氢氯氟烃的制造方法、1
‑
氯
‑
2,3,3
‑
三氟丙烯的制造方法、1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
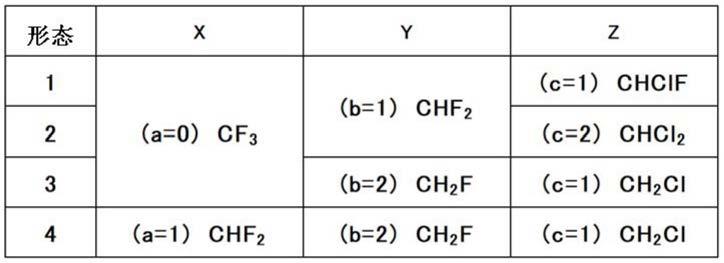
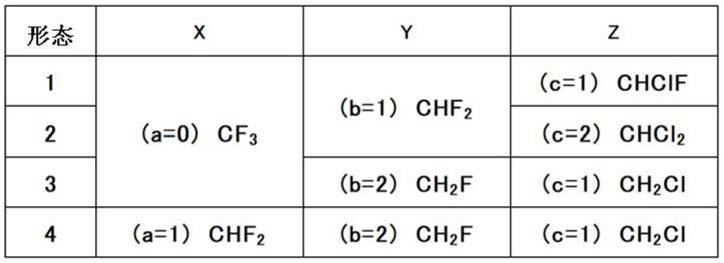
戊烯的制造方法
技术领域
1.本发明涉及氢氯氟烃的制造方法、1
‑
氯
‑
2,3,3
‑
三氟丙烯的制造方法、1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯的制造方法。
背景技术:
2.近年来,氢氯氟烯烃(hcfo)作为对地球环境影响小且可以用于清洗剂、制冷剂、工作流体、喷射剂、热介质、发泡剂、溶剂等各种用途的化合物而受到关注。hcfo中,作为不燃性高且适用于上述用途的化合物,已知1
‑
氯
‑
2,3,3,3
‑
四氟丙烯(cf3‑
cf=cclh、hcfo
‑
1224yd)或1
‑
氯
‑
2,3,3
‑
三氟丙烯(cf2h
‑
cf=cclh,hcfo
‑
1233yd)等碳
‑
碳双键的末端碳结合了氯原子的hcfo。另外,本说明书中,对于卤代烃在化合物名称之后的括号内记载了该化合物的简称,但本说明书中根据需要也会使用其简称来代替化合物名称。此外,作为简称,有时仅使用横线(
‑
)后的数字和小写字母部分(例如对于“hcfo
‑
1233yd”仅使用“1233yd”)。现有技术文献专利文献
3.专利文献1:国际公开第1990/008754号
技术实现要素:
发明所要解决的技术问题
4.本发明人发现,后述的具有特定结构(
‑
cf2‑
ch
b
cl
c
f
(3
‑
b
‑
c)
)的氢氯氟烃(以下也记为“hcfc”)能够高效地合成碳
‑
碳双键的末端碳上结合了氯原子的hcfo。作为上述具有特定结构的hcfc的制造方法,例如专利文献1中记载了使具有二氟亚甲基的hcfc与氟化氢进行反应而将上述hcfc氟化的方法。但是,专利文献1所记载的制造方法难以控制氟化反应,在选择性地获得上述具有特定结构的hcfc上存在困难,因此期待以更高选择率来获得的方法。这样,对于hcfc的制造期待更高效的方法。特别是从工业上的观点考虑,期待能够从比较容易合成的氢氟烃出发以高选择率制造hcfc。本发明的目的在于,提供一种能够以高选择率制造hcfc的hcfc制造方法。此外,本发明的目的还在于,提供1
‑
氯
‑
2,3,3
‑
三氟丙烯的制造方法和1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯的制造方法。解决技术问题所采用的技术方案
5.为了解决上述技术问题,本发明人经过深入研究,发现通过以下技术方案可以解决上述技术问题。
6.(1)一种氢氯氟烃的制造方法,其特征在于,在催化剂的存在下,使后述式(1)所表示的氢氟烃与氯化氢进行反应,以制造后述式(2)所表示的氢氯氟烃。
(2)如(1)所述的制造方法,其中,所述催化剂为含金属催化剂。(3)如(1)或(2)所述的制造方法,其中,所述催化剂包含选自金属氧化物、金属氧化物的部分卤化物和金属卤化物的1种以上,金属氧化物、金属氧化物的部分卤化物和金属卤化物分别包含选自cr和al的至少1种。(4)如(3)所述的制造方法,其中,所述催化剂包含金属氧化物的部分卤化物。(5)如(1)~(4)中任一项所述的制造方法,其中,l为
‑
cf2‑
、
‑
cf2cf2‑
、
‑
cf2‑
cf2‑
cf2‑
、
‑
cf2‑
chf
‑
cf2‑
或
‑
cf2‑
ch2‑
cf2‑
。(6)如(1)~(5)中任一项所述的制造方法,其中,a为1,b为2,c为1。(7)如(1)~(6)中任一项所述的制造方法,其中,在气相下使式(1)所表示的氢氟烃与氯化氢进行反应。(8)如(1)~(7)中任一项所述的制造方法,其中,反应温度为100~450℃。(9)如(1)~(8)中任一项所述的制造方法,其中,氯化氢相对于式(1)所表示的氢氟烃的摩尔比为0.5~2.0。(10)如(1)~(9)中任一项所述的制造方法,其中,使用包含式(1)所表示的氢氟烃的原料组合物与氯化氢进行反应,原料组合物的水分含量为5000质量ppm以下。(11)如(10)所述的制造方法,其中,水分含量为400质量ppm以下。(12)如(1)~(11)中任一项所述的制造方法,其中,式(1)所表示的氢氟烃为1,1,2,2,3
‑
五氟丙烷,式(2)所表示的氢氯氟烃为3
‑
氯
‑
1,1,2,2
‑
四氟丙烷。(13)一种1
‑
氯
‑
2,3,3
‑
三氟丙烯的制造方法,其特征在于,使由(12)所述的方法制造的3
‑
氯
‑
1,1,2,2
‑
四氟丙烷进行脱氟化氢反应,以制造1
‑
氯
‑
2,3,3
‑
三氟丙烯。(14)如(1)~(11)中任一项所述的制造方法,其中,式(1)所表示的氢氟烃为1,1,2,2,3,3,4,4,5
‑
九氟戊烷,式(2)所表示的氢氯氟烃为5
‑
氯
‑
1,1,2,2,3,3,4,4
‑
八氟戊烷。(15)一种1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯的制造方法,其特征在于,使由(14)所述的方法制造的5
‑
氯
‑
1,1,2,2,3,3,4,4
‑
八氟戊烷进行脱氟化氢反应,以制造1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯。发明效果
7.根据本发明,可以提供一种能够以高选择率制造hcfc的hcfc制造方法。此外,根据本发明,还可以提供1
‑
氯
‑
2,3,3
‑
三氟丙烯的制造方法和1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯的制造方法。
附图说明
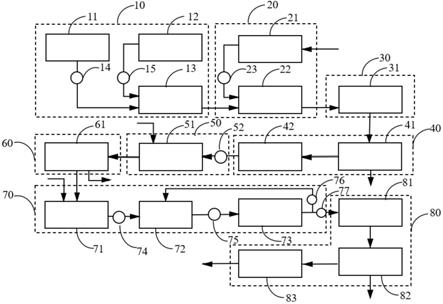
8.图1所示为反应装置的示意图。
具体实施方式
9.本发明的氢氯氟烃的制造方法(以下也简单记为“本发明的制造方法”)是在催化剂的存在下使后述式(1)所表示的氢氟烃(以下也记为“化合物1”)与氯化氢进行反应以制造式(2)所表示的氢氯氟烃(以下也记为“化合物2”)的方法。即,本发明的制造方法是在催
化剂的存在下使化合物1与氯化氢接触以制造化合物2的方法。以下,首先对本发明的制造方法所使用的成分进行详述,之后对制造方法的步骤进行详述。作为关于成分的记载,首先对化合物1和化合物2进行详述。
10.化合物1为式(1)所表示的化合物。式(1)x
‑
l
‑
yx表示
‑
ch
a
f
(3
‑
a)
。a表示0或1。
11.y表示
‑
ch
b
f
(3
‑
b)
。b表示1或2。其中,在a为0的情况下,b表示1或2,在a为1的情况下,b表示2。即,作为化合物1,可列举cf3‑
l
‑
chf2、cf3‑
l
‑
ch2f、chf2‑
l
‑
ch2f。
12.l为
‑
cf2‑
、
‑
cf2cf2‑
、或至少一部分氢原子可以被取代成氟原子的碳数为3~6的氟代亚烷基,其中,碳数为3~6的氟代亚烷基的末端均为
‑
cf2‑
。另外,上述氟代亚烷基的末端均为
‑
cf2‑
是指,
‑
cf2‑
位于氟代亚烷基中的两个末端处,例如,作为碳数为3的上述氟代亚烷基,可列举
‑
cf2‑
cf2‑
cf2‑
、
‑
cf2‑
ch2‑
cf2‑
、
‑
cf2‑
cfh
‑
cf2‑
。作为l,优选
‑
cf2‑
、
‑
cf2cf2‑
、
‑
cf2‑
cf2‑
cf2‑
、
‑
cf2‑
chf
‑
cf2‑
和
‑
cf2‑
ch2‑
cf2‑
,更优选作为全氟亚烷基的
‑
cf2‑
、
‑
cf2cf2‑
和
‑
cf2‑
cf2‑
cf2‑
。
13.以化合物1作为原料制造的化合物为式(2)所表示的化合物。式(2)x
‑
l
‑
zz表示
‑
ch
b
cl
c
f
(3
‑
b
‑
c)
。b表示1或2。c表示1或2。其中,在b为1的情况下,c表示1或2,在b为2的情况下,c表示1。即,作为z,可列举
‑
chclf、
‑
chcl2、
‑
ch2cl。
14.上述a、b、c的关系如以下表1所示。表1中,“x”“y”“z”对应于上述式(1)和式(2)中的“x”“y”“z”。此外,表1中的“x”“y”“z”栏示出a、b、c可以取的数值和此时的结构。例如,“(a=0)cf
3”是指在a为0的情况下x表示cf3。
15.[表1]
[0016]
本发明的制造方法中,在催化剂的存在下,通过使化合物1与氯化氢进行反应,可
以使位于化合物1末端的碳原子上所结合的氟原子被取代成氯原子。此时,可以将两个末端中氟原子取代数较少的末端碳原子上所取代的氟原子选择性地取代成氯原子。本发明的制造方法中,若比较式(1)中的x与y,则y的氟原子取代数少,在y的位置上进行氟原子与氯原子的取代。此外,从氟原子到氯原子的取代数可以通过调整反应条件等来进行控制。例如,如表1所示,在y为chf2的情况下,1个氟原子被取代成氯原子而形成chclf,或2个氟原子被取代成氯原子而形成chcl2。更具体而言,上述a、b、c的关系表示为上述表1的形态1~4的情况。例如,在“形态4”的情况下,“x”栏和“y”栏表示化合物1为chf2‑
l
‑
ch2f,“x”栏和“z”栏表示化合物2为chf2‑
l
‑
ch2cl。即,本发明的制造方法中,a、b、c可以取以下4个形态。(形态1)a为0,b为1,c为1。(形态2)a为0,b为1,c为2。(形态3)a为0,b为2,c为1。(形态4)a为1,b为2,c为1。本发明的制造方法可以特别有利地用于化合物1的仅1个氟原子被取代成氯原子的反应。例如在使用1,1,2,2,3
‑
五氟丙烷作为化合物1的情况下,可以制造3
‑
氯
‑
1,1,2,2
‑
四氟丙烷(hcfc
‑
244ca)作为化合物2。此外,例如在使用1,1,2,2,3,3,4,4,5
‑
九氟戊烷(hfc
‑
449pccc)作为化合物1的情况下,可以制造5
‑
氯
‑
1,1,2,2,3,3,4,4
‑
八氟戊烷(hcfc
‑
448occc)作为化合物2。
[0017]
本发明的制造方法中,如上所述,化合物1对反应起到贡献。可以使用包含化合物1的原料组合物作为原料。化合物1可以是2种以上。原料组合物除了化合物1以外,有时还会包含杂质。作为杂质,可列举化合物1的制造原料或制造化合物1时所产生的化合物1以外的副产物、水等。另外,在原料组合物中包含上述杂质的情况下,可以通过蒸馏、萃取蒸馏、共沸蒸馏、膜分离、二层分离、吸附等已知手段来除去由杂质所产生的副产物。作为杂质,优选在本发明的反应条件下为惰性化合物。
[0018]
从化合物1的转化率更优异的观点考虑,优选在用于反应前将原料组合物脱水。即,反应优选使用实施过脱水处理的原料组合物。反应所使用的上述原料组合物的水分含量(含水量)优选5000质量ppm以下,更优选2000质量ppm以下,进一步优选400质量ppm以下,特别优选200质量ppm以下。作为下限,可列举0质量ppm。本文中,ppm表示百万分率。上述水分含量用卡尔费休水分仪来进行测定。作为卡尔费休水分仪,使用微量水分测定装置(三菱化学分析技术株式会社(三菱化学
アナリテック
社)制,ca
‑
200型),在试样导入部上连接液化气气化装置(同社制,型号:vg
‑
200型),使预先设定好量的脱水处理后的上述原料组合物气化,自动注入到水分仪中。
[0019]
反应所使用的原料组合物中包含化合物1作为主成分。主成分是指化合物1的含量相对于原料组合物的总质量为50质量%以上,优选60质量%以上,更优选75质量%以上。作为上限,可列举100质量%。
[0020]
将原料组合物脱水的方法没有特别限定,例如可列举蒸馏或使用脱水剂的方法。在使用脱水剂的情况下,使脱水剂与原料组合物接触,减少原料组合物中的水分含量。作为脱水剂,可列举沸石、分子筛、氧化铝、氯化钙、硫酸镁、硫酸钠、硫酸钙、碳酸钾。
[0021]
氯化氢在常温常压下为气体。氯化氢可以在操作时溶解在水中来使用。另外,反应优选使用实施过脱水处理的氯化氢。
[0022]
作为本发明的制造方法中的催化剂,优选含金属催化剂。作为含金属催化剂的具体例,可列举金属单体、金属氧化物、金属氧化物的部分卤化物、金属卤化物。上述金属氧化物的部分卤化物是金属氧化物的一部分卤化(f、cl、br、i等)而成的化合物。上述金属卤化物是由金属与卤素构成的化合物。金属氧化物的部分卤化物和金属卤化物可以仅包含1种卤素,也可以包含2种以上的卤素。作为催化剂,可以2种以上组合使用。
[0023]
作为金属氧化物的部分卤化物,优选金属氧化物氟化而成的金属氧化物的部分氟化物。该情况下,金属氧化物的部分氟化物还可以包含氟以外的卤素。此外,作为金属卤化物,优选金属氟化物。金属氟化物可以包含氟以外的卤素。
[0024]
从提升反应性的观点考虑,作为上述金属单体、金属氧化物和金属卤化物中所含的金属元素,优选选自li、na、k、cs、mg、ca、sr、ba、al、cr、zr、fe、ni、co、zn、mn、sb、nb和ta的至少1种,更优选选自al、zn、cr、mg、ca、k、zr和li的至少1种,进一步优选选自al、zn、cr、mg和zr的至少1种。
[0025]
其中,从能够以更高选择率制造化合物2的观点考虑,催化剂优选包含选自金属氧化物、金属氧化物的部分卤化物、或金属卤化物的1种以上,上述金属氧化物、金属氧化物的部分卤化物和金属卤化物分别包含选自cr和al的至少1种金属元素(以下也记为“金属a”)。即,催化剂优选包含选自含金属a的金属氧化物、含金属a的金属氧化物的部分卤化物和含金属a的金属卤化物的1种以上。上述金属氧化物、上述金属氧化物的部分卤化物和上述金属卤化物也可以包含cr和al两者。其中,催化剂更优选包含具有金属a的金属氧化物的部分卤化物。
[0026]
作为包含金属a的催化剂,其中,从使化合物1的转化率更高的观点考虑,优选包含cr的金属氧化物、包含cr的金属氧化物的部分卤化物、或包含cr的金属卤化物。
[0027]
作为催化剂,在使用包含金属a的金属氧化物、金属氧化物的部分卤化物、或金属卤化物的情况下,为了提升耐久性等种种目的,催化剂还可以包含金属a以外的金属。作为其他金属的具体例,可列举na、k、mg、ca、zr、fe、zn、ni、co、mn。其中,从提高催化剂的物理
·
化学耐久性、能够更高效地制造化合物2的观点考虑,优选na、k、mg、zn或mn,进一步优选mg或zn。另外,在催化剂还包含金属a以外的金属的情况下,化合物1的转化率更优异。从进一步提升转化率的观点考虑,作为其他金属,优选na、k、mg、zn或mn,进一步优选mg或zn。
[0028]
在催化剂包含cr的情况下,作为cr的含量,从能够提升化合物1的转化率的观点考虑,相对于催化剂中所含的金属总质量(100质量%),优选1质量%以上,更优选5质量%以上,进一步优选10质量%以上。上限为100质量%。在催化剂包含al的情况下,作为al的含量,从能够提升化合物1的转化率的观点考
虑,相对于催化剂中所含的金属总质量(100质量%),优选5质量%以上,更优选20质量%以上,进一步优选50质量%以上。上限为100质量%。在催化剂还包含金属a以外的金属的情况下,金属a的含量相对于金属总质量(100质量%)优选90~99.9质量%,更优选95~99质量%。而金属a以外的金属相对于金属总质量(100质量%)优选0.1~10质量%,更优选1~5质量%。
[0029]
供于反应的催化剂的水分含量优选较低。具体而言,将催化剂配置在反应器内,向反应器内以每1g催化剂达到3.94nml/分钟的方式供应n2,从反应器出口获得的气体中水分优选为100体积ppm以下,更优选为50体积ppm以下。
[0030]
作为催化剂,具体而言,可列举部分氟化的铬锌复合氧化物(铬锌复合氧化物的部分氟化物)、部分氟化的铬铝镁复合氧化物(铬铝镁复合氧化物的部分氟化物)、部分氟化的氧化铝(氧化铝的部分氟化物)、部分氟化的铬氧化物(铬氧化物的部分氟化物)。其中,从化合物1的转化率优异的观点考虑,优选部分氟化的铬锌复合氧化物(铬锌复合氧化物的部分氟化物)和部分氟化的铬氧化物(铬氧化物的部分氟化物)。
[0031]
为了提升反应性,催化剂可以成形为颗粒状来使用,也可以担载在载体上来使用。作为载体,可列举活性炭、炭黑、碳纤维等碳材料,氧化铝、氧化硅、氧化钛、氧化锆、碱金属氧化物、碱土金属氧化物等氧化物材料,优选活性炭、氧化铝、氧化硅、氧化锆、碱金属氧化物和碱土金属氧化物。其中,从比表面积大、容易担载催化剂的角度考虑,更优选活性炭、氧化铝和氧化锆。
[0032]
作为将催化剂成形为颗粒状的方法,可列举将催化剂碎解为粉末、通过压片机等进行成形的方法。作为颗粒状的催化剂,例如,可以使用成形为直径3.0mm左右、高度4.0mm左右的圆柱状者。此外,根据需要,可以将催化剂与粘合剂混合。粘合剂的使用量相对于催化剂100质量份优选100质量份以下,更优选50质量份以下,进一步优选10质量份以下。该情况下,可以通过压片机等将催化剂与粘合剂的混合物成形为颗粒状的催化剂。另外,作为粘合剂的具体例,可列举碳、纤维素、氧化铝、氧化硅。
[0033]
为了提升反应性,催化剂优选预先在惰性气氛中(例如氮气流中)干燥。从操作的简便化和提升工作效力的观点考虑,催化剂可以在收纳于反应器内的状态下以和上述相同的方式进行干燥。此外,也可以预先在收纳于反应器之前对催化剂进行干燥。
[0034]
催化剂的比表面积取决于各催化剂的种类,一般而言呈现出越小则转化率越低、越大则选择率越低而劣化越快的倾向。例如,在不使用上述粘合剂的情况下,上述金属氧化物和上述金属氧化物的部分卤化物的比表面积优选10~400m2/g,上述金属卤化物的比表面积优选3~300m2/g。在使用上述粘合剂的情况下,取决于粘合剂的比表面积而不能一概而论,例如,在使用高比表面积的碳粘合剂的情况下,催化剂与碳粘合剂的混合物的比表面积优选20~1200m2/g左右。另外,本说明书中,比表面积为bet法所测定的值。
[0035]
此外,从提升反应性的观点(特别是提升化合物1的转化率的观点)考虑,优选对催化剂预先实施活化处理。作为活化处理的方法,可列举在加热下或非加热下使催化剂与活
化处理剂接触的方法。作为活化处理剂,可列举含卤化合物,具体而言,可列举氯化氢、氟化氢、氯代烃、氟代烃、氯氟烃、氢氯烃、氢氟烃、氢氯氟烃、氯代烯烃、氟代烯烃、氯氟烯烃、氢氟烯烃、氢氯烯烃、氢氯氟烯烃。此外,也可以将作为原料的化合物1用作含卤化合物。通过对金属氧化物实施上述活化处理,可以生成金属氧化物的部分卤化物。
[0036]
催化剂可以在收纳于反应器之前进行活化处理,但是考虑到操作简便、工作效率良好,优选在收纳于反应器的状态下进行活化处理。因此,优选将活化处理剂导入到收纳有催化剂的反应器中来进行活化处理。活化处理剂可以在常温下直接导入到反应器中,但是从能够高效地进行活化处理的观点考虑,优选在导入反应器时通过加热等进行温度调节。在导入活化处理剂时,可以与惰性气体(稀释气体)合并导入。作为稀释气体的具体例,可列举氮气、二氧化碳、氦气、氩气。此外,为了提高活化处理的效率,优选在加热的状态下对反应器内进行活化处理。此时,供应活化处理剂时的反应器温度优选50~500℃,更优选100~400℃,进一步优选150~350℃。活化处理剂的滞留时间优选1~1000秒,更优选2~500秒。
[0037]
此外,除了上述的反应前的活化处理以外,还可以对催化剂进行再活化处理。特别是在催化剂的活性降低、化合物1的转化率下降时,可以对催化剂进行再活化处理。籍此,可以使催化剂的活性再生而将催化剂再利用。
[0038]
作为再活化处理的方法,与使用前的活化处理相同,可列举在加热下或非加热下使催化剂与用于再活化处理的处理剂(再活化处理剂)接触的方法。作为再活化处理剂的具体例,可列举氧、氯、氟化氢、氯化氢、含卤烃、全卤代碳。此外,再活化处理中,从抑制副反应和提升催化剂耐久性等观点考虑,可以将氮气、二氧化碳、稀有气体(氦气等)、水蒸汽等惰性气体用于稀释再活化处理剂。
[0039]
在催化剂的活性下降到无法再活化的程度的情况下,或在停止制造化合物2的情况下,有时会进行从反应器中抽出催化剂、重新装填催化剂的操作。在抽出催化剂时,为了除去反应器内残留或附着在催化剂上的有机物或酸成分,优选事先用惰性气体对反应器进行吹扫。作为所使用的惰性气体的具体例,可列举氮气、氦气。空气吹扫可能会在催化剂中形成6价铬等有害物质,因此有时是不优选的。
[0040]
从能够以更高选择率制造化合物2的观点考虑,上述反应(液相反应、气相反应)中,所使用的氯化氢相对于所使用的化合物1的摩尔比(氯化氢的摩尔量/化合物1的摩尔量)优选0.01~100,更优选0.1~10,进一步优选0.3~5,特别优选0.5~2。从抑制副产物的产生、提升反应器的容积效率的观点考虑,更特别优选0.75~1.5,最优选0.8~1.2。其中,在上述摩尔比为0.75以上的情况下,化合物1的转化率更优异。
[0041]
上述反应使用反应器来进行。作为反应器,形状和结构没有特别限定。例如,在后述气相反应的情况下,可列举内部可以填充催化剂的圆筒状的立式反应器。作为圆筒状的立式反应器,可列举多管式反应器。作为反应器的材质的具体例,可列举玻璃、铁、镍、不锈钢、以铁或镍作为主成分的合金。反应器可以在内部具备电热器等加热部。反应器也可以具有插入了用于测定内部温度的温度计的套管(原文:
さや
管)。
在使用多管式反应器作为反应器的情况下,各反应管的压力损失相对于所有反应管的压力损失的平均值优选为
±
20%以内,更优选为
±
15%以内,进一步优选为
±
10%以内。通过调整使各反应管的催化剂填充量一定,可以减小各反应管的压力损失的差异。
[0042]
本发明的制造方法中,化合物1与氯化氢的反应可以是液相反应和气相反应中任一者。液相反应是指,在化合物1与氯化氢分别为液体的状态下使之进行反应。气相反应是指,在化合物1与氯化氢分别为气体的状态下使之进行反应。上述反应可以分批式进行,也可以半连续式、连续流通式进行。
[0043]
下面对液相反应进行详细说明。作为液相反应的具体步骤,例如可列举向存在有液体状态下的化合物1与催化剂的混合物的反应器内连续地或非连续地供应氯化氢、将反应所生成的化合物2从反应器内连续或非连续地抽出的步骤。从反应产率和制造效率的观点考虑,液相反应的反应温度优选20℃以上,更优选30℃以上,进一步优选50℃以上,优选250℃以下,更优选200℃以下,进一步优选150℃以下。从反应产率和制造效率的观点考虑,液相反应的反应时间优选0.1~100小时,更优选0.2~50小时,进一步优选0.5~20小时。反应时间是指反应器内原料的滞留时间。根据需要,液相反应也可以在溶剂的存在下实施。作为溶剂的具体例,可列举cf3(cf2)
m
cf3(式中m表示3~6的整数)所表示的碳数5~8的直链全氟烷基化合物。
[0044]
接着对关于气相反应进行详细说明。作为气相反应的具体步骤,可列举将加热至气体状态下的作为原料的化合物1与氯化氢连续地供应至反应器内使填充于反应器中的催化剂与气体状态下的化合物1和氯化氢接触而获得化合物2的步骤。从对调整流量、抑制副产物、抑制催化剂失活等有效的观点考虑,上述反应中也可以向反应器供应惰性气体(稀释气体)。作为稀释气体的具体例,可列举氮气、二氧化碳、氦气、氩气。
[0045]
从能够更高效地制造化合物2的观点考虑,气相反应的反应温度(反应器内的温度)优选100~450℃,更优选120~380℃,进一步优选140~360℃,特别优选160~340℃。在上述反应温度为100℃以上(优选180℃以上,更优选225℃以上)的情况下,化合物1的转化率提高。此外,在上述反应温度为450℃以下(优选360℃以下)的情况下,化合物2的选择率提高。反应器内的温度可以通过调整向反应器供应的原料的温度和压力来进行控制。根据需要,可以通过电热器或微波发生器等对反应器内进行辅助加热。
[0046]
气相反应的反应时间优选0.1~1000秒,更优选1~800秒,进一步优选5~600秒。上述反应时间相当于原料在反应器内的滞留时间,可以通过调节原料向反应器的供应量(流量)来进行控制。
[0047]
气相反应的反应体系的压力(反应器内的压力)优选0~2.0mpa,更优选0~1.5mpa。也可以是负压。从操作性的观点考虑,反应器内的压力进一步优选0~1.0mpa。本说明书中,只要没有特别限定,压力以表压表示。
[0048]
产物中副产物的含量相对于产物总质量优选10质量%以下,更优选5质量%以下。上述副产物的含量的下限通常为0质量%。本文中,产物是指化合物1与氯化氢反应而生成的卤代烃。在产物包含杂质的情况下,可以实施从所获得的产物中分离化合物2的处理。作为分离处理,可列举蒸馏等公知的方法。
[0049]
接着,参照图1对气相反应的更详细的形态进行说明。图1所示的反应装置20为气相反应所使用的反应装置的一例。反应装置20具备反应器1。反应器1上连接有化合物1的供应管路2、氯化氢的供应管路3和作为稀释气体的氮气的供应管路4。反应器1优选具备电热器等加热部。化合物1的供应管路2和氯化氢的供应管路3可以分别各自与反应器1连接,也可以在反应器1的跟前相连而与反应器1连接。例如,如图1所示,使化合物1的供应管路2、氯化氢的供应管路3和氮气的供应管路4相连。籍此,化合物1与氯化氢与氮气的混合物经由混合物供应管路5向反应器1供应。
[0050]
图1所示的反应装置20中,化合物1的供应管路2、氯化氢的供应管路3和氮气的供应管路4分别设置有具备电热器等的预热器(原文:
プレヒータ
)2a、3a和4a。向反应器1供应的化合物1、氯化氢和氮气优选分别通过预热器2a、3a和4a预热至规定的温度后再向反应器1供应。籍此,能够高效地使化合物1、氯化氢和氮气升温至反应器1内部所规定的反应温度。预热器2a、3a和4a并非必需,但是优选设置。
[0051]
反应器1的出口介由热交换器等冷却部6与出口管路7连接。出口管路7进一步与水蒸汽和酸性液的回收槽8、碱清洗装置9以及脱水塔10依次连接。从反应器1中抽出的反应混合物通过出口管路7以后的处理被除去氯化氢、氟化氢等酸性物质、水蒸汽、水。由此获得的气体以下称为“出口气体”。出口气体中的各成分通过气相色谱法(gc)等分析装置11进行分析和定量。
[0052]
出口气体包含化合物2。作为出口气体中所含的化合物2以外的化合物,可列举未反应原料即化合物1和氯化氢、氟化氢。
[0053]
出口气体中所含的化合物2以外的成分可以通过蒸馏等已知手段来除去至所期望的程度。反应装置20中,可以通过蒸馏等从由反应器1排出的反应混合物或出口气体中分离出未反应原料,使其作为原料的一部分返回反应器。籍此,可以提升化合物2的生产性。
[0054]
本发明的制造方法中,在化合物1为1,1,2,2,3
‑
五氟丙烷(245ca)的情况下,可以获得3
‑
氯
‑
1,1,2,2
‑
四氟丙烷(244ca)作为化合物2。以下对使用245ca作为原料的情况进行详述。
[0055]
245ca可以通过公知的方法来制造,例如,可以通过国际公开第1994/27939号所记载的方法来制造。
[0056]
在使用245ca作为原料通过气相反应制造244ca时,可以作为粗制气体的成分获得244ca。粗制气体中,除了244ca以外,有时还会包含未反应的245ca、2
‑
氯
‑
1,3,3
‑
三氟丙烯(1233xe)、1
‑
氯
‑
2,3,3
‑
三氟丙烯(1233yd)、1,2
‑
二氯
‑
3,3
‑
二氟丙烯(1232xd)、1,3
‑
二氯
‑
2,3
‑
二氟丙烯(1232yd)、2,3
‑
二氯
‑
1,3
‑
二氟丙烯(1232xe)、1,2,3
‑
三氯
‑3‑
氟丙烯
(1231xd)、2,3,3
‑
三氯
‑1‑
氟丙烯(1231xe)、1,3,3
‑
三氯
‑2‑
氟丙烯(1231yd)、1,2,3,3
‑
四氯丙烯(1230xd)、未反应的氯化氢、氟化氢等。上述244ca以外的成分可以通过蒸馏、萃取蒸馏、共沸蒸馏、膜分离、二层分离、吸附等已知手段来除去至所期望的程度。
[0057]
另外,本说明书中,在没有特别区分地使用化合物名或化合物简称的情况下,其表示选自z体和e体中至少1种,更具体而言,其表示z体或e体、或z体与e体的任意比例混合物。在化合物名或化合物简称后附加(e)或(z)的情况下,其分别表示化合物的(e)体或(z)体。例如,1233yd(z)表示z体,1233yd(e)表示e体。
[0058]
可以使所获得的244ca进行脱氟化氢反应,以制造1233yd。作为脱氟化氢反应的步骤,可列举国际公开第2016/136744号等公知的方法。
[0059]
上述244ca的脱氟化氢反应可以是液相反应和气相反应中任一者。另外,液相反应是指使液体状态下或溶解在液体中的244ca进行脱氟化氢反应。气相反应是指使气体状态下的244ca进行脱氟化氢反应。
[0060]
此外,本发明的制造方法中,在化合物1为hfc
‑
449pccc的情况下,可以获得hcfc
‑
448occc作为化合物2。以下,对使用449pccc作为原料的情况进行详述。
[0061]
449pccc可以通过公知的方法制造,例如,可以通过russian journal of applied chemistry vol.5no.7 2002pp.1162
‑
1165(俄罗斯应用化学,第5卷第7期,2002年,第1162
‑
1165页)所记载的方法来制造。
[0062]
在使用449pccc作为原料通过气相反应制造448occc时,可以作为粗制气体的成分获得448occc。粗制气体除了448occc以外,有时还会包含未反应的449pccc、c5h2f
(7
‑
x)
cl
(1 x)
(x表示0~7)等。上述448occc以外的成分可以通过蒸馏、萃取蒸馏、共沸蒸馏、膜分离、二层分离、吸附等已知手段来除去至所期望的程度。
[0063]
可以使所获得的448occc进行脱氟化氢反应,以制造1
‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯(hcfo
‑
1437dycc)。特别优选制造(z)
‑1‑
氯
‑
2,3,3,4,4,5,5
‑
七氟
‑1‑
戊烯(hcfo
‑
1437dycc(z))。作为脱氟化氢反应的步骤,可列举zhurnal organicheskoi khimii,(俄罗斯),1988年,第24巻第8号,第1626
‑
1633页等公知的方法。
[0064]
上述448occc的脱氟化氢反应可以是液相反应和气相反应中任一者。另外,液相反应是指使液体状态下或溶解在液体中的448occc进行脱氟化氢反应。此外,气相反应是指使气体状态下的448occc进行脱氟化氢反应。实施例
[0065]
以下,通过例子对本发明进行详细说明,但是本发明不限于此。后述例1~19相当于实施例。
[0066]
<气相色谱法的条件>以下各种化合物的制造中,使用气相色谱法(gc)对所获得的产物进行组成分析。柱采用db
‑
1301(长60m
×
内径250μm
×
厚1μm,安捷伦科技有限公司(
アジレント
·
テクノロジー
株式会社)制)。
[0067]
<催化剂的配制>用于后述反应的催化剂如下进行配制。使用与基于图1说明的反应装置20同样的反应装置作为各配制例中所使用的反应装置。使用内径16.1mm、长度15cm的sus304制管状反应器作为反应器1。
[0068]
(催化剂配制例1)在反应器中填充铬锌复合氧化物(pricat62
‑
3m,庄信万丰公司(
ジョンソンマッセイ
社))38.1g,一边使氮气(n2)以150nml/分钟流过,一边使反应器内升温至300℃。使反应器内维持在大气压下,同时将所填充的催化剂干燥至从反应器出口获得的气体中的水分达到5体积ppm以下。之后,使反应器升温至350℃,三氟甲烷(hfc
‑
23)以21.9nml/分钟的流量、n2以43.7nml/分钟的流量供应16小时,在反应器内配制部分氟化的铬锌复合氧化物。所获得的催化剂相当于金属氧化物的部分氟化物。另外,上述铬锌复合氧化物(pricat62
‑
3m)中cr的含量相对于催化剂中所含的金属总质量为97.0质量%,zn的含量相对于催化剂中所含的金属总质量为2.9质量%。
[0069]
(催化剂配制例2)除了将铬锌复合氧化物(pricat62
‑
3m,庄信万丰公司制)变更为铬铝镁复合氧化物(n401ag,日挥触媒化成株式会社(日揮触媒化成社)制)33.6g以外,按照与(催化剂配制例1)相同的步骤,获得部分氟化的铬铝镁复合氧化物。所获得的催化剂相当于金属氧化物的部分氟化物。另外,上述铬铝镁复合氧化物(n401ag)中cr的含量相对于催化剂中所含的金属总质量为20.3质量%,al的含量相对于催化剂中所含的金属总质量为76.6质量%,mg的含量相对于催化剂中所含的金属总质量为3.0质量%。
[0070]
(催化剂配制例3)除了将铬锌复合氧化物(pricat62
‑
3m,庄信万丰公司制)变更为氧化铝(n612n,日挥触媒化成株式会社制)25.5g以外,按照与(催化剂配制例1)相同的步骤,进行氟化处理,获得部分氟化的氧化铝。所获得的催化剂相当于金属氧化物的部分氟化物。
[0071]
(催化剂配制例4)除了将反应器的温度从350℃变更为320℃以外,按照与(催化剂配制例1)相同的步骤,配制部分氟化的铬锌复合氧化物。
[0072]
(催化剂配制例5)将1100g的cr(no3)3·
9h2o溶解于2.5升水中,添加28质量%的氢氧化铵水溶液2000g。一边对加热后的4l水进行搅拌,一边将以上述方式获得的水溶液添加至该水中,获得氢氧化物沉淀。之后,将沉淀物过滤,用纯水清洗所获得的固体成分、干燥,将所获得的产物碎解而获得氧化物粉末。在所获得的氧化物粉末中混合3质量%石墨,用压片成型机成形为直径5mm、高度5mm的圆筒状,在氮气中,于420℃下烧成5小时以制成铬氧化物。除了将铬锌复合氧化物(pricat62
‑
3m,庄信万丰公司制)变更为以上述方式获得的铬氧化物36.6g以外,按照与(催化剂配制例1)相同的步骤,获得部分氟化的铬氧化物。
[0073]
(催化剂配制例6)在反应器中填充铬锌复合氧化物(pricat62
‑
3m,庄信万丰公司)38.1g,一边使氮气(n2)以150nml/分钟流过,一边使反应器内升温至300℃。使反应器内维持在大气压下,同
时将所填充的催化剂干燥至从反应器出口获得的气体中的水分达到5体积ppm以下。
[0074]
<例1>使n2以15.6nml/分钟的流量流过包含催化剂配制例1中所配制的催化剂的反应器,将反应器温度设定为250℃。反应器温度稳定后,将245ca以0.047g/分钟、氯化氢以0.013g/分钟的流量供应至反应器中。使反应器出口的粗制气体在水洗后通过碱清洗部、分子筛4a,从而除去酸成分而获得干燥产物。此外,用气相色谱法对反应器出口的粗制气体的各成分进行分析。245ca和氯化氢供应开始10小时后的粗制气体的分析结果示于表2。另外,作为上述245ca,使用实施过脱水处理者。具体而言,在填充有包含245ca的原料组合物1(原料组合物1中245ca的含量:99.9质量%以上)(1kg)的密闭容器中,加入作为脱水剂的分子筛4a(纯正化学株式会社(純正化学社)制)100g,静置3日,实施脱水处理,将脱水处理后的原料组合物2用作上述245ca。上述原料组合物2的水分含量为20质量ppm。水分含量用卡尔费休水分仪来进行测定。作为卡尔费休水分仪,使用微量水分测定装置(三菱化学分析技术株式会社制,ca
‑
200型),在试样导入部上连接液化气气化装置(同社制,型号:vg
‑
200型),使预先设定好量的上述原料组合物2气化,自动注入到水分仪中。
[0075]
<例2>如表2所示,除了变更氯化氢和n2的供应量以外,按照与例1相同的步骤,获得产物。
[0076]
<例3>如表2所示,除了变更催化剂的种类以外,按照与例1相同的步骤,获得产物。
[0077]
<例8>如表2所示,除了变更氯化氢和n2的供应量以外,按照与例1相同的步骤,获得产物。
[0078]
<例9>如表2所示,除了变更反应温度以及245ca、氯化氢和n2的供应量以外,按照与例1相同的步骤,获得产物。
[0079]
<例10>如表2所示,除了变更反应温度以及245ca、氯化氢和n2的供应量以外,按照与例1相同的步骤,获得产物。
[0080]
<例11>如表2所示,除了变更245ca、氯化氢和n2的供应量以外,按照与例1相同的步骤,获得产物。
[0081]
<例12>使n2以81.5nml/分钟的流量流过包含催化剂配制例4中所配制的催化剂的反应器,将反应器温度设定为330℃。反应器温度稳定后,将245ca以0.244g/分钟、氯化氢以0.066g/分钟的流量供应至反应器中。使反应器出口的粗制气体在水洗后通过碱清洗部、分子筛4a,从而除去酸成分而获得干燥产物。此外,用气相色谱法对反应器出口的粗制气体的各成分进行分析。245ca和氯化氢供应开始10小时后的粗制气体的分析结果示于表2。
另外,作为上述245ca,使用实施过与例1同样的脱水处理者。
[0082]
<例13>如表2所示,除了变更反应温度以及245ca、氯化氢和n2的供应量以外,按照与例12相同的步骤,获得产物。
[0083]
<例14>如表2所示,除了变更反应温度以及245ca、氯化氢和n2的供应量以外,按照与例12相同的步骤,获得产物。
[0084]
<例15>除了将催化剂变更为(催化剂配制例5)所配制的催化剂以外,按照与例1相同的步骤,获得产物。
[0085]
<例16>使n2以85.7nml/分钟的流量流过包含催化剂配制例6中所配制的催化剂的反应器,将反应器温度设定为300℃。反应器温度稳定后,将245ca以0.256g/分钟、氯化氢以0.070g/分钟的流量供应至反应器中。使反应器出口的粗制气体在水洗后通过碱清洗部、分子筛4a,从而除去酸成分而获得干燥产物。此外,用气相色谱法对反应器出口的粗制气体的各成分进行分析。245ca和氯化氢供应开始5小时后的粗制气体的分析结果示于表2。
[0086]
<例17>如表2所示,除了变更反应温度以及245ca、氯化氢和n2的供应量以外,按照与例16相同的步骤,获得产物。
[0087]
表2中,转化率表示反应所消耗的245ca的摩尔量相对于反应中所使用的245ca的摩尔量的比例(单位:%)。244ca选择率表示产物中244ca的摩尔量相对于反应所消耗的245ca的摩尔量的比例(单位:%)。1233yd(z)选择率表示产物中1233yd(z)的摩尔量相对于反应所消耗的245ca的摩尔量的比例(单位:%)。其他选择率表示产物中上述成分以外的成分的摩尔量相对于反应所消耗的245ca的摩尔量的比例(单位:%)。另外,例1~3、8~10、14~15中,反应压力(反应器内的压力)均为0mpag,滞留时间均为30秒。例11~13、16~17中,反应压力(反应器内的压力)均为0mpag,滞留时间均为5秒。此外,后述的例4中,反应压力(反应器内的压力)为0mpag,滞留时间为30秒。
[0088]
[表2]
[0089]
如表2所示,根据本发明的制造方法,能够以高选择率制造hcfc。另外,在使用催化剂配制例3所获得的催化剂来代替例1的催化剂配制例1所获得的催化剂的情况下,尽管能够获得规定的hcfc(例4),但是转化率低于例3,选择率也只有
70%左右,稍微有点不佳。通过例1~4的比较,可以确认在催化剂包含cr的情况下,效果更好。
[0090]
通过例1与例8的比较,可以确认在氯化氢相对于化合物1的摩尔比为0.75以上的情况下,转化率更好。通过例1与例9~10的比较,可以确认在反应温度为180℃(优选225℃)以上的情况下,转化率更好。通过例11~14的比较,可以确认在反应温度为360℃以下的情况下,选择性更好。通过例12与例16~17的比较,可以确认在实施了活化处理的情况下(换而言之,在使用金属氧化物的部分卤化物的情况下),转化率更好。
[0091]
<例5>使n2以15.6nml/分钟的流量流过包含催化剂配制例1中所配制的催化剂的反应器,将反应器温度设定为250℃。反应器温度稳定后,将449pccc以0.082g/分钟、氯化氢以0.013g/分钟的流量供应至反应器中。使反应器出口的粗制气体在水洗后通过碱清洗部、分子筛4a,从而除去酸成分而获得干燥产物。此外,用气相色谱法对反应器出口的粗制气体的各成分进行分析。对449pccc和氯化氢供应开始10小时后的粗制气体进行分析的结果为,449pccc的转化率为63.4%,448occc选择率为94.8%,1437dycc(z)选择率为0.1%。另外,上述转化率表示反应所消耗的449pccc的摩尔量相对于反应中所使用的449pccc的摩尔量的比例(单位:%)。448occc选择率表示产物中448occc的摩尔量相对于反应所消耗的449pccc的摩尔量的比例(单位:%)。1437dycc(z)选择率表示产物中1437dycc(z)的摩尔量相对于反应所消耗的449pccc的摩尔量的比例(单位:%)。
[0092]
<例6>在设置有搅拌机、迪姆罗冷凝器的2升四口烧瓶中加入251.31g例1所获得的244ca、2.51g四正丁基氯化铵(tbac),将烧瓶加热至50℃。将反应温度维持在50℃,用5分钟滴加631.55g的34质量%氢氧化钾(koh)水溶液。之后,继续搅拌30小时,回收有机层。将以上述方式回收的有机层水洗后,蒸馏以获得包含1233yd(e)和1233yd(z)的精制1233yd。1233yd(e)的选择率为8.9%,1233yd(z)的选择率为91.0%。
[0093]
<例7>在设置有搅拌机、迪姆罗冷凝器的0.2升四口烧瓶中加入100.7g例7所获得的448occc、1.0g的作为相转移催化剂的四正丁基溴化铵(tbab),将烧瓶冷却至10℃。将反应温度维持在10℃,用30分钟滴加153.9g的34质量%氢氧化钾(koh)水溶液。之后,继续搅拌38小时。将所获得的反应液分离成有机相和水相两相,回收有机相。将所回收的有机相提纯,获得78.6g的纯度99.5%的1437dycc(z)与1437dycc(e)的异构体混合物。另外,异构体混合物中1437dycc(z)与1437dycc(e)的质量比(1437dycc(z)/1437dycc(e))为99/1。
[0094]
<例17>如表3所示,除了原料组合物2的水分含量为198质量ppm以外,按照与例1相同的步骤,获得产物。
[0095]
<例18>
如表3所示,除了原料组合物2的水分含量为395质量ppm以外,按照与例1相同的步骤,获得产物。
[0096]
<例19>如表3所示,除了原料组合物2的水分含量为1995质量ppm以外,按照与例1相同的步骤,获得产物。
[0097]
表3中,“含水量[质量ppm]”栏显示原料组合物2的水分含量。
[0098]
[表3]
[0099]
如表3所示,可以确认根据本发明的制造方法,在原料组合物2的含水量为2000质量ppm以下(优选400质量ppm以下,更优选200质量ppm以下)的情况下,效果优异。这里引用2019年04月25日提出申请的日本专利申请2019
‑
084111号的说明书、权
利要求书、摘要和附图的全部内容作为本发明说明书的揭示。符号说明
[0100]
1:反应器2:化合物1的供应管路2a:预热器3:氯化氢的供应管路3a:预热器4:氮气的供应管路4a:预热器5:混合物供应管路6:冷却部7:出口管路8:水蒸汽和酸性液的回收槽9:碱清洗装置10:脱水塔11:分析装置20:反应装置
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。