。
32.具体的,所述催化剂原位测试系统包括所述二氧化碳还原催化反应系统、质量流量控制器、质量流量监测器、气相色谱仪和电化学工作站。
33.相对于现有技术,本发明的有益效果如下:
34.(1)本发明采用乙酸对反应过程进行调控,使制备的铋基催化剂具有金属铋和三氧化二铋组成的异质结构,其表面丰富的氧空位分布使得催化剂能够在大电压窗口下(
‑
0.7到
‑
1.15v)保持高甲酸选择性,甲酸法拉第效率大于90%,在二氧化碳转化以及甲酸产业领域具备良好前景。
35.(2)本发明提供制备方法主要包括溶剂热反应和煅烧处理两步,其工艺简单,材料易得,能够实现大规模产业化应用。
附图说明
36.图1为实施例1步骤s3制备的粉末的扫描电子显微镜图;
37.图2为实施例1制得的铋基催化剂的扫描电子显微镜图;
38.图3为对比例1步骤s3制备的粉末的扫描电子显微镜图;
39.图4为对比例1制得的铋基催化剂的扫描电子显微镜图;
40.图5为对比例2步骤s3制备的粉末的扫描电子显微镜图;
41.图6为对比例2制得的铋基催化剂的扫描电子显微镜图;
42.图7为实施例1步骤s3制备的粉末x射线衍射谱;
43.图8为实施例1制得的铋基催化剂的x射线衍射谱;
44.图9为对比例1步骤s3制备的粉末和铋基催化剂的x射线衍射谱;
45.图10为对比例2步骤s3制备的粉末和铋基催化剂的x射线衍射谱;
46.图11为实施例1制得的铋基催化剂的高分辨透射电子显微镜图;
47.图12为对比例1制得的铋基催化剂的高分辨透射电子显微镜图;
48.图13为对比例2制得的铋基催化剂的高分辨透射电子显微镜图;
49.图14为实施例1制得的铋基催化剂的电子自旋共振谱图;
50.图15为对比例1制得的铋基催化剂的电子自旋共振谱图;
51.图16为对比例2制得的铋基催化剂的电子自旋共振谱图;
52.图17为实施例1制得的铋基催化剂的x射线电子能谱;
53.图18为对比例1制得的铋基催化剂的x射线电子能谱;
54.图19为对比例2制得的铋基催化剂的x射线电子能谱;
55.图20为应用例2、对比例5和对比例6提供的二氧化碳还原催化反应系统产甲酸法拉第效率对比图;
56.图21为应用例2、对比例5和对比例6提供的二氧化碳还原催化反应系统的co2还原极性曲线。
具体实施方式
57.为了让本领域技术人员更加清楚明白本发明所述技术方案,现列举以下实施例进行说明。需要指出的是,以下实施例对本发明要求的保护范围不构成限制作用。
58.以下实施例中所用的原料、试剂或装置如无特殊说明,均可从常规商业途径得到,或者可以通过现有已知方法得到。
59.实施例1
60.一种铋基催化剂的制备方法,包括以下步骤:
61.s1、将24ml无水乙醇与12ml乙酸混合并用玻璃棒轻轻搅匀。将2mg的五水合硝酸铋粉末加入到上述混合溶液中,然后磁力搅拌5分钟,其转速为500转/分钟,最终的溶液颜色
呈淡黄色。
62.s2、将所得溶液置于反应釜中,在160℃的温度下溶剂热反应390分钟,得到淡黄色沉淀。
63.s3、将所得淡黄色沉淀过滤取出,使用去离子水和无水乙醇交替清洗数遍,最后将沉淀离心分离,并在真空干燥箱于60℃的条件下,烘干十二个小时。最后将其研磨成粉末。
64.s4、先在室温下用250ml/min流速的h2/n2混合气体(h2占10%)通入管式炉3小时,以确保炉管中的残余空气被完全置换掉。然后将粉末分散铺平于刚玉方舟中,将其置于管径为50mm的管式炉中。
65.s5、在持续通入氮氢混合气体(h2占10%)的条件下,同时以5℃每分钟的升温速率将炉体温度从室温升高至200℃。炉体在200℃的条件下持续加热半小时后,自然冷却至室温,并将管式炉中的铋基粉末材料取出,得到铋基催化剂。
66.一种铋基催化剂,由上述方法制备得到,具有金属铋和三氧化二铋组成的异质结构。
67.实施例2
68.一种铋基催化剂的制备方法,包括以下步骤:
69.s1、将24ml无水乙醇与12ml乙酸混合并用玻璃棒轻轻搅匀。将2mg的五水合硝酸铋粉末加入到上述混合溶液中,然后磁力搅拌5分钟,其转速为500转/分钟,最终的溶液颜色呈淡黄色。
70.s2、将所得溶液置于反应釜中,在150℃的温度下溶剂热反应450分钟,得到淡黄色沉淀。
71.s3、将所得淡黄色沉淀过滤取出,使用去离子水和无水乙醇交替清洗数遍,最后将沉淀离心分离,并在真空干燥箱于60℃的条件下,烘干十二个小时。最后将其研磨成粉末。
72.s4、先在室温下用250ml/min流速的h2/n2混合气体(h2占10%)通入管式炉3小时,以确保炉管中的残余空气被完全置换掉。然后将粉末分散铺平于刚玉方舟中,将其置于管径为50mm的管式炉中。
73.s5、在持续通入氮氢混合气体(h2占10%)的条件下,同时以8℃每分钟的升温速率将炉体温度从室温升高至230℃。炉体在230℃的条件下持续加热半小时后,自然冷却至室温,并将管式炉中的铋基粉末材料取出,得到铋基催化剂。
74.实施例3
75.一种铋基催化剂的制备方法,包括以下步骤:
76.s1、将24ml无水乙醇与12ml乙酸混合并用玻璃棒轻轻搅匀。将2mg的五水合硝酸铋粉末加入到上述混合溶液中,然后磁力搅拌5分钟,其转速为500转/分钟,最终的溶液颜色呈淡黄色。
77.s2、将所得溶液置于反应釜中,在170℃的温度下溶剂热反应390分钟,得到淡黄色沉淀。
78.s3、将所得淡黄色沉淀过滤取出,使用去离子水和无水乙醇交替清洗数遍,最后将沉淀离心分离,并在真空干燥箱于60℃的条件下,烘干十二个小时。最后将其研磨成粉末。
79.s4、先在室温下用250ml/min流速的h2/n2混合气体(h2占10%)通入管式炉3小时,以确保炉管中的残余空气被完全置换掉。然后将粉末分散铺平于刚玉方舟中,将其置于管
径为50mm的管式炉中。
80.s5、在持续通入氮氢混合气体(h2占10%)的条件下,同时以4℃每分钟的升温速率将炉体温度从室温升高至180℃。炉体在180℃的条件下持续加热半小时后,自然冷却至室温,并将管式炉中的铋基粉末材料取出,得到铋基催化剂。
81.应用例1
82.一种电极,采用实施例1制备的铋基催化剂制备。
83.具体制备过程如下:
84.s1、取10mg实施例1制备的铋基催化剂分散于950ml无水乙醇中,然后滴加50ml 5wt%的nafion黏连剂,超声30分钟形成催化剂分散液。
85.s2、取200ml催化剂分散液滴加至干净的疏水碳纸(面积为3厘米*1厘米)表面,室温下干燥形成致密膜,得到负载了铋基催化材料的工作电极。
86.应用例2
87.一种气液固双界面的二氧化碳还原催化反应系统,用应用例1制备的电极作为二氧化碳还原工作电极,以铂片作为对电极,以ag/agcl作为参比电极,并用0.5m碳酸氢钾水溶液作为电解液,pe膜为离子交换膜,构建气液固双界面co2还原催化反应系统。
88.对比例1
89.本对比例与实施例1的区别在于,不加入乙酸进行调控。
90.具体地,一种铋基催化剂的制备方法,包括以下步骤:
91.s1、将2毫克的五水合硝酸铋粉末加入到36ml的无水乙醇中,将得到的溶液磁力搅拌5分钟,转速为500转/分钟,最终的溶液颜色呈淡黄色。
92.s2、将所得溶液置于反应釜中,在160℃的温度下溶剂热反应390分钟,得到淡黄色沉淀。
93.s3、将所得淡黄色沉淀过滤取出,使用去离子水和无水乙醇交替清洗数遍,最后将沉淀离心分离,并在真空干燥箱于60℃的条件下,烘干十二个小时。最后将其研磨成粉末。
94.s4、先在室温下用250ml/min流速的h2/n2混合气体(h2占10%)通入管式炉3小时,以确保炉管中的残余空气被完全置换掉。然后将粉末分散铺平于刚玉方舟中,将其置于管径为50mm的管式炉中。
95.s5、在持续通入氮氢混合气体(h2占10%)的条件下,同时以5℃每分钟的升温速率将炉体温度从室温升高至200℃。炉体在200℃的条件下持续加热半小时后,自然冷却至室温,并将管式炉中的铋基粉末材料取出,得到铋基催化剂。
96.对比例2
97.本对比例与实施例1的区别在于,采用乙二醇进行调控。
98.具体地,一种铋基催化剂的制备方法,包括以下步骤:
99.s1、将24ml无水乙醇与12ml乙二醇混合并用玻璃棒轻轻搅匀。将2毫克的五水合硝酸铋粉末加入到上述混合溶液中,然后磁力搅拌5分钟,搅拌的转速为500转/分钟,最终的溶液颜色呈淡黄色。
100.s2、将所得溶液置于反应釜中,在160℃的温度下溶剂热反应390分钟,得到淡黄色沉淀。
101.s3、将所得淡黄色沉淀过滤取出,使用去离子水和无水乙醇交替清洗数遍,最后将
沉淀离心分离,并在真空干燥箱于60℃的条件下烘干十二个小时。最后将其研磨成粉末。
102.s4、先在室温下用250ml/min流速的h2/n2混合气体(h2占10%)通入管式炉3小时,以确保炉管中的残余空气被完全置换掉。然后将粉末分散铺平于刚玉方舟中,将其置于管径为50mm的管式炉中。
103.s5、在持续通入氮氢混合气体(h2占10%)的条件下,同时以5℃每分钟的升温速率将炉体温度从室温升高至200℃。炉体在200℃的条件下持续加热半小时后,自然冷却至室温,并将管式炉中的铋基粉末材料取出,得到铋基催化剂。
104.对比例3
105.一种电极,采用对比例1制备的铋基催化剂制备。
106.具体制备过程如下:
107.s1、取10mg对比例1制备的铋基催化剂分散于950ml无水乙醇中,然后滴加50ml 5wt%的nafion黏连剂,超声30分钟形成催化剂分散液。
108.s2、取200ml催化剂分散液滴加至干净的疏水碳纸(面积为3厘米*1厘米)表面,室温下干燥形成致密膜,得到负载了铋基催化材料的工作电极。
109.对比例4
110.一种电极,采用对比例2制备的铋基催化剂制备。
111.具体制备过程如下:
112.s1、取10mg对比例2制备的铋基催化剂分散于950ml无水乙醇中,然后滴加50ml 5wt%的nafion黏连剂,超声30分钟形成催化剂分散液。
113.s2、取200ml催化剂分散液滴加至干净的疏水碳纸(面积为3厘米*1厘米)表面,室温下干燥形成致密膜,得到负载了铋基催化材料的工作电极。
114.对比例5
115.一种气液固双界面的二氧化碳还原催化反应系统,用对比例3制备的电极作为二氧化碳还原工作电极,以铂片作为对电极,以ag/agcl作为参比电极,并用0.5m碳酸氢钾水溶液作为电解液,pe膜为离子交换膜,构建气液固双界面co2还原催化反应系统。
116.对比例6
117.一种气液固双界面的二氧化碳还原催化反应系统,用对比例4制备的电极作为二氧化碳还原工作电极,以铂片作为对电极,以ag/agcl作为参比电极,并用0.5m碳酸氢钾水溶液作为电解液,pe膜为离子交换膜,构建气液固双界面co2还原催化反应系统。
118.产品效果测试
119.1.对实施例1、对比例1和对比例2制备的铋基催化剂进行进行表征,具体包括采用以下一起或技术进行测试:扫描电子显微镜、x射线衍射仪、高分辨透射电子显微镜、电子自旋共振技术和x射线光电子能谱仪。
120.(1)采用扫描电子显微镜(sem,zeiss)对实施例1、对比例1和对比例2制备中的材料以及最终制备的铋基催化剂进行表征。
121.图1为实施例1步骤s3制备的粉末的扫描电子显微镜图,图2为实施例1制得的铋基催化剂的扫描电子显微镜图,由图1和2可知,添加乙酸后并经过煅烧后处理后形成的铋基催化剂具有纳米级的片层结构。
122.图3为对比例1步骤s3制备的粉末的扫描电子显微镜图,图4为对比例1制得的铋基
催化剂的扫描电子显微镜图。由图3和4可知,不添加乙酸进行调控,无论是否进行煅烧,制备的材料的微观形貌均呈现聚集状态,且无明显的纳米集合形貌特征。
123.图5为对比例2步骤s3制备的粉末的扫描电子显微镜图,图6为对比例2制得的铋基催化剂的扫描电子显微镜图。由图5和6可知,采用乙二醇进行调控,无论是否进行煅烧,制备的材料的微观形貌均呈现聚集状态,且无明显的纳米集合形貌特征。
124.(2)使用x射线衍射仪(xrd,d8 advance,bruker)在40kv,40ma的cuka辐射advance,bruker)在40kv,40ma的cuka辐射下,在2θ=10
‑
80
°
的衍射角范围内分别测试实施例1、对比例1和对比例2制备中的材料以及最终制备的铋基催化剂。
125.图7为实施例1步骤s3制备的粉末的x射线衍射谱,图8为实施例1制得的铋基催化剂的x射线衍射谱,图7和图8中,纵坐标为强度(intensity),横坐标为2θ(2theta)。在图7中,#71
‑
0466和#76
‑
2478为bi2o3的标准卡片;在图8中,#14
‑
0717代表bi smuth oxalate的标准卡,#71
‑
0466和#76
‑
2478代表bi2o3的标准卡片,#65
‑
4028代表bi2o
2.7
的标准卡片;#85
‑
1329代表bi的标准卡片。由图7和图8可知,采用乙酸调控制备的铋基催化剂在煅烧处理前(步骤s3制备的粉末)为三氧化二铋,经过煅烧后形成了铋/三氧化二铋的异质结构。
126.图9为对比例1步骤s3制备的粉末和铋基催化剂的x射线衍射谱,在图9中,pdf#71
‑
0466和pdf#78
‑
1793代表bi2o3的标准卡片,pdf#65
‑
4020代表bio的标准卡片。由图9可知,当不采用乙酸进行调控,在煅烧处理前(步骤s3制备的粉末)为三氧化二铋,而经过煅烧退火处理后有一氧化铋/三氧化二铋结构形成,但无异质结构。图10为对比例2步骤s3制备的粉末和铋基催化剂的x射线衍射谱,在图10中,pdf#71
‑
0466和pdf#45
‑
1344代表bi2o3的标准卡片,pdf#85
‑
1331和pdf#89
‑
2387代表bi的标准卡片。由图10可知,采用乙二醇进行调控,在煅烧处理前为三氧化二铋,经过煅烧后则形成铋/三氧化二铋的异质结构。但采用乙酸调控制备的铋基催化剂所形成的铋/三氧化二铋异质结构更稳定。
127.(3)采用高分辨透射电子显微镜(hrtem,jem
‑
3200fs,jeol)对实施例1、对比例1和对比例2制备的铋基催化剂进行表征。
128.图11为实施例1制得的铋基催化剂的高分辨透射电子显微镜图,由图11中a)可知,经乙酸调控后制备的铋基催化剂,在微观形貌上呈现典型的纳米薄片结构;而由图11中b)、c)可知,铋基催化剂对应的晶格间距测量结果则与金属铋和三氧化二铋相吻合。
129.图12为对比例1制得的铋基催化剂的高分辨透射电子显微镜图,由图12中a)可知,不经乙酸调控制备的铋基催化剂,其微观形貌特征主要为不规则的聚集状态,图12中b)、c)可知,其晶格间距测量结果与一氧化铋和三氧化二铋吻合。
130.图13为对比例2制得的铋基催化剂的高分辨透射电子显微镜图,由图13中a)可知,经乙二醇调控制备的铋基催化剂,其微观形貌为不规则的聚集状态,部分形貌呈现薄片状。图13中b)、c)可知,其晶格间距测量结果与金属铋和三氧化二铋相吻合。
131.(4)采用电子自旋共振技术(esr,ms5000,bruker)对实施例1、对比例1和对比例2制备的铋基催化剂进行表征。图14为实施例1制得的铋基催化剂的电子自旋共振谱图,图15为对比例1制得的铋基催化剂的电子自旋共振谱图,图16为对比例2制得的铋基催化剂的电子自旋共振谱图。在图14
‑
16中,纵坐标为强度(intensity),横坐标为g值(g value),由图14
‑
16可知,经乙酸调控制备的铋基催化剂形成了氧空位,而不经调控或采用乙二醇进行调控,均无法形成氧空位,氧空位的存在有利于催化剂在更大的电压窗口下维持高的甲酸法
拉第效率。
132.(5)采用x射线光电子能谱仪(xps,escalab 250xi,thermo fisher)对实施例1、对比例1和对比例2制备的铋基催化剂进行表征,测得在bi 4f位置的高分辨x射线光电子能谱图。图17为实施例1制得的铋基催化剂的x射线电子能谱,图18为对比例1制得的铋基催化剂的x射线电子能谱,图19为对比例2制得的铋基催化剂的x射线电子能谱。在图17
‑
19中,纵坐标为绝对强度(raw intensity),横坐标为结合能(b.e.ev),由图17
‑
19可知,采用乙酸和乙二醇调控合成的铋基催化剂,其表面铋元素主要存在形式为金属铋(metallic bi)和三价铋(bi
3
),而不经过调控制备的铋基催化剂(对比例1),其表面铋元素主要存在形式为二价铋(bi
2
)和三价铋(bi
3
)。
133.2.对应用例2、对比例5和对比例6提供的二氧化碳还原催化反应系统的性能进行测试,二氧化碳还原催化反应系统连接质量流量控制器、质量流量监测器、气相色谱仪和电化学工作站,测试用电化学工作站为chi电化学工作站(上海辰华)(即一种二氧化碳还原催化反应系统的催化剂原位测试系统)。
134.图20为应用例2、对比例5和对比例6提供的二氧化碳还原催化反应系统产甲酸法拉第效率对比图。图20中,纵坐标为法拉第效率(fe of hcooh/%),横坐标为相对于可逆氢电极的电势(potentialv vs rhe),由图20可知,应用例2提供的二氧化碳还原催化反应系统(含有经乙酸调控制备的铋基催化剂)在更大的电压窗口内(
‑
0.7至
‑
1.15)都保持了一个较高的甲酸法拉第效率水平,甲酸法拉第效率大于90%,最高可达95%。而对比例5和对比例6提供的二氧化碳还原催化反应系统,虽然在
‑
0.7v的电压下具有良好的产甲酸法拉第效率,但是在
‑
1.15的电压下的产甲酸法拉第效率很低,小于70%。
135.图21为应用例2、对比例5和对比例6提供的二氧化碳还原催化反应系统的co2还原极性曲线。图21中纵坐标为电流密度(a/cm2),横坐标为相对于可逆氢电极的电势(potentialv vs rhe),由图21可知,应用例2提供的二氧化碳还原催化反应系统的电流密度远高于对比例5和对比例6,采用乙酸调控制备的铋基催化剂体现出更高的电流密度。
136.由上述试验可知,经乙酸调控制备的铋基催化剂,在更宽的电压窗口下保持更为优异的催化活性。实施例2、3制备的铋基催化剂与实施例1制备的铋基催化剂具有类似的性质。
137.可见,采用本发明提供的铋基催化剂的制备方法制备的铋基催化剂,用于二氧化碳电催化还原过程中时,实现了在更大的电压窗口下维持高的甲酸法拉第效率的目标,催化剂的性能优异;且制备方法简单,易操作,具有广阔的应用前景。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。