经基因组编辑的鸟类
1.序列表声明
2.本技术含有已经以ascii格式电子提交的序列表,并且在此通过引用以其整体并入。创建于2020年2月24日的所述ascii副本命名为p
‑
585110
‑
pc_st25.txt,并且大小为355.8kb。
技术领域
3.本公开涉及dna编辑剂以及其在制备经dna编辑的细胞和鸟类中的用途。本公开进一步涉及对经dna编辑的鸟类的蛋中的雄性胚胎赋予条件致死表型的方法。
背景技术:
4.zw性别决定系统是决定鸟类、某些鱼类和甲壳类、某些昆虫类和某些爬行类的后代的性别的染色体系统。字母z和w用于将所述系统与xy性别决定系统区分开。在zw系统中,卵子决定后代的性别。雄性是同配性别(zz),而雌性是异配性别(zw)。z染色体更大并且具有更多基因,就像xy系统中的x染色体一样。
5.早期性别鉴定和分离是所有禽类商业应用,尤其是食用蛋行业中的重要方面。对于肉鸡和火鸡来说,性别分离允许根据两种性别的不同需求制定更适合的管理和饲养方案。基本上所有商业孵化场都使用鸡群性别分离。商业价值较低的雄性鸡会在孵化场被剔除,而雌性鸡用于生产蛋。
6.目前,存在三种可用于确定家禽的性别的方法。日龄雏鸡的性别可以通过肛门/泄殖腔鉴定或羽毛分型方法确定。可替代地,可以使雄性雏鸡和雌性雏鸡一起生长到第二性别特性变得显而易见为止,然后可以基于性别将雏鸡分离。肛门/泄殖腔分型依赖于性别相关的解剖结构的表观。羽毛分型是基于在雄性雏鸡与雌性雏鸡之间有所不同的羽毛特性,例如,底色图案以及翅膀羽毛的快/慢生长速率。第三种方法依赖于天然的第二性别特性的表观,例如,雄性的鸡冠和垂肉变得比雌性雏鸡大。
7.对日龄雏鸡的肛门/泄殖腔性别确定困难且昂贵。鉴定鸟类的性别需要有较高技能的人员。虽然更易于执行,但羽毛分型的缺点是受限于鸟类的特定遗传杂交。通过第二性别特性进行的性别分型是最容易执行的方法,但具有以下缺点:需要在孵化之后的第一周使两种性别的鸟类一起生长,这是因为对于孵化场来说,饲养成本和饲养转化率考虑因素可能比肛门/泄殖腔分型的费用更昂贵。
8.最重要的是,仅在美国和欧洲,每年通过各种方法有几乎十亿只雄性雏鸡被销毁。这不仅是经济问题,而且日益成为道德问题。
9.在商业孵化场领域中,需要高通量的方法来防止雄性雏鸡的产生,优选地甚至在蛋阶段,由此避免与存活的雄性雏鸡相关联的问题。
技术实现要素:
10.本文提供了一种用于卵内破坏雄性胚胎雏鸡的技术,包含组合物和方法。此类组
合物和方法是有益的,因为其允许如农户等鸟类繁殖领域的技术人员将雄性后代与雌性后代之间的自然1:1比率偏斜,从而有利于更具商业用途的雌性后代。
11.在一个实施例中,本文提供了一种dna编辑剂,所述编辑剂包括多核苷酸盒,所述多核苷酸盒具有式5'
‑
lha(左同源臂)
‑
oie(光遗传学诱导型元件)
‑
lie(致死诱导元件)
‑
rha(右同源臂)
‑
3'或式5'
‑
lha
‑
lie
‑
oie
‑
rha
‑
3',其中(i)所述lha包括第一核苷酸序列,所述第一核苷酸序列与鸟类的染色体z上的第一对应核苷酸序列基本上同源;(ii)所述oie包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;(iii)所述lie包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述致死促进蛋白与所述诱导子激活的位点特异性重组酶的活性操作性地相关;并且(iv)所述rha包括第四核苷酸序列,所述第四核苷酸序列与鸟类的染色体z上的第二对应核苷酸序列基本上同源。
12.在某些实施例中,所述lha和所述rha中的一个或两个与位于鸟类的染色体z上的经开放转录的区域中的对应核苷酸序列基本上同源。例如,所述经开放转录的区域可以位于鸟类的染色体z上的组氨酸三联体核苷酸结合蛋白1
‑
z(hint1z)基因座处或下游。
13.在某些实施例中,所述诱导子激活的位点特异性重组酶可以是cre重组酶(cre)(seq id no:113)或mag(seq id no:114和seq id no:65)。在某些实施例中,所述诱导子激活的位点特异性重组酶的表达由诱导子诱导。在某些实施例中,所述诱导子是电磁能。例如,所述诱导子可以是波长为450
–
485nm的蓝光。
14.在某些实施例中,所述诱导子激活的位点特异性重组酶包括诱导子激活的位点特异性重组酶的非功能性肽片段,所述非功能性肽片段在所述诱导子存在的情况下组合以形成活性诱导子激活的位点特异性重组酶。
15.在某些实施例中,所述致死诱导蛋白质可以是毒素、促凋亡蛋白、无翅/整合(wnt)信号传导通路的抑制剂、骨形态发生蛋白(bmp)拮抗剂、成纤维细胞生长因子(fgf)拮抗剂、野生型胱天蛋白酶3、组成型活性胱天蛋白酶3、头蛋白或上述蛋白质中的任何蛋白质的致死诱导片段。
16.在一个实施例中,本文公开的所述dna编辑剂包括:(i)lha,所述lha包括seq id no:105的序列;(ii)oie,所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:101的序列连接,所述seq id no:101的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:102的序列连接,所述seq id no:102的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:102的序列连接,所述seq id no:102的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:101的序列连接,所述seq id no:101的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:107的序列连接,所述seq id no:107的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:108的序列连接,所述seq id no:108的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列
连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:108的序列连接,所述seq id no:108的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:107的序列连接,所述seq id no:107的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接;(iii)lie,所述lie包括seq id no:92、或seq id no:94、或seq id no:96或seq id no:98的序列;以及(iv)rha,所述rha包括seq id no:106的序列。
17.在某些实施例中,本文公开的多核苷酸盒可以应用如鸡、火鸡、鸭、鹅、鹌鹑、野鸡或鸵鸟等鸟类。
18.在另一个实施例中,本公开进一步提供了含有本文公开的多核苷酸盒的鸟类细胞。在另一个实施例中,提供了一种嵌合鸟类,所述嵌合鸟类包括含有本文公开的多核苷酸盒的鸟类细胞。
19.在另一方面,进一步提供了使用本文公开的dna编辑剂生成嵌合鸟类的方法。在某些实施例中,所述方法包括以下步骤:将鸟类的细胞与本文公开的外源多核苷酸盒接触,由此生成经基因组编辑的鸟类细胞,以及然后将这些经基因组编辑的鸟类细胞转移到接受者鸟类胚胎中。在另一方面,进一步提供了由以上方法生成的嵌合鸟类。
20.在另一方面,进一步提供了一种诱导鸟类的雄性胚胎致死的方法,所述方法包括以下步骤:向鸟类细胞群体施用本文公开的dna编辑剂,由此生成经基因组编辑的鸟类细胞;将这些经基因组编辑的鸟类细胞转移到接受者鸟类胚胎中;以及将所述胚胎暴露于引发由所述dna编辑剂编码的致死促进蛋白的表达的诱导子中,由此诱导所述鸟类的雄性胚胎致死。
21.在另一个实施例中,本文公开的所述dna编辑剂进一步包括安全锁定元件,所述安全锁定元件插入在所述oie中的启动子下游但插入在对所述诱导子激活的位点特异性重组酶进行编码的序列上游。所述安全锁定元件包括防止由所述oie编码的所述诱导子激活的位点特异性重组酶的转录的核苷酸序列(stop元件)。在一个实施例中,所述stop元件侧接有两个frt位点。在一个实施例中,具有seq id no:120
‑
127之一的序列的dna编辑剂中的每种dna编辑剂包括安全锁定元件。在另一方面,进一步提供了一种使用含有安全锁定元件的dna编辑剂来生成嵌合鸟类的方法。
22.在另一个实施例中,本文提供了一种使用含有安全锁定元件的dna编辑剂诱导鸟类的雄性胚胎致死的方法,所述方法包括以下步骤:向鸟类细胞群体施用此dna编辑剂,由此生成经基因组编辑的鸟类细胞;将这些经基因组编辑的鸟类细胞转移到接受者鸟类胚胎中;以及将所述胚胎暴露于从所述dna编辑剂中去除stop元件的药剂,由此引发由所述dna编辑剂编码的致死促进蛋白的表达并且诱导所述鸟类的雄性胚胎致死。
23.除非另外定义,否则本文所使用的所有技术性和/或科学性术语具有与方面或实施例所属领域的普通技术人员通常所理解的相同的含义。尽管类似或等同于本文所述的那些的方法和材料可以用于实践或测试方面的实施例,但下文描述了示例性方法和/或材料。在冲突的情况下,应以本发明说明书,包含定义为准。另外,材料、方法和实例仅是说明性的并且不意在是必然进行限制。
附图说明
24.本文参考附图仅通过举例的方式来描述一些实施例。现在详细地具体参考附图,应当强调的是所示出的细节是通过举例并且出于某些实施例的说明性讨论的目的。在这点上,利用附图进行的说明使得对本领域的技术人员来说,可以如何实践实施例将是显而易见的。
25.本说明书的结论部分中特别指出并明确要求保护本文所提供的主题。然而,当与附图一起阅读时,可以通过参考以下详细描述最好地理解本实施例的组织和操作方法两者连同其目的、特征和优点,在附图中:
26.图1是展示了生成将孵化仅雌性产蛋雏鸡的光遗传学诱导型鸡系的实施例的动画图。通过将野生型公鸡(zz)与经遗传修饰的母鸡(zw)杂交,所有雌性受精的蛋都将携带野生型zw染色体。所有雄性受精的蛋将携带zz染色体,其中经遗传修饰的z衍生自经遗传修饰的母鸡的基因组。在对受精的蛋的蓝光照射下,此经遗传修饰的染色体上的光遗传学系统将变得有活性并且将激活死亡机制,所述死亡机制将在产卵后很快导致早期雄性胚胎死亡。不会受蓝光照射影响的雌性将孵化、成长到成年期并且将产下用作食物的未受精的蛋。
27.图2展示了用于通过蓝光照射的手段控制基因表达的策略的实施例。产生了两种融合蛋白:具有cre的非活性n末端的cry2(cry2
‑
cren
‑
末端)和与cre的非活性c末端融合的cibn(cibn
‑
cre
‑
c
‑
末端)。在没有蓝光照射的情况下,cre无活性。在蓝光照射下,cry2和cibn形成复合物,并且cre的两个部分结合在一起以形成活性cre酶。
28.图3展示了染色体z上的同源臂的实施例。描绘了hint1z基因座下游的基因组区域。5'臂和3'臂分别是ha
‑
1(左同源臂;lha)和ha
‑
2(右同源臂;rha)。用于扩增臂的引物由空心箭头指示(fwd ha5'p1和rev ha3'p2)。在同源臂之间,在两条dna链上,都存在crispr
‑
cas9的序列(开放框,crispr向导1和3)。图3的下部部分高度详细地示出了两个同源臂之间的区域。展示了seq id no:1中所示的序列,包含lha
‑
crispr
‑
rha片段。
29.图4a
‑
4c展示了靶向载体或dna编辑剂的不同实施例。图4a示出了含有3个主要元件的靶向载体。第一元件是用于同源重组(hr)的侧接整个外源插入盒的5'和3'同源臂(ha)。第二元件是光诱导型系统,在此情况下是cry2
‑
cren和cibn
‑
cre
‑
c。第三元件是致死基因盒。在单个靶向载体策略的一个实施例中,5'ha其后是驱动由自切割肽p2a分离的cry2
‑
cren和cibn
‑
cre
‑
c基因的表达的pgk启动子。此元件其后是含有pgk启动子的其后是loxp
‑
stop
‑
loxp位点(lsl)的外源致死基因盒,所述lsl其后是致死诱导基因。此外源盒其后是3'ha。在光诱导下,cry2
‑
cren和cibn
‑
cre
‑
c二聚化以形成活性形式的cre。后者然后切除lsl元件,由此允许致死诱导基因的表达,所述致死诱导基因导致携带此载体的所有胚胎的胚胎死亡。图4b示出了替代性方法。与使用lsl元件相反,使用dio
‑
lox翻转策略。在dio
‑
lox位点之间,gfp其后是聚腺苷酸化位点#1(pa1),并且致死基因其后是处于相反朝向的不同聚腺苷酸化位点#2(pa2)。在此情况下,在光激活之前,pgk启动子驱动gfp的表达。在光激活下,dio
‑
lox位点之间的盒翻转,并且致死基因现在处于待表达的正确朝向上,而gfp现在放置在相反朝向上并且其不再有活性。图4c示出了又另一个实施例。在激活cre后,去除lsl,并且使cas9和单向导rna(sgrna)表达。这导致在必需基因的编码区域中引入了错义突变(由sgrna靶向),由此诱导胚胎致死。
30.图5a
‑
5f:pgc系衍生和表征。图5a,pgc培养;图5b,左,如所指示的不同多能和生殖
细胞标记物的mrna表达。右,对雌性pgc(左,核糖体s18和w染色体的两种pcr产物)和雄性pgc(右,仅核糖体s18)的性别鉴定的代表性表征。图5c,用抗ssea1抗体进行的pgc染色。图5d,使用lipofectamine 2000试剂用pcagg
‑
gfp质粒对pgc进行的转染。图5e,使用电穿孔用pcagg
‑
gfp质粒对pgc进行的转染。图5f,移植有gfp表达的经培养的pgc之后10天,胚胎的性腺(睾丸)。
31.图6a
‑
6c.设计用于经crispr介导的靶向的sgrna位点。图6a示出了z染色体上针对潜在crispr靶向位点的基因组区域的实例。图6b示出了前12个sgrna序列(向导#1
‑
#12)。选择部分重叠的处于相反朝向的向导#1和#3以进行另外的实验。3
‑
核苷酸pam序列不是向导序列的一部分,并且pam序列不包含在seq id no:66
‑
77中。图6c中示出了向导#1的潜在脱靶的前10个研究结果。seq id no:78
‑
87不包含3
‑
核苷酸pam序列。
32.图7a
‑
7c.使用核酸内切酶测定来验证crispr活性。图7a.使用经退火的wt 320bp pcr产物和经突变的产物在crispr1的预测的切割位点处以所指示的比率进行的核酸内切酶测定的阳性对照。图7b.对用crispr1质粒转染的12个集落的核酸内切酶测定。图7c.对用crispr3质粒转染的12个集落的核酸内切酶测定。crispr1与crispr3的两个预测切割位点之间存在12bp距离。
33.图8a
‑
8d.使用dna测序来验证crispr活性。图8a.crispr1的预测的切割位点处的野生型(wt)基因组区域的dna色谱图,其示出了作为阴性对照的正常序列。图8b.wt和经人工突变的pcr产物的混合物的示出在预测的切割位点后出现双峰(箭头)的,作为阳性对照的序列。图8c.对示出了正常序列的阴性集落的测序。图8d.阳性集落的序列,其示出了在crispr1切割位点之后出现双峰(箭头)。
34.图9a
‑
9f.构建用于将基因组整合到z染色体中的靶向载体。图9a
‑
使用基因组dna作为用于用引物p1和p2进行的pcr反应的模板,所述引物位于侧接含crispr位点的区域的5'ha和3'ha区域(用虚线标出)。图9b
‑
位于hint1z基因座下游的约3kb产物与穿梭载体pjet1.2连接。使用此质粒作为pcr与引物p3和p4的模板。这些引物具有与pcagg
‑
neo
‑
ires
‑
gfp片段上的等效区域相对应的延伸突出序列(用波形括号标出)。图9c
‑
含有两个同源臂的,不包含含crispr位点的区域的,侧接在gibson反应期间结合pcagg
‑
neo
‑
ires
‑
gfp盒的端部的序列的线性化产物(载体)。图9d
‑
使用了pcagg
‑
neo
‑
ires
‑
gfp质粒作为用于用引物p5和p6进行的pcr反应的模板。这些引物含有与同源臂的边缘上的等效区域相对应的延伸突出序列(用波形括号标出)。图9e
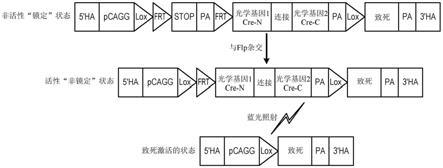
‑
侧接结合同源臂的端部的序列的线性化插入盒。通过gibson组装反应将载体和插入物缝合在一起,以产生如图9f所示的最终靶向载体质粒。
35.图10a
‑
10d.将靶向载体和crispr质粒共转染成到pgc。图10a.用crispr1和hr靶向载体质粒进行的到pcg的经脂质转染介导的pgc共转染。图10b.在g
‑
418选择后两周,>99%的抗性pgc对于gfp为阳性。图10c.在将靶向的pgc注射到宿主胚胎后十天,发现大量细胞定位在性腺(睾丸)中。图10d.对性腺进行解剖,用抗gfp抗体进行免疫染色,并且使用共聚焦显微镜(gfp抗体染色为绿色,并且用4',6二脒基
‑2‑
苯基吲哚(dapi)复染的核为蓝色)进行扫描。
36.图11a
‑
11d.对经facs分选的pgc中的hr整合的验证。图11a.对g
‑
418抗性pgc的facs分选。facs门控被设计成对被分选为池的单个(sin)gfp阳性细胞或96孔板中的单独细胞进行分选。图11b.对于pcr分析,设计了两组用于5'整合位点(p7和p8)和3'整合位点(p9
和p10)的引物。图11c.将从经池化的细胞中提取的基因组dna用作pcr的模板,并且wt dna充当阴性对照。对于分别在5'和3'区域中的正确hr整合,预测的1.6kb和1.8kb带是明显的。图11d.将从雄性和雌性细胞集落中提取的源自经单细胞facs分选的pgc的基因组dna用作pcr的模板,并且wt dna充当阴性对照。对于分别在5'和3'区域中的正确hr整合,预测的1.6kb和1.8kb带是明显的。
37.图12a
‑
12d.hr整合的southern印记分析。图12a
‑
对wt等位基因和经历hr整合的等位基因的southern印记分析中的预期的经bglii切割的产物的示意表示。用于5'、3'整合位点和用于neo的探针被标记为短条。示出了在bglii消化后每个dna探针的预期的产物大小。图12b
‑
通过pcr制备dig标记的探针。在琼脂糖凝胶上对dig标记的探针( )或未标记的(
‑
)进行分析。应注意的是dig标记的产物移位高于其实际大小,证实了dig标记的核苷酸会整合。指示了用于扩增探针的引物组。图12c
‑
利用5'和3'探针对从雄性来源的pgc的经池化的且纯的集落中提取的dna进行southern印记分析。在hr之前,从原始系中提取的wt dna充当阴性对照。图12d
‑
用5'和neo探针对雌性来源的pgc进行的southern印记分析。单个7.5kb带在两种情况下都显而易见,这指示发生了正确的hr并且仅靶向载体的单个副本进行了整合。
38.图13.体外对hek293细胞中的光遗传学系统进行的验证。用pmcherry
‑
cry2
‑
cren、pmcherry
‑
cibn
‑
cre
‑
c和pb
‑
rage
‑
gfp质粒进行的三重转染。转染后二十四个小时,将实验组中的细胞暴露于蓝光照射持续15秒,同时将对照细胞保持在黑暗中(上排)。在照射(下排)之后,将细胞进一步培育24小时。在这些细胞中,gfp表达显而易见,这证实了在蓝光照射下会激活cre酶。
39.图14.对电穿孔之前培育54
‑
60小时的雏鸡胚胎中的卵内光遗传学系统的验证。用pmcherry
‑
cry2
‑
cren、pmcherry
‑
cibn
‑
crec和pb
‑
rage
‑
gfp质粒对鸡胚胎进行的三重电穿孔。电穿孔之后十二个小时,将实验组胚胎卵内暴露于蓝光照射持续15秒,同时将对照胚胎保持在黑暗中(上排)。在照射(下排)之后,将来自两个组的胚胎培育另外12小时。培育之后,在经照射的组中明显可见gfp表达细胞,由此证实了在鸡胚胎中卵内蓝光照射下会激活光遗传学系统和cre酶。
40.图15a
‑
15f.构建cagg启动子下的单个光学基因表达载体。图15a.将光学基因质粒pmcherry
‑
cibn
‑
crec和pmcherry
‑
cry2
‑
cren用作模板以分别使用p40
‑
p41和p42
‑
p43引物扩增光学基因融合蛋白。两种产物共享p2a位点处的引入在引物p41和p42中的重叠序列。这允许单循环突出延伸pcr将两个片段(参见图15b)连接成单件,所述单件与如图15c所示的pjet1.2穿梭载体连接。使用分别含有具有smai和nhei限制位点的尾部的引物p44和p45,生成了图15d中的产物。使用适当的限制酶对此产物进行消化,并且与用相同的酶进行消化的pcagg
‑
ires
‑
gfp(图15e)连接以获得如图15f所示的载体。
41.图16.验证pcagg
‑
光学基因质粒在hek293细胞中的活性。用pcagg
‑
光学基因质粒和pb
‑
rage
‑
mcherry质粒进行的共转染。转染之后二十四个小时,在将阴性对照组保持在黑暗中(上排)的同时,将实验组细胞暴露于蓝光照射(下排)持续15秒。在照射(下排)之后,将细胞进一步培育24小时。在这些细胞中,mcherry表达显而易见(白色箭头),这证实了在蓝光照射下通过pcagg
‑
光学基因质粒会激活cre酶。
42.图17.使用pcagg
‑
光学基因质粒卵内验证单载体策略。用pcagg
‑
光学基因和pb
‑
rage
‑
mcherry质粒对处于阶段14
‑
16h&h的鸡胚胎进行共电穿孔。后一种质粒充当用于光遗传学系统的活性的报告基因。电穿孔之后十二个小时,将实验组胚胎(下排)卵内暴露于蓝光照射持续15秒,同时将对照胚胎保持在黑暗中(上排)。将胚胎进一步培育12小时。培育之后,在两个组中明显可见gfp表达细胞,这表明电穿孔成功,然而,仅在经照射的组中明显可见mcherry表达细胞,这证实了在鸡胚胎中卵内蓝光照射下会激活光遗传学系统和cre酶。
43.图18.在pgk启动子下dta的表达抑制卵内蛋白质合成。用pgk
‑
ires
‑
gfp(上排)或pgk
‑
dta
‑
ires
‑
gfp(下排)表达载体对阶段14
‑
16h&h胚胎进行电穿孔。阴性对照胚胎广泛表达gfp(上排,箭头),这表明蛋白质合成正常。dta表达细胞未示出gfp表达(下排),这表明这些胚胎中的蛋白质合成受到抑制。呈现了仅gfp、仅明场和gfp叠加在明场上的图像。
44.图19a
‑
19b.靶向载体的实施例。在这些载体中,激活酶(例如cre)与致死基因盒分离。在图19a中,将激活酶插入到母鸡妈妈的基因组中,并且将非活性致死盒插入在公鸡的z染色体上,所述公鸡是此等位基因的纯合子。在此情况下,通过将两种转基因亲本杂交而无需光诱导来执行激活雄性胚胎致死。所有雄性的cre去除了母体来源的z染色体上的lsl,由此允许致死基因得以表达,而雌性胚胎携带非活性致死盒,由此其不受影响。图19b.可替代地,用含有在向右方向上的flp重组酶和其后的在反向朝向上的致死基因的由cagg启动子驱动的dio
‑
lox翻转盒对母鸡妈妈的z染色体进行靶向。公鸡再次是与用侧接frt位点的cagg
‑
cre盒靶向的z染色体的纯合子。在将两个转基因亲本杂交时,雄性胚胎将表达位于亲本z染色体上的cre,dio
‑
lox盒翻转并且致死基因变得有活性,由此导致雄性胚胎的胚胎致死。来自此杂交的雌性胚胎的合子含有在卵子发生期间产生的flp重组酶的母体贡献。此母体蛋白从z染色体中去除cagg
‑
cre盒,使雌性胚胎存活,在z染色体上仅存在frt“疤痕”。
45.图20.对来自源自整个新鲜产下的胚盘(bl)和pgc的总rna提取物的cdna进行的rt
‑
pcr,其中hint1z和gapdh的引物作为阳性对照(gapdh引物:正向
–
(seq id no:90);反向
–
(seq id no:91),93bp)。预测大小为153bp的带指示在两个样品中,对位于z染色体上的hint1z进行了转录。
46.图21.含有经修饰的光遗传学系统的两个质粒(pcagg
‑
cibn
‑
cre
‑
c
‑
p2a
‑
cry2
‑
cre
‑
n和pcagg
‑
cry2
‑
cre
‑
n
‑
p2a
‑
cibn
‑
cre
‑
c)的示意图。
47.图22.验证经培养的hek293细胞中的光遗传学系统。将光遗传学质粒pcagg
‑
cibn
‑
crec
‑
ires
‑
cry2
‑
cren
‑
ires
‑
gfp与pb
‑
rage
‑
mcherry共转染。与上述pb
‑
rage
‑
gfp载体类似,pb
‑
rage
‑
mcherry含有多终止密码子序列,所述多终止密码子序列在mcherry编码区域上游侧接有loxp位点。在cre激活下,去除终止密码子,由此允许mcherry得以表达。尽管在共转染并保持在黑暗中的hek293细胞中不存在mcherry阳性细胞,但在暴露于蓝光照射的经共转染的hek293细胞中许多细胞表达mcherry,这证实了pcagg
‑
光学基因的单载体策略保留了所述系统的光遗传学性质。
48.图23.通过电穿孔对鸡胚胎中的光遗传学系统的验证。对还对gfp进行编码的pcagg
‑
cibn
‑
ires
‑
cry2连同pb
‑
rage
‑
mcherry一起进行电穿孔,当存在活性cre重组酶时这会发出红色荧光。当用蓝光进行诱导时,cry2
‑
cren和cibn
‑
crec二聚化,由此激活cre活性。白色圆圈指示重叠荧光区域。
49.图24a
‑
24b.使用dta(图24a)或胱天蛋白酶3(图24b)诱导pgc的细胞死亡。图24a.呈现了dta对pgc的细胞死亡的影响。用对照1pgk
‑
ires
‑
gfp、对照2pcagg
‑
gfp或pgk
‑
dta
‑
ires
‑
gfp将pgc与pcagg
‑
gfp质粒一起进行转染,并且培育24小时、48小时和72小时。使用gfp和pi的流式细胞术评估细胞死亡。结果呈现了gfp pi与仅gfp细胞之间的比率。图24b.呈现了casp的细胞死亡对pgc的细胞死亡的影响。用对照pgk
‑
ires
‑
gfp、pgk
‑
wt胱天蛋白酶3
‑
ires
‑
gfp或pgk
‑
ca胱天蛋白酶3
‑
ires
‑
gfp质粒对pgc进行转染,并且在分析前如图24a所展示的培育24小时、48小时和72小时。
50.图25.含有所有元件并且在蓝光照射下激活致死诱导盒的靶向载体的示意说明。
51.图26a
‑
26b.含有如实例3所解释的“安全锁定”机制的靶向载体的示意图说明。
52.图27a
‑
27d.验证经培养的hek293细胞中的靶向载体。对于体外验证,用单独的tv4(图27a),用pcagg
‑
cre(图27b),或用pcagg
‑
flpo质粒(图27c
‑
27d)对hek293细胞进行转染。将细胞保持在黑暗中(图27c)或在转染(图27d)之后24小时暴露于蓝光持续15秒。在照射之后,将细胞进一步培育持续24小时。
53.图28a
‑
28d.通过电穿孔对鸡胚胎中的靶向载体的验证。将鸡胚胎与质粒一起注射到神经管中并且如本文所述的进行电穿孔。白线表示出于定向目的的神经管和肢芽的背中线。对四个处理组进行了测试:1.单独的tv4的表达(图28a),2.tv4和作为阳性对照的pcagg
‑
cre质粒的共电穿孔(图28b),3.tv4和pcagg
‑
flpo质粒的共电穿孔。将细胞保持在黑暗中(图28c),以及4.在tv4和pcagg
‑
flpo质粒共电穿孔之后暴露于蓝光持续15秒,并且进一步培育12小时(图28d)。
54.图29a
‑
29b.致死诱导基因头蛋白的光依赖性活性。在神经管中用靶向载体tv1、pcagg
‑
flpo和pcagg
‑
ires
‑
gfp质粒对雏鸡胚胎进行电穿孔。靶向载体tv1含有作为致死诱导元件的头蛋白的编码序列。图29a示出了无光诱导的结果;上排,背侧视图;下排,右侧视图。图29b示出了无蓝光诱导的结果。
55.图30a
‑
30e.头蛋白能够组织在胚盘胚胎阶段的胚胎发育。用外源来源的头蛋白对胚盘进行处理。将质粒pcagg
‑
头蛋白
‑
ires
‑
gfp或pcagg
‑
ires
‑
gfp(作为阴性对照)转染至hek293细胞。图30a示出了用抗头蛋白抗体和抗α
‑
微管蛋白
‑
hrp抗体通过western印迹对从经转染的细胞中提取的总蛋白进行了分析。将来自对照和头蛋白表达细胞的条件培养基注射到新鲜产下的受精的蛋中,随后将所述受精的蛋温育24小时(图30b
‑
c)或54小时(图30d
‑
e)。
56.图31a
‑
31c.用tv1在z染色体上进行hr的pgc将性腺成功地定殖在雏鸡胚胎中。图31a示出了用tv1进行hr的纯雌性pgc系和所表达的gfp。图31b示出了pgc注射之后5天胚胎的腹侧视图。pgc定殖于生殖脊,所述生殖脊是性腺的原基(图31b,箭头)。在孵化后第10天处死雌性雏鸡以对卵巢进行分析。图31c示出了含有大量gfp阳性pgc的卵巢(用线描绘)。
57.应当理解,为了说明的简单和清楚起见,图中所示的元件不一定按比例绘制。例如,为了清楚起见,一些元件的尺寸可能相对于其它元件被夸大。进一步地,在认为适当的情况下,可以在附图中重复附图标记以指示对应或相似的元件。
具体实施方式
58.在以下详细描述中,阐述了许多具体细节以便提供对本文所提供的组合物和方法的透彻理解。然而,本领域的技术人员应了解,本公开可以在没有这些具体细节情况下实践。在其它情况中,未对众所周知的方法、程序和组成进行详细描述,以免模糊本文的组合
物和方法。
59.在详细解释本公开的至少一个实施例之前,应当理解本公开在其应用方面不必局限于以下描述中阐述的或通过举例例示的细节。本公开涵盖其它实施例或可以以各种方式实践或执行。
60.术语“包括(comprises)”、“包括(comprising)”、“包含(includes)”、“包含(including)”、“具有(having)”和其同源词涵盖“包含但不限于”。
61.除非上下文另外清楚地指示,否则如本文所使用的单数形式“一个(a)”、“一种(an)”和“所述(the)”包含复数指示物。例如,术语“化合物”或“至少一种化合物”可以包含多种化合物,包含其混合物。
62.在一些实施例中,术语“约”是指与所指示的数字或数字范围存在介于0.0001
‑
5%之间的偏差。在一些实施例中,术语“约”是指与所指示的数字或数字范围存在介于1
‑
10%之间的偏差。在一些实施例中,术语“约”是指与所指示的数字或数字范围存在至多25%的偏差。在一些实施例中,术语“约”是指
±
10%。
63.贯穿本技术,各个实施例可以以范围格式呈现。应当理解的是,范围形式的描述仅仅是为了方便和简洁起见,并且不应被解释为对某些实施例的范围的不灵活限制。因此,对范围的描述应被视为已经具体公开了所有可能的子范围以及所述范围内的单独数值。例如,对范围如1到6的描述应被视为已经具体地公开了如1到3、1到4、1到5、2到4、2到6、3到6等子范围,以及所述范围内的单独数字,例如1、2、3、4、5和6。无论范围的广度如何,这都适用。
64.每当在此指示数值范围时,这意味着包含所指示范围内的任何引用的数字(分数或整数)。短语第一指示数字与第二指示数字“之间的变动范围/范围”以及第一指示数字“到”第二指示数字的“变动范围/范围”在此可互换使用,并且意指包含所述第一指示数字和第二指示数字以及其之间的所有分数和整数。
65.在一个实施例中,本文提供的技术、产物和方法生成了其中仅雌性蛋鸡将孵化,而雄性胚胎将在受精后不久停止发育的鸡品种。因此,消除了剔除雄性雏鸡的需要,并且孵化场节省了50%的宝贵温育空间。重要的是,雌性和衍生自本文公开的方法的蛋两者在每个方面都与产蛋母鸡和目前公众食用的食用蛋相同。
66.如本文所用,术语“鸟类”或“鸟类物种”是指任何禽类物种,包含但不限于鸡、火鸡、鸭、鹅、鹌鹑、野鸡和鸵鸟。在某些实施例中,鸟类是家养鸟类。在某些实施例中,鸟类是原鸡(gallus gallus)。在某些实施例中,鸟类是家养原鸡。在某些实施例中,鸟类是泰和鸡(gallus gallus domesticus)。
67.在某些实施例中,鸟类是雌性。在某些实施例中,鸟是雄性。在某些实施例中,鸟类是肉鸡。在某些实施例中,鸟是母鸡。在某些实施例中,鸟是产蛋母鸡。在某些实施例中,鸟类是家养鸡。在某些实施例中,鸟类是泰和鸡产蛋母鸡。
68.如本文所用,术语“蛋”是指含有存活的或活胚胎的鸟类的禽类蛋。在一个实施例中,术语“蛋”旨在指代受精的禽类蛋。在一个实施例中,蛋是含有能够经历正常胚胎发生的禽类胚胎的蛋。
69.基因组编辑
70.使用经工程化的核酸内切酶进行的基因组编辑是指使用核酸酶在基因组中期望
的位置(例如,在鸟类的z染色体上)处进行切割并产生特异性双链断裂,然后通过如同源导引的修复(hdr)和非同源端部连接(nhej)等细胞内源性过程进行修复的遗传学方法。nhej直接将dna端部接合在双链断裂中,同时hdr利用同源序列作为用于在断裂点处再生缺失dna序列的模板。为了将特定核苷酸修饰引入到基因组dna,在hdr期间必须存在含有期望的序列的dna修复模板。基因组编辑不能使用传统限制核酸内切酶执行,因为大部分限制酶将dna上的仅几个碱基对识别为其靶标,并且所识别的碱基对组合将存在于跨基因组的许多位置中的概率非常高,从而导致多个切口不限于期望的位置。为了克服这一挑战并且产生位点特异性单链或双链断裂,已经发现了几种不同类别的核酸酶并且迄今为止进行了生物工程化。这些核酸酶包含大范围核酸酶、锌指核酸酶(zfn)、转录激活子样效应核酸酶(talen)和crispr/cas系统。
71.巨型核酸酶
–
其通常分组为四个家族:laglidadg家族、giy
‑
yig家族、his
‑
cys盒家族和hnh家族。这些家族的特征在于影响催化活性和识别序列的结构基序。例如,laglidadg家族的成员的特征在于具有保守的laglidadg基序的一个或两个复本。大范围核酸酶的四个家族在保守的结构元件方面彼此明显不同,并且因此,在dna识别序列特异性和催化活性方面明显不同。大范围核酸酶通常存在于微生物物种中,并且其独特性质是具有非常长的识别序列(>14bp),由此使其天然地对在期望的位置处进行切割具有特异性。此大范围核酸酶可以用于在基因组编辑中产生位点特异性双链断裂。本领域的技术人员可以使用这些天然存在的大范围核酸酶,然而此类天然存在的大范围核酸酶的数量有限。为了克服此挑战,已经使用了诱变和高通量筛选方法来产生识别独特序列的大范围核酸酶变体。例如,已经将多种大范围核酸酶融合以产生识别新序列的杂交酶。可替代地,可以对大范围核酸酶的dna相互作用氨基酸进行更改以设计序列特异性大范围核酸酶(例如美国专利8,021,867)。大范围核酸酶可以使用以下中描述的方法进行设计:例如certo,mt等人,《自然方法(nature methods)》(2012)9:073
‑
975;美国专利第8,304,222号;第8,021,867号;第8,119,381号;第8,124,369号;第8,129,134号;第8,133,697号;第8,143,015号;第8,143,016号;第8,148,098号;或第8,163,514号。
72.zfn和talen
–
两种不同类别的经工程化的核酸酶——锌指核酸酶(zfn)和转录激活子样效应核酸酶(talen)——两者都被证明在产生靶向的双链断裂时是有效的。基本上,zfn和talen限制核酸内切酶技术利用与特异性dna结合结构域(分别为一系列锌指结构域或tale重复序列)连接的非特异性dna切割酶。通常,选择其dna识别位点和切割位点彼此分离的限制酶。将切割部分分离并且然后与dna结合结构域连接,由此产生对于期望的序列具有非常高的特异性的核酸内切酶。具有此类性质的示例性限制酶是fokl。另外,fokl的优点是需要二聚化以具有核酸酶活性,并且这意味着特异性随着每个核酸酶伴侣识别独特的dna序列而显著增加。为了增强此作用,已将fokl核酸酶工程化为仅可以充当异源二聚体并具有增加的催化活性。异源二聚体功能的核酸酶避免了不希望的同源二聚体活性的可能性并且由此增加了双链断裂的特异性。
73.因此,例如,可以将zfn和talen构建为核酸酶对,其中所述对的每个成员被设计成在靶向的位点处结合相邻的序列。在细胞中瞬时表达时,核酸酶与其靶位点结合,并且foki结构域异源二聚化以产生双链断裂。这些双链断裂通过非同源端部连接(nhej)通路进行的修复最通常产生插入缺失,所述插入缺失是较少缺失或较少序列插入。因为通过nhej进行
的每种修复是独特的,所以使用单个核酸酶对可以在靶位点处产生具有一系列不同缺失的等位基因系列。缺失的长度的范围在别处通常为几个碱基对到几百个碱基对,但是在细胞培养中通过同时使用两个核酸酶对已经成功生成了更大的缺失(参见例如carlson等人,2012,《美国国家科学院院刊(proc natl acad sci u s a)》;109(43):17382
‑
7;lee等人,2010,《生物技术趋势(trends biotechnol)》;28(9):445
‑
6)。此外,当与靶向的区域具有同源性的dna的片段结合核酸酶对引入时,可以通过同源导引的修复来修复双链断裂以生成特异性修饰(参见例如li等人,2011,《核酸研究(nucleic acids res)》39(1):359
‑
72;miller等人,2010,《自然结构与分子生物学(natstruct mol biol)》17(9):1144
‑
51;urnov等人,2005,《自然(nature)》435(7042):646
‑
51)。
74.尽管zfn和talen两者的核酸酶部分具有相似的性质,但这些经工程化的核酸酶之间的差异在于其dna识别肽。zfns依赖于cys2
‑
his2锌指,并且talen依赖于tale。这两个dna识别肽结构域具有其以组合形式天然存在于其蛋白质中的特性。cys2
‑
his2锌指通常存在于隔开3bp的重复序列中,并且以不同组合的形式存在于多种核酸相互作用蛋白中。另一方面,tale存在于氨基酸与所识别的核苷酸对之间的识别比为一比一的重复序列中。因为锌指和tale两者都以重复模式发生,所以可以尝试不同组合以产生各种序列特异性。用于制备位点特异性锌指核酸内切酶的方法包含,例如,模块组装(其中与三联体序列相关的锌指以排的形式连接以覆盖所需的序列)、open(肽结构域对核苷酸三联体的低严格性选择之后是肽组合对细菌系统中的最终靶标的高严格性选择)和锌指文库的细菌单杂交筛选等。
75.crispr
‑
cas系统
‑
许多细菌和古细菌含有可以使入侵噬菌体和质粒的核酸降解的基于内源性rna的适应性免疫系统。这些系统由产生rna组分的成簇的、规律间隔的短回文重复序列(crispr)基因和对蛋白质组分进行编码的crispr相关(cas)基因组成。crispr rna(crrna)含有与特定病毒和质粒同源的较短延伸并且充当用于导引cas核酸酶以使对应病原体的互补核酸降解的向导。对化脓性链球菌(streptococcus pyogenes)的ii型crispr/cas系统的研究示出,以下三种组分形成rna/蛋白质复合物并且一起足以具有序列特异性核酸酶活性:cas9核酸酶、含有与靶序列同源的20个碱基对的crrna和反式激活crrna(tracrrna)(jinek等人,《科学(science)》(2012)337:816
–
821)。进一步证明了由crrna与tracrrna之间的融合物构成的合成嵌合向导rna(grna)可以导引cas9以切割在体外与crrna互补的dna靶标。还证明了可以使用cas9结合合成grna的瞬时表达以产生多种不同物种中的靶向的双链制动(例如,cho等人,2013,《自然生物技术(nat biotechnol)》31(3):230
‑
2;cong等人,2013,《科学》339(6121):819
‑
23;dicarlo等人,2013,《核酸研究》41(7):4336
‑
43;hwang等人,2013,《自然生物技术》31(3):227
‑
9;jinek等人,2013,《e生命(elife)》2013年1月29日;2:e00471;mali等人,2013,《自然方法》10(10):957
‑
63)。
76.众所周知,用于基因组编辑的cripsr/cas系统含有两个不同组分:向导rna(grna)和如cas9等核酸内切酶。grna通常为对靶同源序列(crrna)与内源细菌rna的组合进行编码的20核苷酸序列,所述内源细菌rna将crrna与单个嵌合转录物中的cas9核酸酶(tracrrna)连接。通过grna序列与补体基因组dna之间的碱基配对将grna/cas9复合物募集到靶向序列。为了成功结合cas9,基因组靶序列还必须含有紧跟靶序列的正确的前间区序列邻近基序(pam)序列。grna/cas9复合物的结合将cas9定位到基因组靶序列,使得cas9可以切割dna的两个链,从而引起双链断裂。正如zfn和talen一样,通过crispr/cas产生的双链断裂可以
经历同源重组或nhej。在某些实施例中,crispr/cas系统包括单个向导rna(sgrna)和cas蛋白。在某些实施例中,crispr/cas系统包括单个向导rna(sgrna)和cas蛋白的复合物。在某些实施例中,crispr/cas系统的cas包括单个多肽。在某些实施例中,crispr/cas系统是核酸内切酶。在某些实施例中,crispr/cas是crispr/cas9。
77.cas9核酸酶具有两个功能结构域:ruvc和hnh,其各自切割不同的dna链。当这两个结构域都有活性时,cas9引起基因组dna中的双链断裂。crispr/cas的显著优势在于此系统的高效率加上能够容易产生合成grna使得能够同时靶向多个基因。grna序列与基因组dna靶序列之间的碱基配对相互作用中的明显灵活性允许与待由cas9切割的靶序列不完美匹配。
78.含有单个非活性催化结构域,ruvc
‑
或hnh
‑
,的cas9酶的经修饰版本被称为“切口酶”。在仅一个活性核酸酶结构域的情况下,cas9切口酶切割仅靶dna的一个链,从而产生单链断裂或“切口”。单链断裂或切口通常使用完整的互补dna链作为模板,通过hdr路径快速修复。然而,将由cas9切口酶引入的两个邻近的相对链切口处理为双链断裂,因为其通常被称为“双切口”crispr系统。双切口可以通过nhej或hdr修复,这取决于对基因靶标的期望的作用。因此,如果特异性和减小的脱靶作用至关重要,则使用cas9切口酶以通过将两个grna设计成具有紧挨着的靶序列并且位于基因组dna的相对链上来产生双切口将降低脱靶作用,因为单独的grna将产生不会改变基因组dna的切口。
79.含有两个非活性催化结构域的cas9酶的经修饰版本(死cas9或dcas9)在仍能够基于grna特异性与dna结合的同时不具有核酸酶活性。可以利用dcas9作为dna转录调节子的平台,以通过将非活性酶融合到已知调节结构域来激活或抑制基因表达。例如,单独的dcas9与基因组dna中的靶序列的结合可能干扰基因转录。在某些实施例中,crispr/cas是crispr/dcas9。
80.为了使用crispr系统,应在靶细胞中表达grna和cas9两者。插入载体在单个质粒上可以含有两个盒,或所述盒由两个单独质粒表达。crispr质粒可公开获得,如来自addgene公司的px330质粒。另外,可以将对cas9编码的mrna和grna引入到靶细胞以及与grna的复合物中的重组cas9蛋白质中(即,将rnp复合物插入到细胞中)。
81.在某些实施例中,crispr/cas系统是1类crispr/cas系统。在某些实施例中,1类crispr/cas系统包括多亚基crrna
–
效应复合物。在某些实施例中,crispr/cas系统是i型crispr
–
cas系统。在某些实施例中,crispr/cas系统是iii型crispr/cas系统。在某些实施例中,crispr/cas系统是iv型crispr
–
cas系统。
82.在某些实施例中,crispr/cas系统是2类crispr/cas系统。在某些实施例中,2类crispr/cas系统包括单亚基crrna
–
效应模块。在某些实施例中,crispr/cas系统是ii型crispr
–
cas系统。在某些实施例中,crispr/cas系统是v型crispr/cas系统。
83.在某些实施例中,2类crispr/cas系统中的cas可以是cas9、cpf1、c2c1、c2c2或c2c3。本领域的普通技术人员将理解crispr/cas系统的分类,因为这是本领域所熟知的(例如《自然评论微生物学(nat rev microbiol)》2017年3月,15(3):169
–
182;《自然评论微生物学》2015年11月,13(11):722
–
736),并且此分类会随着时间的推移而演变(《分子细胞(mol cell)》2015年11月5日,60(3):385
–
397)。在一些实施例中,crispr/cas是本领域已知的任何crispr相关蛋白(cas)核酸内切酶。
84.使用重组腺相关病毒(raavs)进行基因组编辑是基于raav载体,所述载体使得能够在活哺乳动物细胞的基因组中插入、删除或取代dna序列。raav基因组是阳性或阴性感测的单链脱氧核糖核酸(ssdna)分子,其长度为约4.7kb。这些单链dna病毒载体具有较高转导速率并且具有在基因组中不存在双链dna断裂的情况下刺激内源性同源重组的独特性质。本领域的技术人员可以将raav载体设计成靶向所期望的基因组基因座并且在细胞中执行总体和/或细微的内源基因改变两者。raav基因组编辑的优势在于其靶向单个等位基因并且不会导致任何脱靶的基因组改变。
85.dna编辑剂
86.本文描述的技术在某些方面和实施例中提供了一种dna编辑剂。所述dna编辑剂可以使用本领域的技术人员众所周知的重组dna技术来构建。
87.在一个实施例中,本文公开的dna编辑剂可以包括在单个核酸构建体中,或者包括在核酸构建体的组合中。在一个实施例中,所述dna编辑剂包括如下所述的至少两种关键元件:
88.第一元件是核苷酸序列盒,所述核苷酸序列盒将被稳定地整合到鸟类的基因组中的特定位置。当将第一元件整合到鸟类的基因组中时,其会改变鸟类的基因型,但在某些实施例中并且在某些条件下,还会改变鸟类的表型。相比于其它鸟类的表型,所述鸟类的表型改变是本文提供的技术的目的。简而言之,并且如本文提供的实施例所详细描述的,改变的表型可用于防止从雄性胚胎发育出存活的雄性雏鸡。此防止可以为农户和孵化场减轻重大的经济负担,以及防止处死存活的雄性雏鸡。
89.第二元件是侧接第一元件的第一核苷酸靶向序列和第二核苷酸靶向序列。第二元件负责确定第一元件稳定地整合到其中的鸟类的基因组中的位置。如果将外来dna随机整合到任何生物体的基因组中会干扰负责基本细胞功能的基因,这将是有害的。可替代地,如果外来dna整合到dna的非活性片段,则随机整合可能无关紧要。第二元件执行引导将第一元件整合到鸟类的dna中所定义的和预定的区域中的重要功能。在本公开的一个实施例中,第二元件引导将第一元件并入到鸟类的染色体z中的经开放转录的区域中,而对基本细胞功能不会产生任何负作用。
90.本领域的技术人员将理解,术语“dna编辑剂”通常是指促进如鸟类等生物体的基因组改变的任何分子,如核苷酸序列或酶。所述改变可以是例如通过整合到dna的药剂进行的对dna的添加,例如通过同源重组进行的对dna序列的替代或dna的缺失。
91.在一个实施例中,所述dna编辑剂可以在病毒载体中构建(例如,使用单个载体或多个载体)。此类载体通常用于基因转移和基因疗法应用中。不同的病毒载体系统有其独特的优点和缺点。可用于将某些实施例的第一核苷酸序列整合到鸟类的z染色体中的病毒载体包含但不限于腺病毒载体、腺相关病毒载体、甲病毒载体、单纯疱疹病毒载体、逆转录病毒载体或慢病毒载体。
92.如逆转录病毒构建体的病毒构建体包含至少一种转录启动子/增强子或基因座限定元件,或通过如选择性剪接、核rna输出或信使的翻译后修饰等其它手段来控制基因表达的其它元件。除非已经存在于病毒构建体中,否则此类载体构建体还包含包装信号、长末端重复序列(ltr)或其部分,以及适合于所使用的病毒的正链和负链引物结合位点。另外,此构建体典型地包含用于从肽所放置的宿主细胞中分泌肽的信号序列。在某些实施例中,信
id no:108的序列与seq id no:103中所示的序列连接,所述seq id no:103的序列与seq id no:107中所示的序列连接。
100.在某些实施例中,(i)所述lha包括seq id no:105中所示的序列,(ii)所述oie包括seq id no:100中所示的序列,所述seq id no:100的序列与seq id no:116中所示的序列连接,所述seq id no:116的序列与seq id no:101中所示的序列连接,所述seq id no:101的序列与seq id no:103中所示的序列连接,所述seq id no:103的序列与seq id no:102中所示的序列连接,所述seq id no:102的序列与seq id no:104中所示的序列连接,所述seq id no:104的序列与seq id no:116中所示的序列连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:102的序列连接,所述seq id no:102的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:101的序列连接,所述seq id no:101的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,或所述oie包括seq id no:100中所示的序列,所述seq id no:100的序列与seq id no:116中所示的序列连接,所述seq id no:116的序列与seq id no:107中所示的序列连接,所述seq id no:107的序列与seq id no:103中所示的序列连接,所述seq id no:103的序列与seq id no:108中所示的序列连接,所述seq id no:108的序列与seq id no:104中所示的序列连接,所述seq id no:104的序列与seq id no:116中所示的序列连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:108的序列连接,所述seq id no:108的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:107的序列连接,所述seq id no:107的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,(iii)所述lie包括seq id no:92中所示的序列或包括seq id no:94中所示的序列或seq id no:96中所示的序列或seq id no:98中所示的序列,所述seq id no:98的序列与seq id no:104中所示的序列连接,并且(iv)rha包括seq id no:106中所示的序列。
101.在某些实施例中,所述dna编辑剂包括seq id no:120
‑
127之一的序列。
102.dna编辑剂可以对可易于通过其存在或活性检测到的报告蛋白进行编码,所述报告蛋白包含但不限于萤光素酶、荧光蛋白(例如,绿色荧光蛋白)、氯霉素乙酰转移酶、β
‑
半乳糖苷酶、分泌的胎盘碱性磷酸酶、β
‑
内酰胺酶、人生长激素和其它分泌的酶报告子。总体上,报告基因对另外不是由宿主细胞产生的多肽进行编码,所述多肽可通过细胞的分析,例如通过细胞的直接荧光、放射性同位素或分光光度分析并且通常在不需要杀死细胞的情况下监测以供信号分析。在某些实施例中,报告基因对产生宿主细胞的荧光性质的变化的酶进行编码,所述酶可通过定性、定量或半定量功能或转录激活来检测。示例性酶包含酯酶、β
‑
内酰胺酶、磷酸酶、过氧化物酶、蛋白酶(组织纤溶酶原激活子或尿激酶)和其功能可以通过本领域的技术人员已知或未来开发的适当显色或荧光底物检测到的其它酶。报告基因可以就构建体成功地整合到z染色体中进行报告。
103.在某些实施例中,dna编辑剂可以包括对报告多肽进行编码的核苷酸序列。在某些实施例中,报告多肽可以是绿色荧光蛋白(gfp)(seq id no:115)或mcherry/rfp(seq id no:119)。
104.在某些实施例中,dna编辑剂进一步包括用于有效地选择与构建体一起经历同源
重组事件的转移的细胞的阳性和/或阴性选择标记物。阳性选择提供了用于富集已经吸收外来dna的克隆群体的手段。此阳性标记物的非限制性实例包含谷氨酰胺合成酶、二氢叶酸还原酶(dhfr)、如新霉素、潮霉素、嘌呤霉素和杀稻瘟菌素s抗性盒等赋予抗生素抗性的标记物。阴性选择标记物需要针对标记物序列(例如,阳性标记物)的随机整合和/或消除进行选择。此类阴性标记物的非限制性实例包含将更昔洛韦(ganciclovir,gcv)转化为细胞毒性核苷类似物的单纯疱疹胸苷激酶(hsv
‑
tk)、次黄嘌呤磷酸核糖转移酶(hprt)、白喉毒素(dt)和腺嘌呤磷酸核糖转移酶(arpt)。
105.在某些实施例中,对dna编辑剂的蛋白质进行编码的密码子是“经优化的”密码子,即所述密码子是经常出现在例如鸟类物种中的高度表达的基因中的密码子,而不是经常由例如流感病毒使用的那些密码子。此密码子使用提供了蛋白质在禽类细胞中的高效表达。密码子使用模式是许多物种的高度表达的基因的文献中已知的(例如,nakamura等人,1996,《核酸研究》24(1):214
‑
5;mcewan等人,1998,《生物技术(biotechniques)》24(1):131
‑
6,138)。
106.在某些实施例中,dna编辑剂可以进一步包含自切割肽,如2a,包含但不限于p2a、t2a、e2a(wang等人,《科学报告(scientific report)》,5,文章16273(2015)),或内部核糖体进入位点(ires)序列。
107.左同源臂和右同源臂
108.通常认为hr的同源臂的大小应与臂之间的插入物的大小成比例。本领域的普通技术人员将容易地确定和构建具有合适长度的同源臂。在一个实施例中,同源臂可以短至50个碱基。在某些实施例中,(i)lha的长度为约0.5到约5千碱基(kb);(ii)rha的长度为约0.5到约5kb;或(iii)(i)和(ii)的任何组合。在某些实施例中,(i)lha的长度为约1.5kb;(ii)rha的长度为约1.5kb;或(iii)(i)和(ii)的任何组合。在某些实施例中,lhr和/或rha可以短至50个碱基。
109.在某些实施例中,lha的长度为约0.5到约5千碱基(kb)。在某些实施例中,lha的长度为约0.5kb。在某些实施例中,lha的长度为约1kb。在某些实施例中,lha的长度为约1.5kb。在某些实施例中,lha的长度为约2kb。在某些实施例中,lha的长度为约2.5kb。在某些实施例中,lha的长度为约3kb。在某些实施例中,lha的长度为约3.5kb。在某些实施例中,lha的长度为约4kb。在某些实施例中,lha的长度为约4.5kb。在某些实施例中,lha的长度为约5kb。在某些实施例中,lhr可以短至50个碱基。
110.在某些实施例中,rha的长度为约0.5到约5千碱基(kb)。在某些实施例中,rha的长度为约0.5kb。在某些实施例中,rha的长度为约1kb。在某些实施例中,rha的长度为约1.5kb。在某些实施例中,rha的长度为约2kb。在某些实施例中,rha的长度为约2.5kb。在某些实施例中,rha的长度为约3kb。在某些实施例中,rha的长度为约3.5kb。在某些实施例中,rha的长度为约4kb。在某些实施例中,rha的长度为约4.5kb。在某些实施例中,rha的长度为约5kb。在某些实施例中,rha可以短至50个碱基。
111.在某些实施例中,lha和rha中的每一个的长度为约0.5kb。在某些实施例中,lha和rha中的每一个的长度为约1kb。在某些实施例中,lha和rha中的每一个的长度为约1.5kb。在某些实施例中,lha和rha中的每一个的长度为约2kb。在某些实施例中,lha和rha中的每一个的长度为约2.5kb。在某些实施例中,lha和rha中的每一个的长度为约3kb。在某些实施
例中,lha和rha中的每一个的长度为约3.5kb。在某些实施例中,lha和rha中的每一个的长度为约4kb。在某些实施例中,lha和rha中的每一个的长度为约4.5kb。在某些实施例中,lha和rha中的每一个的长度为约5kb。在某些实施例中,lhr和rha可以短至50个碱基。
112.在某些实施例中,左同源臂和右同源臂中的每个同源臂的长度足以允许特异性重组到鸟类的染色体dna中。在一个实施例中,lha和/或rha的长度为至少500个核苷酸,例如长度介于500个核苷酸与3000个核苷酸之间。典型地,lha和/或rha同源臂的期望的大小依赖于侧接有这些臂的盒的长度。较小的盒需要较短的臂并且反之亦然。在一个实施例中,同源臂可以短至50个碱基。
113.在某些实施例中,(i)所述lha与位于鸟类的染色体z上的经开放转录的区域中的对应第一核苷酸序列基本上同源;(ii)所述rha与位于鸟类的染色体z上的经开放转录的区域中的对应第二核苷酸序列基本上同源;或(iii)(i)和(ii)两者。
114.对本领域的技术人员来说显而易见的是,如果第一序列和第二序列在序列方面相似或相同,则第一序列与第二序列“基本上同源”,条件是第一序列和第二序列可以通过同源重组彼此替代。用于测试和鉴定同源重组的方法是本领域众所周知的。
115.在某些实施例中,基本上同源是至少50%相同。在某些实施例中,基本上同源是至少60%相同。在某些实施例中,基本上同源是至少70%相同。在某些实施例中,基本上同源是至少80%相同。在某些实施例中,基本上同源是至少90%相同。在某些实施例中,基本上同源是至少95%相同。在某些实施例中,基本上同源是至少99%相同。
116.在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面50%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面80%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面85%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面90%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面95%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面99%到100%相同。在某些实施例中,lha中的第一核苷酸序列与染色体z上的第一对应核苷酸序列在序列方面100%相同。
117.在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面50%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面80%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面85%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面90%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面95%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面99%到100%相同。在某些实施例中,rha中的第四核苷酸序列与染色体z上的第二对应核苷酸序列在序列方面100%相同。
118.在某些实施例中,lha和/或rha与鸟类的z染色体内的充当整合位点的靶基因座内的至少一个核苷酸序列同源或示出与所述核苷酸序列的同源性或同一性为约70%、71%、72%、73%、74%、75%、76%、77%、78%、79%、80%、81%、82%、83%、84%、85%、86%、
87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%。
119.本领域的技术人员将理解,术语“染色体中的经开放转录的区域”通常是指包含以足以允许其它基因也容易地转录的水平转录的基因的染色体的区域。经开放转录的区域的非限制性实例是靠近在细胞或生物体的生命期间高度转录的管家基因的区域。经开放转录的区域的其它非限制性实例是基因座之间的区域(例如染色质调节元件、非编码dna、“垃圾dna”等)。经不良转录的区域的非限制性实例是位于每个染色体的被称为端粒的端部处的区域,所述区域在细胞或生物体的生命期间不进行转录。在某些实施例中,经开放转录的区域位于鸟类的染色体z上的hint1z基因处或下游。
120.在一个实施例中,lha和/或rha对应于存在于鸟类的z染色体上的基因组序列。在某些实施例中,基因组序列位于有转录活性的基因处或下游(例如,hint1z基因(基因id:395424)处或下游)。另一个设想的靶标是也位于染色体z上的从胚胎发生的早期阶段开始表达的isl1(基因id 369383)。图3展示了染色体z上的位于hint1z基因下游的同源臂的实施例。
121.可以选择lha和/或rha靶向序列,使得例如通过自发同源重组或通过同源性导引的修复(hdr)将lha和/或rha靶向序列特异性地整合到z染色体中,而不是细胞的任何其它染色体中。同源重组可以自发地发生。此外,lha和/或rha靶向序列可以取决于在将第一靶向序列整合到染色体中所依赖的方法进行选择。将核苷酸序列整合到染色体中的方法是本领域所熟知的,包含靶向同源重组、位点特异性重组酶和通过经工程化的核酸酶进行的基因组编辑(参见例如menke d.《genesis》(2013)51:
‑
618;capecchi,《科学》(1989)244:1288
‑
1292;santiago等人,《美国国家科学院院刊》(2008)105:5809
‑
5814;国际专利申请第wo 2014085593号、第wo 2009071334号和第wo 2011146121号;美国专利第8771945号、第8586526号、第6774279号和美国专利申请公开第20030232410号、第20050026157号、第20060014264号)。还设想了pb转座酶。可以通过可公开获得的资源设计用于将核酸改变引入到所关注基因中的药剂。
122.在某些实施例中,lha的第一5'核苷酸对应于染色体z,位置44,914,961。在某些实施例中,lha的第一对应核苷酸序列位于染色体z上,位置44,914,961至位置44,916,456。
123.在某些实施例中,lha的第一5'核苷酸对应于泰和鸡染色体z,组装件grcg6a,nc_006127.5,位置44,914,961。在某些实施例中,lha的第一对应核苷酸序列位于泰和鸡染色体z上,组装件grcg6a,nc_006127.5,位置44,914,961至位置44,916,456。
124.在某些实施例中,rha的第一5'核苷酸对应于染色体z,位置44,916,480。在某些实施例中,rha的第二对应核苷酸序列位于染色体z上,位置44,916,480到位置44,918,043。
125.在某些实施例中,rha的第一5'核苷酸对应于泰和鸡染色体z,组装件grcg6a,nc_006127.5,位置44,916,480。在某些实施例中,rha的第二对应核苷酸序列位于泰和鸡染色体z上,组装件grcg6a,nc_006127.5,位置44,916,480到位置44,918,043。
126.在某些实施例中,(i)所述lha包括seq id no:105中所示的核苷酸序列或其片段,例如seq id no:105中所示的核苷酸序列的至少50个或至少500个连续核苷酸;(ii)所述rha包括seq id no:106中所示的核苷酸序列或其片段,例如seq id no:106中所示的核苷酸序列的至少50个或至少500个连续核苷酸;或(iii)(i)和(ii)两者。在某些实施例中,(i)所述lha包括seq id no:105中所示的核苷酸序列或seq id no:105中所示的核苷酸序列的
至少1000个连续核苷酸的片段;(ii)所述rha包括seq id no:106中所示的核苷酸序列或seq id no:106中所示的核苷酸序列的至少1000个连续核苷酸的片段;或(iii)(i)和(ii)两者。
127.在某些实施例中,所述lha包括seq id no:105中所示的核苷酸序列。在某些实施例中,所述lha包括来自seq id no:105中所示的核苷酸序列的至少50个或至少500个连续核苷酸。在某些实施例中,所述lha包括来自seq id no:105中所示的核苷酸序列的至少1000个连续核苷酸。在某些实施例中,所述lha包括来自seq id no:105中所示的核苷酸序列的500个连续核苷酸。在某些实施例中,所述lha包括来自seq id no:105中所示的核苷酸序列的1000个连续核苷酸。
128.在某些实施例中,所述rha包括seq id no:106中所示的核苷酸序列。在某些实施例中,所述rha包括来自seq id no:106中所示的核苷酸序列的至少50个或至少500个连续核苷酸。在某些实施例中,所述rha包括来自seq id no:106中所示的核苷酸序列的至少1000个连续核苷酸。在某些实施例中,所述rha包括来自seq id no:106中所示的核苷酸序列的500个连续核苷酸。在某些实施例中,所述rha包括来自seq id no:106中所示的核苷酸序列的1000个连续核苷酸。
129.在某些实施例中,lha包括seq id no:105中所示的核苷酸序列或seq id no:105中所示的核苷酸序列的至少50个或至少500个连续核苷酸的片段;并且所述rha包括seq id no:106中所示的核苷酸序列或seq id no:106中所示的核苷酸序列的至少50个或至少500个连续核苷酸的片段。在某些实施例中,lha包括seq id no:105中所示的核苷酸序列或seq id no:105中所示的核苷酸序列的至少1000个连续核苷酸的片段;并且所述rha包括seq id no:106中所示的核苷酸序列或seq id no:106中所示的核苷酸序列的至少1000个连续核苷酸的片段。在某些实施例中,lha包括seq id no:105中所示的核苷酸序列;并且所述rha包括seq id no:106中所示的核苷酸序列。
130.光遗传学诱导型元件
131.启动子
132.如本领域的技术人员显而易见的,“与核苷酸序列功能性地连接的启动子”涵盖启动子位于核苷酸序列的上游并且顺式参与核苷酸序列的转录。与核苷酸序列功能性地连接的启动子的非限制性实例是oie中的第一启动子,所述第一启动子驱动对诱导子激活的位点特异性重组酶进行编码的核苷酸序列在oie中的转录。
133.在某些实施例中,第一启动子是鸟类的组成型启动子。如本领域的技术人员显而易见的,“组成型启动子”涵盖允许其相关核苷酸序列或基因连续转录的启动子。
134.在某些实施例中,第一启动子是鸟类的诱导型启动子。如本领域的技术人员显而易见的,“诱导型启动子”涵盖允许其相关核苷酸序列或基因非连续转录的启动子。在某些实施例中,非连续转录由诱导子调节。在某些实施例中,诱导子对鸟类的细胞是外源的。
135.在某些实施例中,第一启动子可以是pcagg(seq id no:100)、pgk(seq id no:109)、pcmv(seq id no:110)、phsyn(seq id no:111)或pefl
‑
a(seq id no:112)。
136.诱导子激活的位点特异性重组酶
137.在某些实施例中,所述诱导子激活的位点特异性重组酶可以是cre重组酶(cre)(seq id no:113)或mag(seq id no:114和seq id no:65)。
138.根据某些实施例,诱导子启动或增加对诱导子激活的位点特异性重组酶进行编码的核苷酸序列的转录。在其它实施例中,诱导子启动或增加对诱导子激活的位点特异性重组酶的mrna编码的翻译。在又其它实施例中,诱导子启动或增加诱导子激活的位点特异性重组酶的活性。根据某些实施例,诱导子通过使位点特异性重组酶的非功能性片段彼此复合来启动或增加功能性诱导子激活的位点特异性重组酶的形成。如本领域的技术人员显而易见的,酶的多个非功能性片段(肽)可以共同相互作用以形成功能性酶,其中酶在自然界以多肽形式存在,但是每个片段(肽)不与其它片段(肽)共价连接,如图2所展示的。在某些实施例中,所述诱导子激活的位点特异性重组酶包括诱导子激活的位点特异性重组酶的非功能性肽片段,所述非功能性肽片段在诱导子存在的情况下组合以形成活性诱导子激活的位点特异性重组酶。
139.如本文所使用的术语“诱导型”可以涵盖开关的所有方面,不论所涉及的分子机制如何。因此,开关可以包含但不限于基于抗生素的诱导型系统、基于电磁能的诱导型系统、基于小分子的诱导型系统、基于核受体的诱导型系统和基于激素的诱导型系统。在一些实施例中,开关是光诱导型系统、四环素(tet)/dox诱导型系统、脱落酸(aba)诱导型系统、cumate阻遏子/操纵子系统、40ht/雌激素诱导型系统、基于蜕皮激素的诱导型系统或fkbp12/frap(fkbp12
‑
雷帕霉素复合物)诱导型系统。在向未孵化的蛋中施用诱导子的某些实例中,诱导子能够穿透蛋的壳。在某些实例中,诱导子对蛋内的雌性胚胎没有毒性,并且不会改变蛋内的雌性胚胎的发育。
140.如本文所用,术语“开关”是指以协调方式起作用以影响变化的单个组分或一组组分,涵盖生物功能的所有方面,如所述功能的激活、抑制、增强或终止。在一个实施例中,开关涉及用于基因调节的诱导型和/或阻遏型系统。通常,除非存在允许基因表达的一些分子或能量形式(被称为诱导子),否则诱导型系统可能“闭合”。所述分子据说可“诱导表达”。所述闭合发生的方式取决于对照机制以及细胞类型的差异。除非存在某种抑制基因表达的分子或能量形式(被称为抑制剂),否则阻遏型系统“闭合”。所述闭合发生的方式取决于对照机制以及细胞类型的差异。
141.示例性光遗传学开关在图4a
‑
4c中展示,其中每个开关利用来自拟南芥的光敏二聚化蛋白结构域隐花色素2(cry2)和cib1以及作为效应分子的位点特异性重组酶。cry2在框中与cre重组酶,即分裂重组酶的一半融合,而cib1在框中与cre重组酶的另一半融合。因此,当提供诱导子(蓝光)时,cry2和cib1异源二聚化以产生能够执行位点特异性重组的功能性cre重组酶。
142.在某些实施例中,所述诱导子激活的位点特异性重组酶的表达由诱导子诱导。在某些实施例中,所述诱导子是电磁能。在某些实施例中,所述电磁能是波长为380
–
740nm的可见光,或可见光的组分。在某些实施例中,可见光的组分是波长为450
–
485nm的蓝光。
143.在一个实施例中,所述诱导子激活的位点特异性重组酶使用电磁能诱导。可见光的组分的波长可以在450nm
‑
700nm的范围内或介于450nm
‑
500nm之间,即蓝光。蓝光的强度可以为至少0.2mw/cm2,或至少4mw/cm2。可见光的组分的波长可以在620nm
‑
700nm的范围内,即红光。设想了呈任何次序并且呈任何组合形式的可见光的单个或多个应用。可见光可以以单个应用或多个连续应用形式或以脉冲形式递送(脉冲式递送)。
144.此类光遗传学开关的实例描述于muller等人,《生物化学杂志(biol,chem)》2015
年2月,396(2):145
‑
52.doi:10.1515/hsz
‑
2014
‑
0199;motta mena等人,《自然化学生物学(nat chem biol)》2014年3月,10(3):196
–
202;和wo 2014/018423。
145.衍生自p1细菌噬菌体的cre重组酶和衍生自酵母酿酒酵母的flp重组酶是各自识别独特的34个碱基对dna序列的位点特异性dna重组酶(分别被称为“lox”和“frt”)。侧接有lox位点或frt位点的序列可以在cre或flp重组酶分别表达时通过位点特异性重组容易地去除。
146.在某些实施例中,重组酶识别位点可以是lox511、lox5171、lox2272、m2、lox71、lox66、frt、f1、f2、f3、f4、f5、frt(le)、frt(re)、attb、attp、attl或attr。
147.例如,lox序列由侧接有13个碱基对反向重复序列的不对称八碱基对间隔子区域构成。cre通过与13个碱基对反向重复序列结合来重组34碱基对lox dna序列,并且催化间隔子区域内的链切割和重新连接。由6个碱基对将由cre在间隔子区域中进行的交错dna切割分离,以得到充当同源性传感器的重叠区域,以确保仅具有相同重叠区域的重组位点重组。
148.在某些实施例中,在同源重组之后使用位点特异性重组酶系统以从鸟类的染色体中去除dna,例如选择盒。值得注意的是,cre和flp重组酶留下34个碱基对的lox或frt“疤痕”。保留的lox或frt位点通常保留在经修饰的基因座的内含子或3'utr中,并且这些位点通常不会显著干扰基因功能。所述系统还允许生成条件更改的等位基因,所述条件更改的等位基因可以以时间或组织特异性方式失活或激活。
149.因此,cre/lox和flp/frt重组可以涉及引入具有3'和5'同源臂的靶向载体,所述靶向载体含有所关注的突变、两个lox或frt序列和通常置于两个lox或frt序列之间的可选盒。应用阳性选择并且鉴别含有靶向突变的同源重组物。cre或flp的瞬时表达结合阴性选择导致切除选择盒,并且选择其中盒已经丢失的细胞。最终靶向的等位基因含有外源序列的lox或frt疤痕。
150.在某些实施例中,诱导子是热、超声、电磁能或化学品。在某些实施例中,诱导子在产卵前在鸟类体内产生蛋的过程期间递送到蛋中。
151.虽然不希望的雄性胚胎可以自由地暴露于日光(或例如,蓝光),但可能需要在特殊条件下保持转基因细胞和生物体,以防止不希望地激活本文提供的光遗传学系统。并且,众所周知,当在绿/红光条件下培养时雌性鸟类的生产力更高。
152.在某些实施例中,本文提供的方法在绿光下执行。在某些实施例中,本文提供的细胞和生物体保持在绿光下。在某些实施例中,绿光的波长为500
–
565nm。
153.在某些实施例中,本文提供的方法在红光下执行。在某些实施例中,本文提供的细胞和生物体保持在红光下。在某些实施例中,红光的波长为625
–
740nm。
154.设想了几种能量激活方法,例如,具有类似作用的电场能和/或超声。如有必要,可以改变和/或修改开关的蛋白质配对以通过另一种能源来实现最大效果。
155.电场能可以基本上如本领域中所描述的在体内条件下使用约1伏/cm到约10千伏/cm的一个或多个电脉冲施用。代替脉冲或者除了脉冲之外,可以以连续方式递送电场。电脉冲可以在1毫秒与500毫秒之间,或1毫秒与100毫秒之间施加。电场可以连续地或以脉冲方式施加持续约5分钟。如本文所用,“电场能”是细胞暴露于其中的电能。在某些实施例中,电场的强度在体内条件下为约1伏/cm到约10千伏/cm或更大(参见例如wo 97/49450)。
156.如本文所用,术语“电场”包含可变电容和电压下的一个或多个脉冲,并且包含指数和/或方波和/或调制波和/或调制方波形式。应使对电场和电的引用包含引用细胞环境中存在电势差。如本领域已知的,此环境可以通过静电、交流电(ac)、直流电(dc)的方式等来建立。电场可以是均匀的,不均匀的或另外的,并且可以以时间依赖性方式在强度和/或方向上变化。
157.呈任何次序并且呈任何组合形式的电场的单个或多个应用以及超声的单个或多个应用也是可能的。超声和/或电场可以以单个或多个连续应用形式或以脉冲形式递送(脉冲式递送)。
158.致死诱导元件
159.如本领域的技术人员显而易见的,“与第二蛋白质的活性操作性地相关的第一蛋白质”涵盖第二蛋白质反式控制第一蛋白质的运作。与第二蛋白质的活性操作性地相关的第一蛋白质的非限制性实例是在lie中编码的致死促进蛋白,所述致死促进蛋白通过在oie中编码的诱导子激活的位点特异性重组酶的活性而变得有活性。术语“顺式”和“反式”为本领域的普通技术人员所公认和理解的。
160.由于诱导子激活的位点特异性重组酶反式激活致死促进蛋白,因此在不同的实施例中,oie和lie在同一分子或不同分子中的相应位置可能不同。在某些实施例中,oie和lie处于同一分子中。在某些实施例中,oie和lie处于不同分子中。
161.在某些实施例中,激活酶(例如,重组酶,如cre)与致死基因盒分离。在此情况下,将激活酶插入到雄性或雌性鸟类的基因组中,并且将非活性致死盒插入在对应性别的鸟类的z染色体上。在此情况下,仅通过将两个转基因亲本杂交来执行雄性胚胎致死的激活。图19a
‑
19b是靶向载体的实施例,其中激活酶(例如cre)与致死基因盒分离。
162.在某些实施例中,lie进一步包括第二启动子,所述第二启动子与对致死促进蛋白进行编码的第三核苷酸序列功能性地连接。在某些实施例中,第二启动子是鸟类的组成型启动子。在某些实施例中,第二启动子是鸟类的诱导型启动子。在某些实施例中,第二启动子可以是pcagg(seq id no:100)、pgk(seq id no:109)、pcmv(seq id no:110)、phsyn(seq id no:111)或pefl
‑
a(seq id no:112)。
163.致死促进蛋白
164.如本文所用,术语“致死促进蛋白”是指对禽类胚胎(例如雄性胚胎)是致死的,从而防止活雄性鸟类从蛋中孵化的蛋白质。
165.在某些实施例中,可以干扰早期胚胎发生的基本阶段的致死诱导蛋白,如n
‑
钙粘蛋白,以及干扰基本信号传导通路的蛋白质,如由骨形态发生蛋白(bmp)或成纤维细胞生长因子(fgf)介导的那些。
166.在某些实施例中,所述致死诱导蛋白可以是毒素、促凋亡蛋白、无翅/整合(wnt)信号传导通路的抑制剂、骨形态发生蛋白(bmp)拮抗剂、成纤维细胞生长因子(fgf)拮抗剂或其致死诱导片段。在某些实施例中,致死诱导蛋白是毒素或其致死诱导片段。在某些实施例中,致死诱导蛋白是促凋亡蛋白或其致死诱导片段。在某些实施例中,致死诱导蛋白是wnt信号传导通路的抑制剂或其致死诱导片段。在某些实施例中,致死诱导蛋白是bmp拮抗剂或其致死诱导片段。在某些实施例中,致死诱导蛋白是fgf拮抗剂或其致死诱导片段。如本领域的技术人员显而易见的,分子的“致死诱导片段”是足以诱导致死的分子的任何片段。
167.在某些实施例中,致死诱导蛋白可以是白喉毒素a(dta)(seq id no:93)、野生型胱天蛋白酶3(seq id no:95)、组成型活性胱天蛋白酶3(seq id no:97)或头蛋白(seq id no:99)。在某些实施例中,致死诱导蛋白可以是假单胞菌外毒素(genbank登录号abu63124)、白喉毒素(genbank登录号aav70486)或蓖麻毒素(genbank登录号eef27734)。在某些实施例中,致死诱导蛋白可以是白介素2(genbank登录号caa00227)、cd3(genbank登录号p07766)、cd16(genbank登录号np_000560.5)、白介素4(genbank登录号np_000580.1)或白介素10(genbank登录号p22301)。
168.在某些实施例中,致死由rna引导的dna核酸内切酶介导。在某些实施例中,所述dna编辑剂进一步包括对靶向所述鸟类的必需基因的向导rna进行编码的核苷酸序列,此核苷酸序列与所述诱导子激活的位点特异性重组酶的活性操作性地相关。在某些实施例中,所述必需基因可以是骨形态发生蛋白受体ia型(bmpr1a,基因id:396308),骨形态发生蛋白2(bmp2,基因id:378779),骨形态发生蛋白4(bmp4,基因id:396165),或成纤维细胞生长因子受体1(fgfr1,基因id:396516)。
169.如本领域的技术人员显而易见的,“rna引导的dna核酸内切酶”涵盖解开dna并寻找与向导rna分子互补的位点的dna核酸内切酶。在某些实施例中,向导rna分子包括seq id no:66
‑
77之一中所示的核苷酸序列。
170.安全锁定元件
171.在一个实施例中,本文公开的dna编辑剂可以进一步包括“安全锁定”元件,所述安全锁定元件确保光遗传学致死机制在移除安全锁定元件之前为非活性的。此元件通过默认基本上使光遗传学致死系统为非活性的。仅当经基因组编辑的细胞暴露于可以从dna编辑剂中去除安全锁定元件的药剂时,光遗传学致死系统才会变得有活性。此“安全锁定”机制确保在整个生产过程中,不需要使经历hr的细胞免受光,因为光遗传学系统基本上为非活性的。
172.在一个实施例中,将所述安全锁定元件插入在所述oie中的启动子下游但插入在对诱导子激活的位点特异性重组酶进行编码的序列上游。所述安全锁定元件包括防止由oie(参见图26a,非活性“锁定”状态)编码的诱导子激活的位点特异性重组酶转录的核苷酸序列(stop元件)。在一个实施例中,所述安全锁定元件包括蛋白质和其后的聚腺苷酸化位点的编码序列。在其它实施例中,也可以使用将防止下游编码序列转录的任何其它序列作为安全锁定元件。在一个实施例中,所述安全锁定元件的侧接有两个frt位点。因此,此安全锁定元件可以在flp重组酶表达时去除。一旦移除了安全锁定元件,在oie中编码的光学基因就可以进行转录并且以光依赖方式变得有活性(图26a,活性“非锁定”状态)。
173.在另一个实施例中,安全锁定元件和对诱导子激活的位点特异性重组酶进行编码的序列侧接有lox序列(图26b,非活性“锁定”状态)。在表达cre重组酶时,可以去除安全锁定元件以及对诱导子激活的位点特异性重组酶进行编码的序列,由此允许在lie中编码的致死促进蛋白进行表达(图26b,致死激活的状态)。
174.嵌合雏鸡
175.如本文所用,术语“嵌合”、“嵌合体”或“嵌合雏鸡”是指含有本文公开的dna编辑剂的鸟类细胞,或具有含有本文公开的dna编辑剂的细胞的鸟类。还应当注意,嵌合体胚胎或嵌合体成年鸟类也可以被称为“替代者”;因此,这些术语可以互换使用。嵌合鸟类细胞的代
表性实例包含但不限于鸟类原始生殖细胞(pgc),如性腺pgc、血液pgc、生殖新月pgc或含有本文公开的dna编辑剂的配子。嵌合鸟类的代表性实例包含但不限于具有含有本文公开的dna编辑剂的细胞的鸡、火鸡、鸭、鹅、鹌鹑、野鸡或鸵鸟。
176.如本文所使用的,术语“方法”是指用于完成给定任务的方式、手段、技术和程序,包含但不限于化学、药理学、生物学、生物化学以及医学领域的从业者已知的或容易依据已知方式、手段、技术和程序开发的那些方式、手段、技术和程序。
177.在一个实施例中,提供了一种鸟类细胞,所述鸟类细胞包括本文公开的外源多核苷酸盒,所述外源多核苷酸盒包括式5'
‑
oie
‑
lie
‑
3'或式5'
‑
lie
‑
oie
‑
3',其中(i)所述oie是光遗传学诱导型元件,其包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;并且(ii)所述lie是致死诱导元件,其包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述第三核苷酸序列与诱导子激活的位点特异性重组酶的活性操作性地相关。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。
178.在另一方面,进一步提供了一种鸟类细胞群体,所述鸟类细胞群体包括鸟类细胞,所述鸟类细胞包括如本文公开的外源多核苷酸盒。所述多核苷酸盒包括式5'
‑
oie
‑
lie
‑
3'或式5'
‑
lie
‑
oie
‑
3',其中(i)所述oie是光遗传学诱导型元件,其包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;并且(ii)所述lie是致死诱导元件,其包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述第三核苷酸序列与诱导子激活的位点特异性重组酶的活性操作性地相关。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。
179.在某些实施例中,鸟类细胞包括外源多核苷酸盒,所述外源多核苷酸盒包括式5'
‑
lha
‑
oie
‑
lie
‑
rha
‑
3'或式5'
‑
lha
‑
lie
‑
oie
‑
rha
‑
3',其中(i)所述lha是左同源臂,其包括第一核苷酸序列,所述第一核苷酸序列与鸟类的染色体z上的第一对应核苷酸序列基本上同源;(ii)所述oie是光遗传学诱导型元件,其包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;(iii)所述lie是致死诱导元件,其包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述第三核苷酸序列与诱导子激活的位点特异性重组酶的活性操作性地相关;并且(iv)所述rha是右同源臂,其包括第四核苷酸序列,所述第四核苷酸序列与鸟类的染色体z上的第二对应核苷酸序列基本上同源。
180.在某些实施例中,包括外源多核苷酸盒的鸟类细胞包括鸟类原始生殖细胞(pgc)。在某些实施例中,鸟类pgc可以是性腺pgc、血液pgc或生殖新月pgc。
181.如本文所用,术语“原始生殖细胞”和“pgc”是指存在于早期胚胎中并且在成年鸟类体内可以分化/发育成单倍体配子(即精子和卵子)的二倍体细胞。pgc也可以在早期发育阶段从胚盘中获得。
182.如本领域的技术人员已知的,原始生殖细胞可以从不同发育阶段并且从发育中的禽类胚胎的各个位点分离,所述各个位点如但不限于生殖嵴、发育中的性腺、血液和生殖新月(chang等人,《国际细胞生物学(cell biol int)》21:495
‑
9,1997;chang等人,《国际细胞生物学》19:143
‑
9,1995;allioli等人,《发育生物学》165:30
‑
7,1994;swift,《美国生理学杂志(am j physiol)》15:483
‑
516;pct国际公开第wo 99/06533号)。生殖嵴是本领域的普
通技术人员已知的发育中的胚胎的区段(strelchenko,《兽医产科学(theriogenology)》45:130
‑
141,1996;lavoir,《生殖与发展杂志(j reprod dev)》37:413
‑
424,1994)。通常,pgc可以通过碘酸
‑
雪夫(periodic acid
‑
schiff,pas)技术进行阳性染色。在几种物种中,pgc可以使用抗ssea抗体鉴定(一个明显的例外是火鸡,来自火鸡的pgc不显示ssea抗原)。用于分离和纯化pgc的各种技术是本领域已知的,包含使用ficoll密度梯度离心对来自血液的pgc进行浓缩(yasuda等人,《生殖与生育杂志(j reprod fertil)》96:521
‑
528,1992)。
183.可以使用含有鸡和牛血清、条件培养基、饲养细胞和如fgf2等生长因子的培养基进行对pgc的体外培养(van de lavoir等人,2006,《自然》441:766
–
769.doi:10.1038/nature04831;choi等人,2010,《公共科学图书馆
·
综合(plos one)》5:e12968.doi:10.1371/journal.pone.0012968;macdonald等人,2010《公共科学图书馆
·
综合》5:e15518.doi:10.1371/journal.pone.0015518)。已经示出,可以使用含有用于激活fgf、胰岛素和tgf
‑
β信号传导通路的生长因子的饲养剂替代培养基来繁殖pgc(whyte等人,2015,《干细胞报道(stem cell rep)》5:1171
–
1182.doi:10.1016/j.stemcr.2015.10.008)。
184.可以提供原始生殖细胞(pgc)并通过任何合适的技术调配以执行本发明公开的主题,并在使用前如期望的进行储存、冷冻、培养等。例如,原始生殖细胞可以在适当的胚胎阶段从供体胚胎中收集。禽类发育的阶段在本文中两个领域公认的分期系统之一中所指代的:耶尔基勒迪和柯查夫系统(eyal
‑
giladi&kochav system,eg&k;eyal
‑
giladi和kochav,《发育生物学》49:321
‑
327,1976),所述系统使用罗马数字来指代发育的前原始条纹阶段;以及汉布格尔和汉密尔顿分期系统(hamburger&hamilton staging system,h&h;hamburger&hamilton,《形态学杂志(j morphol)》88:49
‑
92,1951),所述系统使用阿拉伯数字来引用产蛋后的阶段。除非另有说明,否则本文所提到的阶段是根据h&h分期系统的阶段。在某些实施例中,pgc衍生自从阶段14(h&h)胚胎分离的血液。在某些实施例中,pgc衍生自从阶段15(h&h)胚胎分离的血液。在某些实施例中,pgc衍生自从阶段16(h&h)胚胎分离的血液。
185.在一个实施例中,pgc可以在阶段4或生殖新月阶段直至阶段30分离,其中细胞在随后阶段从血液、生殖嵴或性腺中收集。原始生殖细胞的大小通常是体细胞的两倍,并且可以基于大小容易地进行区分和分离。雄性(或同配)原始生殖细胞(zz)可以通过任何合适的技术与异配原始生殖细胞(zw)区分开,如从特定供体收集生殖细胞并对来自所述供体的其它细胞进行分型,所收集的细胞的染色体类型与分型后的细胞相同。
186.使用pgc的替代方案是使用本文公开的dna编辑剂对精子直接进行转染(cooper等人,2016《转基因研究(transgenic res)》26:331
–
347,doi:10.1007/s11248
‑
016
‑
0003
‑
0)。
187.在一个实施例中,为了从在体外编辑的pgc中产生嵌合鸟类,将经外源编辑的细胞在其内源pgc迁移到生殖嵴的阶段静脉内注射到替代宿主胚胎中。“供体”pgc可以属于与替代宿主胚胎相同的物种或属于不同物种。经编辑的“供体”pgc必须保持是活的,并且在一个实施例中,如果其要将形成中的性腺定殖并且通过种系传递经编辑的染色体,则会比过内源pgc。为了提供具有优势的供体pgc,可以通过化学或遗传消融减少内源pgc的数量(smith等人,2015,《男科学(andrology)》3:1035
–
1049doi:10.1111/andr.12107)。已示出将替代胚胎的胚盘暴露于经乳化的白消安中可将供体pgc的种系传递增加到90%以上,但是如果pgc已经进行了培养或冷冻保存,则此比率会大大降低(nakamura等人,2008,《生殖、生育力
和发育(reprod fertil dev)》20:900
–
907.doi:10.1071/rd08138;naito等人,2015,《动物繁殖科学(anim reprod sci)》153:50
–
61.doi:10.1016/j.anireprosci.2014.12.003)。使经编辑的pgc与天然pgc的比率便偏移的其它方法在美国申请第2006/0095980号中进行了描述。
188.在某些实施例中,可以将经遗传修饰的pgc移植到本领域已知的成年性腺中(trefil等人,2017,《科学报告》,10月27日;7(1):14246doi:10.1038/s41598
‑
017
‑
14475
‑
w)。
189.可以通过将细胞解离(例如通过机械解离)并将细胞与药学上可接受的载体(例如,磷酸盐缓冲盐溶液)紧密掺合来调配经遗传修饰的细胞(例如pgc)以施用于其它鸟类。在一个实施例中,原始生殖细胞是性腺原始生殖细胞或血液原始生殖细胞(“性腺”或“血液”是指原始胚胎供体来源的组织)。所施用的原始生殖细胞可以是异配的(zw)或同配的(zz)。在一个实施例中,pgc可以在ph为约6到约8或8.5的生理学上可接受的载体中,以实现期望的作用的合适的量(例如,每胚胎100到30,000个pgc)施用。pgc可以在无其它成分或细胞的情况下施用,或者可以将其它细胞和成分与pgc一起施用。
190.可以在pgc仍可以迁移到发育中的性腺中的任何合适的时间执行卵内向接受者动物施用原始生殖细胞。在一个实施例中,所述施用从根据耶尔基勒迪和柯查夫(eg&k)分期系统的约阶段ix到根据胚胎发育的汉布格尔和汉密尔顿(eg&k)分期系统的约阶段30执行,或在另一个实施例中,在阶段15执行。因此,对于鸡而言,施用的时间为在胚胎发育的第1天、第2天、第3天或第4天期间,例如,第2天到第2.5天。施用通常通过注射到任何合适的靶位点,如由羊膜(包含胚胎)、卵黄囊等限定的区域中完成。在一个实施例中,细胞注射到胚胎本身(包含胚胎体壁)中。在替代性实施例中,可以采用血管内或体腔内注射到胚胎中。在其它实施例中,执行注射到心脏中。当前公开的主题的方法可以在对接受者鸟类在卵内进行预先灭菌的情况下执行(例如,通过使用白消安进行的化学处理或通过γ射线或x射线照射)。如本文所用,术语“灭菌”是指使部分地或完全地不能产生衍生自内源pgc的配子。当从此接受者收集供体配子时,所述供体配子可以作为与供体和接受者的配子的混合物来收集。此混合物可以直接使用,或者所述混合物可以进一步进行处理以丰富其中的供体配子的比例。
191.可以通过任何合适的技术以手动方式或自动方式执行对原始生殖细胞的卵内施用。在一个实施例中,卵内施用通过注射执行。卵内施用的机制并不重要,但所述机制不应过度损伤胚胎的组织和器官或其周围的胚外膜,使得处理不会过度降低孵化率。装配有规格为约18到26的针的皮下注射器适合于所述目的。可以使用开口直径为约20
‑
50微米的锐化拉制玻璃移液管。取决于发育的精确阶段和胚胎的位置,一英寸的针将终止于雏鸡上方的流体中或雏鸡本身中。可以在插入针之前穿过壳冲出或钻出导向孔以防止针损坏或钝化。如果期望,则可以利用如蜡等基本上细菌不可渗透的密封材料将蛋密封,以防止不期望的细菌随后进入。设想了禽类胚胎的高速注射系统将适合于实践本发明公开的主题。如适配成用于实践本文公开的方法的所有此类装置包括含有如本文所述的原始生殖细胞的调配物的注射器,所述注射器被定位成对由设备携带的蛋进行注射。另外,可以提供操作性地连接到注射设备的密封设备,以用于在注射后密封蛋中的孔。在另一个实施例中,可以使用拉制玻璃微量移液管将pgc引入到蛋内的适当位置中,例如直接引入到静脉或动脉血流中,
或者直接引入到心脏中。
192.一旦用经修饰的pgc对蛋进行注射中,就培育嵌合胚胎直到孵化为止。在一个实施例中,将雏鸡饲养到性成熟,其中嵌合鸟类产生衍生自供体pgc的配子。
193.在某些实施例中,鸟类的细胞包括鸟类配子。然后,使用来自嵌合体(或来自已直接遗传操纵的材料,如上文所述)的配子(卵或精子)来饲养创始者鸡(f1)。可以使用本领域已知的分子生物学技术(例如pcr和/或southern印记)来证实种系传递。f1鸡可以反向杂交以生成纯合的zz载体雄性和载体雌性(f2)。然后可以使用创始者鸡f2的配子来扩大繁殖集落。通常使集落生长到性成熟为止。可以通过暴露于引发致死表型的诱导子(例如蓝光)来对从这些鸡群中获得的受精的蛋的雄性早期胚胎死亡率进行测试。诱导后(例如通过蓝光照射),将蛋温育(例如8天)并进行筛选(例如通过烛光)以检测早期胚胎死亡率。
194.在一个实施例中,提供了一种生成嵌合鸟类的方法,所述方法包括向鸟类细胞群体施用外源多核苷酸盒,所述外源多核苷酸盒具有式5'
‑
oie
‑
lie
‑
3'或式5'
‑
lie
‑
oie
‑
3',其中(i)所述oie是光遗传学诱导型元件,其包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;并且(ii)所述lie是致死诱导元件,其包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述第三核苷酸序列与诱导子激活的位点特异性重组酶的活性操作性地相关。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。然后将这些经基因组编辑的细胞注射到接受者鸟类胚胎中。
195.在另一个实施例中,提供了一种生成嵌合鸟类的方法,所述方法包括向鸟类细胞群体施用外源多核苷酸盒,所述外源多核苷酸盒具有式5'
‑
lha
‑
oie
‑
lie
‑
rha
‑
3'或式5'
‑
lha
‑
lie
‑
oie
‑
rha
‑
3',其中(i)所述lha是左同源臂,其包括第一核苷酸序列,所述第一核苷酸序列与鸟类的染色体z上的第一对应核苷酸序列基本上同源;(ii)所述oie是光遗传学诱导型元件,其包括第一启动子,所述第一启动子与第二核苷酸序列功能性地连接,所述第二核苷酸序列对诱导子激活的位点特异性重组酶进行编码;(iii)所述lie是致死诱导元件,其包括第三核苷酸序列,所述第三核苷酸序列对致死促进蛋白进行编码,所述第三核苷酸序列与诱导子激活的位点特异性重组酶的活性操作性地相关;并且(iv)所述rha是右同源臂,其包括第四核苷酸序列,所述第四核苷酸序列与鸟类的染色体z上的第二对应核苷酸序列基本上同源。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。然后将这些经基因组编辑的细胞注射到接受者鸟类胚胎中。
196.在某些实施例中,所述方法进一步包括在卵内培育嵌合鸟类胚胎,直到孵化为止。在某些实施例中,所述方法进一步包括将嵌合鸟类饲养到性成熟,其中嵌合鸟类产生衍生自所施用的细胞的配子。
197.在某些实施例中,通过卵内注射施用经基因组编辑的细胞。在另一个实施例中,可以将胚胎从蛋壳中取出,“卵外”进行注射,并且然后放回在替代蛋壳中。在某些实施例中,所施用的细胞群体衍生自与接受者鸟类胚胎相同的禽类物种。在某些实施例中,所施用的细胞群体衍生自与接受者鸟类胚胎不同的禽类物种。
198.在另一个实施例中,可以通过将经基因组编辑的pgc注射到胚盘来生成嵌合鸟类。通常,可以将经基因组编辑的pgc返回到或注射回到内源pgc所位于的地方。在一个实施例中,可以将经基因组编辑的pgc返回到胚盘中。可替代地,可以将经基因组编辑的pgc返回到
生殖新月、血液、胚胎性腺或甚至成年性腺中。
199.在某些实施例中,经基因组编辑的鸟类细胞群体当接受者胚胎处于根据耶尔基勒迪和柯查夫分期系统的约阶段ix时施用。在某些实施例中,所述鸟类细胞群体当接受者胚胎处于根据汉布格尔和汉密尔顿分期系统的约阶段30时施用。在某些实施例中,所鸟类细胞群体在以下时施用:当接受者胚胎处于根据耶尔基勒迪和柯查夫分期系统的约阶段ix时;并且当受体胚胎处于根据汉布格尔和汉密尔顿分期系统分期系统的约阶段30时。在某些实施例中,所述鸟类细胞群体当接受者胚胎处于根据汉布格尔和汉密尔顿分期系统分期系统的阶段14后施用。
200.在某些实施例中,经基因组编辑的鸟类细胞群体在胚胎辐照后施用。在某些实施例中,辐照包括γ辐照或x射线辐照。在某些实施例中,辐照包括600
‑
800拉德的γ辐照。在某些实施例中,辐照包括600
‑
800拉德的辐照。在某些实施例中,辐照包括400
‑
1000拉德的辐照。在某些实施例中,辐照包括200
‑
1200拉德的辐照。
201.在另一方面,进一步提供了一种可以从上述方法获得的嵌合鸟类。
202.使用方法
203.在一个实施例中,提供了一种生成鸟类细胞的方法,所述方法包括将鸟类细胞与具有如本文所述的式5'
‑
oie
‑
lie
‑
3'或式5'
‑
lie
‑
oie
‑
3'的外源多核苷酸盒接触的步骤。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。在另一个实施例中,所述方法包括将鸟类细胞与具有如本文所述的式5'
‑
lha
‑
oie
‑
lie
‑
rha
‑
3'或式5'
‑
lha
‑
lie
‑
oie
‑
rha
‑
3'的外源多核苷酸盒接触的步骤。所述外源多核苷酸盒稳定地整合到细胞的z染色体中。
204.在另一个实施例中,提供了一种诱导鸟类的受精的蛋的雄性胚胎致死的方法,所述方法包括以下步骤:向鸟类细胞群体施用本文公开的dna编辑剂以生成经基因组编辑的鸟类细胞;将这些经基因组编辑的鸟类细胞转移到接受者鸟类胚胎中;以及将所述胚胎暴露于引发由所述dna编辑剂编码的致死促进蛋白的表达的诱导子,由此诱导鸟类的受精的蛋的雄性胚胎致死。上文已经讨论了dna编辑剂的各种元件,如光遗传学诱导型元件、诱导子激活的位点特异性重组酶、诱导子、致死促进蛋白。
205.在一个实施例中,以上诱导雄性胚胎致死的方法中使用的dna编辑剂包括:(i)lha,所述lha包括seq id no:105的序列,(ii)oie,所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:101的序列连接,所述seq id no:101的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:102的序列连接,所述seq id no:102的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,或所述oie包括seq id no:100的序列,所述seq id no:100的序列与seq id no:116的序列连接,所述seq id no:116的序列与seq id no:107的序列连接,所述seq id no:107的序列与seq id no:103的序列连接,所述seq id no:103的序列与seq id no:108的序列连接,所述seq id no:108的序列与seq id no:104的序列连接,所述seq id no:104的序列与seq id no:116的序列连接,(iii)lie,所述lie包括seq id no:92、或seq id no:94、或seq id no:96或seq id no:98的序列,以及(iv)rha,所述rha包括seq id no:106的序列。
206.在另一个实施例中,提供了一种诱导鸟类的受精的蛋的雄性胚胎致死的方法,所述方法包括以下步骤:向鸟类细胞群体施用含有本文公开的安全锁定元件的dna编辑剂,由
此生成经基因组编辑的鸟类细胞;将这些经基因组编辑的鸟类细胞转移到接受者鸟类胚胎中;将胚胎暴露于从所述stop元件中去除所述stop元件的药剂,由此引发由所述dna编辑剂编码的致死促进蛋白的表达并且诱导鸟类的受精的蛋的雄性胚胎致死。上文已经讨论了dna编辑剂的各种元件,如光遗传学诱导型元件、诱导子激活的位点特异性重组酶、诱导子、安全锁定元件、致死促进蛋白等。在一个实施例中,诱导雄性胚胎致死的上述方法中使用的dna编辑剂包括seq id no:120
‑
127之一的序列。
207.在一个实施例中,从dna编辑剂中去除stop元件需要flp在stop元件侧接有两个frt位点时表达(见图26a,非活性“锁定”状态)。在一个实施例中,flp的表达可以通过将经基因组编辑的鸟类细胞与对flp蛋白进行编码的核苷酸序列(例如seq id no:129或seq id no:131)接触来实现。在表达flp重组酶时,去除插入在启动子与在ole中对诱导子激活的位点特异性重组酶进行编码的序列之间的stop元件,由此允许诱导子激活的位点特异性重组酶进行表达(图26a,活性“非锁定”状态)。当进一步暴露于诱导子(例如蓝光)时,将激活诱导子激活的位点特异性重组酶,从而导致去除ole并且在lie中编码的致死促进蛋白得以表达(图26a,致死激活的状态)。
208.在另一个实施例中,stop元件的去除可以通过将经基因组编辑的鸟类细胞与对cre蛋白进行编码的核苷酸序列(例如seq id no:128或seq id no:132)接触来实现。在一个实施例中,安全锁定元件和对诱导子激活的位点特异性重组酶进行编码的序列侧接有lox序列(图26b,非活性“锁定”状态)。在表达cre重组酶时,去除安全锁定元件以及对诱导子激活的位点特异性重组酶进行编码的序列,由此允许在lie中编码的致死促进蛋白进行表达(图26b,致死激活的状态)。
209.根据本文提供的技术原理,致死诱导的步骤可以在任何发育阶段完成,例如在产卵后不久。越早诱导死亡,胚胎死亡就越早。因此,之后实现了胚胎死亡。如果使用了毒素或促凋亡剂来诱导胚胎死亡,则胚胎在诱导后不久就会死亡。.如果lie基于如bmp等破坏基本信号传导通路的基因,例如通过表达bmp4拮抗剂头蛋白,将在所述通路有活性且需要的发育时间点处诱导细胞死亡。例如,bmp在包括囊胚形成、原肠胚形成、神经胚形成、器官发生的阶段期间有活性。在某些实施例中,致死在产卵期间诱导,并且胚胎在暴露于头蛋白后约36小时内死亡。作为一个实例,如果光遗传学系统在产卵(阶段x
‑
xiii eg&k)后不久激活,则胚胎将在所述阶段死亡。bmp4敲除小鼠胚胎在性交后6.5天(dpc)到约8.5
‑
9dpc,相当于在鸡胚胎发育中温育的前30
‑
36小时,在子宫内死亡。在某些实施例中,致死在从受精到孵化的21天时间段期间诱导。在某些实施例中,致死在从受精到孵化的21天期间诱导不止一次。在某些实施例中,致死在被称为阶段x
‑
xiii eg&k(eyal
‑
giladi和kochav,1976)的早期囊胚形成阶段之前诱导。
210.在某些实施例中,所述方法在体内执行。在某些实施例中,所述方法离体执行。在某些实施例中,所述方法在卵内执行。在某些实施例中,所述方法在体外内执行。
211.应当理解,出于清楚的目的在单独实施例的上下文中描述的某些实施例的某些特征还可以以组合形式提供在单个实施例中。相反,为简洁起见,在单个实施例的上下文中描述的某些实施例的各种特征也可以单独地或以任何适合的子组合形式提供或在适当情况下在其它所述的实施例中提供。在各个实施例的上下文中描述的某些特征不应被视为那些实施例的基本特征,除非所述实施例在不具有那些元件的情况下无效。
212.如在上文所描绘的以及如在以下权利要求书章节中所要求保护的各个实施例和方面在以下实例中找到实验支持。
213.实例
214.现在参考以下实例,所述实例与以上描述一起以非限制性的方式展示了一些实施例。
215.一般来说,本文所使用的命名和所利用的实验室程序包含分子技术、生物化学技术、微生物学技术和重组dna技术。文献中透彻地解释了此类技术。参见例如《分子克隆:实验室手册(molecular cloning:a laboratory manual)》,sambrook等人,(1989);《当代分子生物学实验指南(current protocols in molecular biology)》,第i
‑
iii卷,ausubel,r.m.编辑(1994);ausubel等人,《当代分子生物学实验指南》,约翰
·
威利父子出版公司(wiley and sons),巴尔的摩,马里兰州(baltimore,maryland)(1989);perbal,《分子克隆实用指南(a practical guide to molecular cloning)》,约翰
·
威利父子出版公司,纽约(1988);watson等人,《重组dna(recombinant dna)》,科学美国人书籍(scientific american books),纽约;birren等人(编辑)《基因组分析:实验室手册系列(genome analysis:a laboratory manual series)》,第1
‑
4卷,冷泉港实验室出版社(cold spring harbor laboratory press),纽约(1998);如以下中所述的方法:美国专利第4,666,828号;第4,683,202号;第4,801,531号;第5,192,659号和第5,272,057号;《细胞生物学:实验室手册(cell biology:a laboratory handbook)》,第i
‑
iii卷,cellis,j.e.编辑(1994);《动物细胞培养:基础技术手册(culture of animal cells
‑
a manual of basic technique)》,freshney,威利
‑
里斯公司(wiley
‑
liss),纽约(1994),第三版;《当代免疫学实验手册(current protocols in immunology)》,第i
‑
iii卷,coligan j.e.编辑(1994);stitesx等人(编辑),《基础和临床免疫学(basic and clinical immunology)》(第8版),阿普尔顿&兰格公司(appleton&lange),康涅狄格州诺沃克(norwalk,ct)(1994);mishell和shigii(编辑),《细胞免疫学选定方法(selected methods in cellular immunology)》,弗里曼公司(w.h.freeman and co.),纽约(1980);专利和科学文献中广泛描述了可用的免疫测定,参见例如,美国专利第3,791,932号;第3,839,153号;第3,850,752号;第3,850,578号;第3,853,987号;第3,867,517号;第3,879,262号;第3,901,654号;第3,935,074号;第3,984,533号;第3,996,345号;第4,034,074号;第4,098,876号;第4,879,219号;第5,011,771号和第5,281,521号;《寡核苷酸合成(oligonucleotide synthesis)》gait,m.j.编辑(1984);《核酸杂交(nucleic acid hybridization)》hames,b.d.和higgins s.j.编辑(1985);《转录和翻译(transcription and translation)》hames,b.d.和higgins s.j.编辑(1984);《动物细胞培养(animal cell culture)》,freshney,r.i.编辑(1986);《固定化细胞和酶(immobilized cells and enzymes)》irl出版社,(1986);《分子克隆实用指南》,perbal b.(1984)和《酶学方法(methods in enzymology)》第1
‑
317卷,学术出版社(academic press);《pcr方案:方法和应用指南(pcr protocols:a guide to methods and applications)》,学术出版社,加利福尼亚州圣地亚哥(san diego,ca)(1990);marshak等人,《蛋白质纯化和表征策略——实验过程指南(strategies for protein purification and characterization
‑
a laboratory course manual)》cshl出版社(1996);所述文献全部如本文完全阐述的通过引用并入。贯穿本文件提供了其它的一般参考文献。据信,本文中
microscopy sciences)的histomount)封固并覆盖。
224.pgc转染、选择和facs分选:使用脂质转染或电穿孔完成pgc的质粒转染。对于脂质转染,根据制造商的方案使用了lipofectamine 2000。在96孔板中将3
‑5×
105个细胞接种在含有neaa、丙酮酸盐、维生素、cacl2和生长因子(激活素a、hfgf和卵转移蛋白)的akodmem中。将100ng的质粒和0.25μl的lipofectamine 2000(英杰公司(invitrogen))单独地稀释在一起培育20分钟并且移液在细胞上的20μl的opti
‑
mem混合物中。对于电穿孔,在akodmem中洗涤3
×
105到1.5
×
106个细胞,并在neon电穿孔仪(英杰公司)上在1000v、12毫秒的3个脉冲下进行电穿孔,并且立即分别在96孔板或48孔板中接种在无抗生素的pgc培养基中。1
‑
3小时后更换培养基。72小时后开始用25
‑
100μg/ml g418进行的选择,持续2
‑
4周。选择后,手动或通过facs分选单独分离细胞。对于facs分选,轻轻完成细胞移液,并在pgc培养基中对细胞进行分选。将阳性gfp细胞用facs aria ii分选到新的96孔板中,每孔单个细胞,或者进行池化。(使用bd facs aria ii流式细胞仪(美国碧迪公司(bd,usa))执行facs分析)。
225.质粒制备
226.crispr质粒的克隆:使用麻省理工学院张实验室(zhang lab,mit)的crispr设计工具对crispr序列进行了设计,将px330
‑
gfp质粒(由addgene质粒#42230进行修饰)使用bbsi限制酶进行切割,并且用作crispr位点插入的骨架,以形成sgrna。将sgrna crispr位点——crispr1、crispr3的寡核苷酸(分别为寡核苷酸p34
‑
p35和p36
‑
p37)在95℃下变性30秒,缓慢退火并连接到bbsi切割质粒上,转移到大肠杆菌中,纯化并如所述的进行序列验证(cong l等人,《科学》2013年1月3日10.1126/science.1231143pubmed 23287718)。
227.pjet
‑
has质粒的克隆:用p1和p2引物,使用pcr(kapa公司(kapa)、roche公司(roche))从pgc dna中对染色体z上的hint1z基因座下游的含有5'ha和3'ha两者的基因组区域进行扩增。根据制造商方案对pcr产物进行纯化并将其连接到pjet1.2质粒中(英杰公司)。
228.靶向载体的构建:使用p5
‑
p6引物将pcagg
‑
ires
‑
neo
‑
gfp质粒用作pcr的模板,以对插入物pcagg
‑
ires
‑
neo
‑
gfp进行扩增。使用p3
‑
p4引物将pjet
‑
ha质粒用作pcr模板,以对含有5'ha和3'ha的载体进行扩增。对分别采取0.03pm、0.06pm线性化产物的经纯化的载体和插入物pcr产物进行gibson组装反应。将gibson组装反应产物转移到用于质粒制备的进行序列验证的大肠杆菌(e.coli)中。
229.pcagg
‑
光学基因载体的构建:为了生成pcagg
‑
光学基因载体,将光学基因质粒pmcherry
‑
cibn
‑
crec和pmcherry
‑
cry2
‑
cren11用作模板,以使用p40
‑
p41和p42
‑
p43引物对光学基因进行扩增,这分别得到1.3kb和2.1kb产物。这两种产物共享p2a位点处的引入在引物p41和p42中的重叠序列。使用单循环突出延伸pcr以将两个片段连接成单个3.5kb产物,由琼脂糖凝胶对所述产物进行清洁。使用引物p44和p45将所述产物与用作pcr的模板的pjet1.2穿梭载体连接,所述引物分别含有具有smai和nhei限制位点的尾部。使用适当的限制酶对此产物进行消化,并且将其用作用于连接的插入片段,以连接到用作载体的经smai和nhei消化的pcagg
‑
ires
‑
gfp质粒。将连接产物转移到大肠杆菌细菌中,并对繁殖的质粒进行测序验证。
230.pgk
‑
dta
‑
ires
‑
gfp载体的构建:为了生成pgk
‑
dta
‑
ires
‑
gfp,利用引物p46和p47将表达载体psk bs
‑
pgk
‑
dta用作pcr的模板,所述引物分别含有xmai和nhei限制位点的延
伸序列。用相应酶对0.65kb产物进行消化,并且将所述产物用作插入物以连接到用作用于连接的载体的pgk
‑
ires
‑
gfp质粒中的xmai
‑
nhei互补位点。将连接产物转移到大肠杆菌中,并对繁殖的质粒进行测序验证。
231.卵内电穿孔:基本上如前所述进行卵内电穿孔。将受精的蛋在37.8℃下温育56
‑
60小时,对蛋壳开窗,并且使用开口直径为10
‑
15μm的锐化微量移液管将浓度为约2pg/pl的质粒dna注射到神经管中。使用ecm 830方波电穿孔系统(btx)递送25v、30毫秒的三个脉冲。电穿孔后,用石蜡膜将蛋壳密封,并在分析之前进一步培育胚胎。
232.核酸内切酶测定:使用lipofectamine 2000试剂用crispr1或crispr3质粒对pgc进行转染。四十八小时后,将单独的gfp阳性细胞分离到96孔板中并生长以形成纯集落。收集dna,并且用p38
‑
p39引物对侧接crispr位点的350bp区域进行pcr扩增。pcr产物在95℃下经历变性,并且缓慢退火,并且与t7核酸内切酶在37℃下温育1小时。出于校准目的并作为阳性对照,将350bp pcr产物亚克隆到pjet1.2,并且使用位点导引的诱变对crispr位点进行突变。引入的突变用ataccagataacgtaatccttatttggccgtt(seq id no:3)替代wt序列ataccagataacgtgccttatttggccgtt(seq id no:2)。此人工突变作为用于核酸内切酶分析(图7a)和对照测序(图8b)的阳性对照。
233.southern印记测定:使用dig dna标记混合物(罗氏公司),通过分别用引物p13
‑
p14、p15
‑
p16和p11
‑
p12进行pcr扩增(新英格兰生物实验室公司(neb)的longamp)制备用于5'ha、3'ha和neo基因探针的dig标记。在37℃下用bglii限制酶对十五μg的基因组dna进行消化过夜。通过在0.8%(w/v)琼脂糖凝胶(20v,12小时)上进行电泳将dna片段分离,并且转移到带正电荷的尼龙膜(通用电气医疗集团(ge healthcare))中。在转移之后,使用设定为254nm的uv光在每一侧上对潮湿膜进行交联持续3分钟,并且然后用2xssc冲洗。使用dig easy
‑
hyb杂交溶液(罗氏公司)在42℃下对膜进行预杂交持续2小时。通过加热到95℃持续5分钟对探针(50ng/ml)进行变性,并且立即投入到冰中。将经变性的探针添加到10ml温暖的dig easy
‑
hyb溶液中,并在42℃下杂交持续12小时。在室温下,在搅拌下在2xssc、0.1%sds中将膜洗涤两次,持续10分钟,并且然后在65℃下,在搅拌下在0.2xssc、0.1%sds中洗涤3次,持续30分钟。根据其方案,用dig洗涤和阻断缓冲液组(罗氏公司)完成另外的洗涤和阻断。使用抗digoxigenin
‑
ap抗体1:10000(罗氏公司)然后使用cdp
‑
star试剂(罗氏公司)进行化学发光反应来检测dig标记。使用g:box凝胶成像系统(syngene公司)提取图像。
234.胚胎的pgc注射和整体封固染色:在37.8℃下在55%湿度的情况下,用尖压端部将新鲜产下的蛋温育持续58
‑
62小时。温育后,在蛋壳中打开4
‑
8mm的窗口,并且使用开口为约30
‑
40μm的锐化微量移液管将3000
‑
8000个pgc注射到血流中。用白色蛋膜覆盖窗口,并且进一步用石蜡膜(parafilm)或leukoplast(bsn医疗gmbh)胶带密封。培育胚胎直到孵化。将注射的胚胎的一些性腺进行分离,并且进行完整封固gfp染色。将性腺固定在4%pfa中,用利用含5%正常驴血清的pbs 1%triton封闭的pbs洗涤2小时,并且在1:20稀释的含小鼠抗ssea1抗体或兔抗gfp抗体1:500(艾碧康公司(abcam))的阻断缓冲液染色过夜。在用pbs 1%triton洗涤2小时后,在阻断缓冲液中添加二级驴抗小鼠cy3抗体1:500(杰克逊免疫研究实验室公司(jackson immunoresearch laboratories))或二级alexa488抗兔抗体1:500(美国分子探针公司(molecular probes))持续3小时。用dapi(西格玛公司)对组织进行复染并将其封固在甘油中,并且通过共聚焦显微镜(德国韦茨拉尔的莱卡公司(leica,
wetzlar,germany)的tcs spe)成像。
235.引物p1到p32的序列列示于seq id no:4
‑
35中,引物p34到p47的序列列示于seq id no:36
‑
49中。
236.质粒的序列列示如下:
237.1.px330
‑
gfp(seq id no:50);2.crispr1(seq id no:51);3.crispr3(seq id no:52);4.pjet
‑
has(seq id no:53);5.pcagg
‑
neo
‑
ires
‑
gfp(seq id no:54);6.靶向载体(seq id no:55);7.pmcherry
‑
cry2
‑
cren(seq id no:56);8.pmcherry
‑
cibn
‑
crec(seq id no:57);9.pb
‑
rage
‑
gfp(seq id no:58);10.pcagg
‑
ires
‑
gfp(seq id no:59);11.pcagg
‑
光学基因(seq id no:60);12.pb
‑
rage
‑
mcherry(seq id no:61);13.psk bs
‑
pgk
‑
dta(seq id no:62);14.pgk
‑
ires
‑
gfp(seq id no:63);15.pgk
‑
dta
‑
ires
‑
gfp(seq id no:64)。
238.结果
239.pgc系衍生和表征
240.在胚胎发育的最早阶段期间,在产卵后不久并且在原肠胚形成发起之前,pgc喙侧迁移到位于胚胎外中胚层的前部部分处的生殖新月区域。认为此迁移“保护”pgc免于经历如体细胞所经历的分化过程。直到血管暗域(area opaca vasculosa)、血液和心跳在培育约2.5天(第14
‑
17h&h阶段)后形成,细胞才通过血流返回到胚胎中并且定殖于将产生性腺的生殖嵴。在这些阶段,使用约40
‑
60μm直径开口的微量移液管,从胚胎的脉管系统中收集1
‑
3μl的血液,并将其转移到在48孔板中含有pgc培养基的孔中。pgc培养基允许pgc快速分裂(20
‑
24小时的细胞周期),同时在无饲养剂的条件下保持其未分化状态。培养2
‑
3周后,血细胞降解并消失。在另一1
‑
2个多周内,所培养的pgc变得汇合(图5a)。这些细胞可以进一步生长以供基因修饰,或者可以成功地冷冻和解冻以供以后的修饰。在文献中,使用形态学特征、蛋白质和mrna表达模式,以及最后通过当注射回到阶段匹配的接受者胚胎的脉管系统中时其性腺迁移的能力,对培养中的鸡pgc进行了广泛表征。在所生产的pgc细胞培养物中检查了这些特性,以显示其保留了良好确立的pgc特征。在形态学上,pgc是含有大核的直径为约15
‑
20μm的大的略微微粒化的细胞。pgc是全能细胞,因此其表达如cpouv、sox2、klf4和nanog等多能标记物和两个独特的生殖细胞标记物
–
cvh和dazl。对于每个pgc系,使用作为阳性对照的核糖体s18的引物(p19
‑
p20,256bp产物大小)和w染色体的引物(p17
‑
p18,415bp产物大小)提取dna以供性别决定,以鉴定雌性(图5b)。另外,pgc表达膜ssea
‑
1抗原4(图5c)。
241.由雄性系和雌性系两者的产蛋鸡和肉鸡建立十个pgc系。使用阳离子脂质转染试剂lipofectamine 2000执行质粒转染,所述试剂与带负电的dna相互作用,从而允许其渗透到细胞中。用gfp编码质粒(pcagg
‑
gfp)转染导致约15
‑
20%的转染效率(图5d)。进一步地,使用电穿孔转染pgc导致至多90%的更高效率(图5e)。为了证明所培养的pgc成功地定殖于性腺,将gfp表达pgc注射到阶段14
‑
16h&h的血流中,并将胚胎培育10天。解剖胚胎,鉴定性腺中的gfp阳性细胞(图5f)。
242.设计z染色体上的crispr
‑
cas9靶标
243.在一个实施例中,使用crispr
‑
cas9和同源重组过程完成了将dna编辑到z染色体中。虽然crispr
‑
cas9系统将直接在z染色体的特异性位点处切割dna,但使用同源重组过程的内源修复系统将允许将所期望的dna靶向插入到精确位置中。出于此目的,需要构建含有
对应于z染色体上的插入位点的同源臂的靶向载体质粒。选择位于编码基因hint1z下游的z染色体处的dna插入位点。在许多研究中已经示出使用crispr系统可改善直接dna插入事件。广泛用于所述目的,px330质粒包含sgrna位点和cas9酶。sgrna位点含有将cas9酶导引到靶位点并且导致特异性基因组靶向的dsdb的独特序列。使用crispr设计引擎工具,如图6a所示鉴定了sgrna的独特序列。图6b中根据其评分描绘了前12个向导。向导1到12的序列在seq id no:66
‑
77示出。
244.通过二级结构的常规相似性选择了向导#1和#3,并将其用于检查鸡基因组中的可能脱靶位点,通过错配程度对所述向导进行了评分。图6c和seq id no:78
‑
87中示出了针对向导#1的潜在脱靶的研究的前10个结果。值得注意的是,前6个脱靶具有4个错配,这突出了此向导的特异性。
245.通过切割经修饰的px330质粒执行了dna序列插入,所述质粒含有与cas
‑
9的c末端融合的框内gfp。如前所述,将含有sgrna序列的退火的引物与bbsi限制酶连接。(图6b)。将连接产物转移到大肠杆菌中,对质粒进行纯化,并且通过测序验证sgrna插入。
246.crispr
‑
cas9系统的活性验证
247.通过使pgc在无饲养剂的培养基中生长,获得了源自单个细胞的纯集落,由此允许表征crispr
‑
cas9系统的效率。为此,用px330
‑
gfp
‑
crispr1和px330
‑
gfp crispr3质粒对pgc进行转染,并且使克隆集落生长。从源自表达gfp的单个细胞的集落中提取总基因组dna。通过核酸内切酶测定对dna进行分析并测序。对于核酸内切酶测定,设计阳性对照。此对照是在crispr
‑
cas9活性的预测位点处具有插入的突变的320bp pcr产物。将此产物与类似长度的wt产物分别以突变:wt的不同比率
–
1:15、1:7、1:1混合,并且使退火的混合物经受核酸内切酶活性(图7a)。在1:7和1:1的比率下预测大小为136bp和184bp的两个短带清晰可见,这指示测定正常工作。类似地,对从用crispr1和crispr3质粒转染的12个集落获得的基因组dna执行了相同的测定(图7b、7c)。在12个集落中的9个集落中,观察到了预测大小的清晰双峰。这指示crispr1和crispr3质粒两者均在预测位点处有效地生成了dsdb。
248.对于测序分析,还对用于核酸内切酶分析的pcr产物(图7a
‑
7c)进行了测序(图8a
‑
8d)。对wt阴性对照的测序揭示了crispr1的预测切割位点(图8a)。对作为阳性对照的wt和经人工突变的产物的混合物进行测序,揭示了dna色谱图上紧接着预测的切割位点出现了双峰(箭头,图8b)。对经转染的集落中的同一基因组区域的类似测序揭示阴性(图8c)和阳性(箭头,图8d)集落两者,而后者的情况下为>70%。
249.构建基因组整合的靶向载体
250.为了证明使用hr将靶向的基因组整合到z染色体中,设计了靶向载体(图9a
‑
9f)。载体含有pcagg启动子,其后是新霉素选择基因、内部核糖体进入位点(ires)、gfp和兔β珠蛋白聚腺苷酸化位点。此盒在5'端部和3'端部处分别侧接有约1.5kb同源臂。为了生成此载体,使用引物p1和p2对含有两个同源臂的约3kb dna片段进行了扩增,并且将其与穿梭载体pjet1.2连接。发现此片段的完全测序与鸡基因组序列相同。将此质粒——pjet
‑
has用作模板来生成含有两个在其之间不包含23bp序列的分离的同源臂的线性化pcr产物,所述序列含有crispr sgrna位点。使用p3和p4引物完成了扩增,所述引物在其5'端部处含有对应于pcagg
‑
neo
‑
ires
‑
gfp盒的边缘的序列。此线性pcr产物被称为“载体”。将pcagg
‑
neo
‑
ires
‑
gfp质粒用作模板以生成线性pcr产物。使用含有分别对应于5'ha端部和3'ha端部处的3'端
部和5'端部的序列的引物p5和p6对此片段进行扩增。此产物被称为“插入物”。使用gibson组装反应将载体和插入物缝合在一起,以产生最终的靶向载体。
251.使用靶向载体和crispr质粒对z染色体进行的同源重组
252.能够从单个细胞中获得纯pgc集落使得能够使用如pcr和southern印记等方法鉴定经历正确插入的hr的阳性集落。对于pgc转染,使用了转染效率为5
‑
10%(图10a)的脂质转染或效率>40%的电穿孔。用两个质粒、靶向载体和上述两个crispr质粒之一(crispr1或crispr3)执行转染。转染后,使细胞恢复24小时,并且转移到含g
‑
418的培养基中以供选择。两周的选择之后,仅g
‑
418抗性细胞存活,其中>99%为gfp阳性(图10b)。为了验证细胞保留了其定殖于性腺中的能力,将其注射到宿主胚胎中,如上图1f所述(图10c)。用抗gfp抗体对性腺进行免疫染色,并且使用共聚焦显微镜验证gfp阳性pgc细胞在性腺中的定殖(图10d)。
253.g
‑
418抗性、gfp阳性细胞由潜在的非均质群体组成。因此,为了验证hr整合,并且为了获得纯的同质群体,使用facs分选将单个gfp阳性细胞分离到96孔板中(图11a)。产生了纯集落并且提取了基因组dna以用于pcr和southern印记分析。并行地,对经池化的gfp阳性细胞进行facs分选。对于pcr分析,设计了两组引物。第一组,5'ha上游的正向p7和来自cagg启动子的反向p8(1.6kb的产物大小),以及第二组,来自兔β珠蛋白聚腺苷酸化位点的正向p9和3'ha下游的反向p10(1.8kb的产物长度,图11b)。在经池化的细胞(图11c)和纯的集落(图11d)两者中,检测到了5'和3'的预期产物,这指示在这些细胞中已经发生了正确的hr整合。
254.为了进一步验证正确的hr整合以及证实靶向载体的仅单个拷贝整合到了基因组中,执行了southern印记分析。对来自雄性供体和雌性供体的两种pgc细胞系进行了分析。值得注意的是,雌性系具有z染色体的仅单个拷贝。设计了三种洋地黄毒标记的(dig标记的)dna探针(参见图12a和图12b)。使用长度各自为500bp的引物p11
‑
p12和p13
‑
p14扩增的前两个探针分别位于5'ha和3'ha的上游和下游。使用长度为704bp的引物p15
‑
p16扩增的第三探针被设计为检测靶向载体内的neo基因,由此其允许证实整合了载体的仅单个拷贝。使用了bglii限制酶来切割基因组dna以供分析。彼此间隔约6.5kb的两个限制位点在wt染色体上分别位于5'探针和3'探针的上游和下游。另外的bglii位点位于靶向载体中,从而产生了用于鉴定正确的hr整合的预测的7.5kb和3.3kb片段。对从雄性pgc系提取的基因组dna进行的southern印记分析的结果揭示了分别对于5'位点和3',2个带为预测大小,对于wt等位基因为6.5kb并且对于经历了正确的hr整合的等位基因为7.5kb和3.3kb。这证实了来自经池化的细胞的dna以及纯集落两者(图12c)。对雌性pgc细胞系执行了类似分析。在此情况下,发现5'整合位点的单个带的预测大小为7.5kb。由于雌性基因组含有z染色体的仅单个拷贝,因此未检测到wt等位基因(6.5kb)。探测neo基因,揭示了单个带的预测大小为7.5kb,这证实了靶向载体的仅单个拷贝整合到了基因组中(图12d)。
255.hek293细胞中的光遗传学系统的体外验证和鸡胚胎中的光遗传学系统的卵内验证.
256.为了体内验证诱导型系统的活性并且卵内验证鸡胚胎中的诱导型系统的活性,将三种质粒:pmcherry
‑
cry2
‑
cren、pmcherry
‑
cibn
‑
crec和报告子pb
‑
rage
‑
gfp转染到hek293细胞(图13)和雏鸡胚胎(图14)。前两个光遗传学质粒对证实了成功转染的报告基因mcherry进行编码。pb
‑
rage
‑
gfp表达载体含有在gfp编码区域上游侧接有loxp位点的多终
止密码子的序列。在cre激活下,去除stop密码子,由此允许gfp进行表达。而在进行三重转染并且保持在黑暗中的阴性对照hek293细胞中,不存在gfp阳性细胞(图13,上排)。在暴露于蓝光照射的细胞中,转染后24小时,许多细胞表达gfp(图13,下排),这证实了激活了这些细胞中的光遗传学系统。
257.为了卵内验证光遗传学系统的活性,执行了在阶段16h&h,用pmcherry
‑
cry2
‑
cren、pmcherry
‑
cibn
‑
crec和pb
‑
rage
‑
gfp质粒通过电穿孔三重转染到鸡胚胎神经管中。电穿孔后十二小时,使实验组胚胎经受15秒的蓝光照射,而将阴性对照胚胎保持在黑暗中。将胚胎培育另外12小时,并且在荧光立体镜(图14)下检查gfp表达。虽然在保持在黑暗中的胚胎中(图14,上排),但仅mcherry进行表达,由此证实了电穿孔成功,在实验组的胚胎中,gfp阳性细胞明显可见(图9,下排),这证实了光诱导型cre被激活。
258.光学基因质粒pmcherry
‑
cry2
‑
cren和pmcherry
‑
cibn
‑
crec使用cmv启动子驱动基因的表达,这在鸡细胞中是不利的。为了克服此问题并且为了将两个基因组质粒组合成单个载体,设计了驱动由p2a自切割肽连接的其后是ireg
‑
gfp的cagg启动子下的cibn
‑
crec和cry2
‑
cren表达的质粒载体,所述质粒载体在鸡细胞中具有较高的活性。pcagg
‑
cibn
‑
crec
‑
p2a
‑
cry2
‑
cren
‑
ires
‑
gfp的合成基于对kennedy等人,2010(《自然方法》2010年12月;7(12):973
–
975)中描述的原始光学基因质粒的修饰。这些质粒中的每个质粒对其后是具有cibn
‑
crec(与cre酶的c末端融合的cib1的截短形式)或cry2
‑
cren(与cre酶的n末端融合的隐花色素2,图15a)的ires序列的mcherry进行编码。接下来的克隆的目的是,将具有自切割肽p2a的cagg启动子下的其后是ires
‑
gfp的两个融合光学基因连接。为此,使用了cibn
‑
crec质粒作为pcr的利用p40和p41引物的模板,并且使用了cry2
‑
cren质粒作为pcr的利用p42和p43引物的模板(图15a)。值得注意的是,含有p2a切割位点的引物p41和p42共享重叠序列,所述重叠序列允许两个产物通过单循环突出延伸pcr合并(图15b)。将此含有cibn
‑
crec
‑
p2a
‑
cry2
‑
cren的产物连接到穿梭载体pjet1.2上,并且进行序列验证(图15c)。此质粒用作pcr的利用引物p44和p45的模板,所述引物分别在5'端部和3'端部上向产物添加了smai和nhei限制位点(图15d)。用限制酶消化此产物,并且将其与也使用相同的酶切割的pcagg
‑
ires
‑
gfp质粒连接(图15e)。此连接产物含有cagg启动子,其后是cibn
‑
crec、p2a自切割肽、cry2
‑
cren、ires、gfp和兔β珠蛋白多腺苷酸化位点(在本文中也称为pcagg
‑
光学基因),并进行测序验证(图15f)。
259.为了体外验证pcagg
‑
光学基因载体的活性,将表达作为用于成功转染的报告子的gfp的质粒与pb
‑
rage
‑
mcherry共转染到hek293细胞中。与上述pb
‑
rage
‑
gfp载体(图13)类似,pb
‑
rage
‑
mcherry含有多终止密码子序列,所述多终止密码子序列在mcherry编码区域上游侧接有loxp位点。在cre激活下,去除终止密码子,由此允许mcherry得以表达(图16)。而在进行共转染并且保持在黑暗中的hek293细胞中,不存在mcherry阳性细胞(图16,上排)。相比于暴露于蓝光照射的细胞,许多细胞表达mcherry(图16,下排),这证实了pcagg
‑
光学基因的单载体策略保留了系统的光遗传学性质。
260.为了卵内验证pcagg
‑
光学基因载体在活雏鸡胚胎中的活性,通过电穿孔将质粒与pb
‑
rage
‑
mcherry一起共转染到阶段14
‑
16h&h雏鸡胚胎。电穿孔后十二个小时,将阴性对照组蛋保持在黑暗中,同时将实验组胚胎暴露于蓝光持续15秒(图17)。将两个组进一步温育12小时,并在荧光立体镜下检查。温育后,两个组均显示出高水平的gfp表达,这指示电穿孔
成功。然而,仅在光暴露的组中(图17,下排),鉴定出mcherry阳性表达细胞,这指示使用pcagg
‑
光学基因的单载体策略的光遗传学系统是以光诱导型方式激活的。
261.诱导雏鸡胚胎致死
262.为了证明使用毒素引起死亡的可行性,将通常用作阴性选择标记物的dta12的编码区域克隆到含有其后是ires gfp的pgk启动子(pgk
‑
ires
‑
gfp)的表达载体中。此质粒也用作阴性对照。将dta编码区域克隆到ires序列的上游,从而产生当在细胞中表达时会抑制导致细胞死亡的蛋白质合成的pgk
‑
dta
‑
ires
‑
gfp。
263.为了测试dta表达在鸡胚胎中的作用,用作为阴性对照的pgk
‑
ires
‑
gfp或用pgk
‑
dta
‑
ires
‑
gfp载体对阶段14
‑
16h&h胚胎进行电穿孔。电穿孔后十二个小时,在荧光显微镜下对胚胎的gfp的表达进行分析(图18)。尽管在对照胚胎中,gfp在神经管(图18)中广泛表达,但在dta表达的胚胎中,未检测到gfp表达,这指示这些细胞中的蛋白质合成被阻断。
264.实例2
265.本文所述的实验的目的是将致死诱导盒引入到雄性或雌性来源的pgc的z染色体。最终目标是获得在z染色体上携带基因组插入的母鸡。此染色体将仅分离到下一代雄性胚胎,所述雄性胚胎在蓝光诱导下将激活致死诱导盒,由此雄性胚胎将在胚胎发生的早期阶段死亡。
266.盒aka——靶向载体(tv)具有如本文所述的3个元件。第一元件是“同源臂”。第二元件是“光遗传学诱导型元件”。第三元件是“致死诱导盒”。在一个实施例中,两个约1.5kb的同源臂位于tv的5'端部和3'端部上。这被设计成将同源重组(hr)导引到位于z染色体上的hint1z基因座下游。选择了hr的此位点,因为hint1z基因是在pgc和以及整个胚盘(这些是新鲜产下的雏鸡胚胎)中转录的。z染色体上所有其它经开放转录的区域也是出于此目的的潜在良好候选区。
267.图20示出了在来自pgc和整个胚盘的cdna上使用fwd引物(seq id no:88)和rev引物(seq id no:89)的rt
‑
pcr。产物预测大小为153bp。
268.第二元件是“光遗传学诱导型元件”。在一个实施例中,光遗传学系统基于在某些光波长下被激发并且改变允许其二聚化的构象的蛋白质。可能的是将这些蛋白质与另外的蛋白质融合,使得仅在两个光遗传学蛋白质二聚化时才会存在靶基因的转录。例如,如果第一蛋白质与如gal4
‑
bd(gal4结合结构域)等dna靶向结构域融合,并且第二蛋白质与如gal4
‑
ad(gal4激活结构域)等转录激活子融合,则可能激活下游基因。类似地,可以使用阻遏结构域来抑制基因表达。然而,光学基因的问题在于其具有某种量的基底二聚化,并且因此无论光诱导如何,基因激活都会发生。在对诱导的敏感性与基底二聚化之间找到正确的平衡是使用光学基因的关键。使用了由kennedy、m.j.等人(“活细胞中的蛋白质相互作用的快速蓝光介导的诱导(rapid blue
‑
light mediated induction of protein interactions in living cells.)”《自然方法》7 973
–
975,2010)开发的系统的修改版本。原始光学基因质粒是使用cmv启动子驱动基因表达的pmcherry
‑
cry2
‑
cren和pmcherry
‑
cibn
‑
crec,这在鸡细胞中是不利的。为了克服此问题并将这两个原始光学基因质粒与单个载体组合,将质粒载体设计成使用在鸡细胞中有高度活性的cagg启动子来驱动由ires序列连接并且其后是ireg
‑
gfp的cibn
‑
crec和cry2
‑
cren的表达。pcagg
‑
cibn
‑
crec
‑
ires
‑
cry2
‑
cren
‑
ires
‑
gfp的合成基于对kennedy等人,2010(《自然方法》2010,7(12):973
–
975)中描述
的原始光基因质粒的修饰。克隆的目的是将两个融合光学基因与cagg启动子下的其后是ires
‑
gfp的ires连接起来。也成功地执行了对其中光学基因的顺序,即cry2
‑
cren其后是cibn
‑
crec的类似质粒的构建并且产生了类似的结果。图21示出了上述质粒的示意表示。
269.为了体外验证pcagg
‑
光学基因载体的活性,将表达作为用于成功转染的报告子的gfp的质粒与报告质粒pb
‑
rage
‑
mcherry共转染到hek293细胞中。此质粒含有位于pcagg启动子与mcherry报告基因之间的lox
‑
stop
‑
lox(lsl,rage)元件。因此,当与光学基因质粒共转染时,在蓝光照射和cre激活下,去除lsl并表达mcherry。在黑暗中,当系统处于非活性状态时,仅表达来自光学基因质粒的报告gfp,如图22所示。
270.为了卵内验证活的雏鸡胚胎的光遗传学系统的活性,将光遗传学质粒和报告pb
‑
rage
‑
mcherry质粒共电穿孔到经56
‑
60小时培育的鸡胚胎的神经管中。使用锐化微量移液管将质粒dna混合物注射到神经管的管腔中,并且使用2个间隔开3
‑
5mm的钨电极(电穿孔仪btx830)应用电穿孔。递送各自30v、45毫秒的四个脉冲,每个脉冲之间的间隔为745毫秒。电穿孔后,将蛋密封并温育12
‑
18小时。然后,将对照组中的蛋保持在黑暗中,通过蛋壳将实验组中的蛋暴露于1分钟的蓝光照射下。在分析之前,将两个组放回在温育箱中持续另一个12
‑
18小时。
271.如图23所示,在所有的实验组中,使报告gfp在所有胚胎中高度且广泛地表达,这指示质粒成功地进行了电穿孔和表达。保持在黑暗中的蛋中不存在mcherry表达。相比之下,使mcherry在通过蓝光照射的蛋中在神经管中进行表达。
272.可以通过修饰cry2来制成cibn
‑
cry2的改进的系统。此修饰包含在截短的cry2基因中插入l348f突变,所述基因含有仅前535个氨基酸
‑
cry2
‑
δ535
‑
l348f,如《自然化学生物学》第12卷,第425
–
430页(2016)中所发表的。
273.作为cibn
‑
cry2光遗传学系统的替代方法,可以使用另一个概念类似的系统来实现相同的目标。此系统基于magnet系统,其中两种光遗传学蛋白质——带正电的p
‑
mag和带负电的n
‑
mag在蓝光照射下二聚化。这些蛋白质中的每个蛋白质与cre重组酶的非活性部分融合,并且在两个光学基因二聚化时,会产生活性形式的cre(《自然化学生物学》第12卷,1059
–
1064 2016)。
274.第三元件是“致死诱导盒”。在一个实施例中,致死诱导盒包括通过细胞死亡或严重干预胚胎发生(例如bmp4)的早期阶段所需的分子信号传导通路来驱动早期胚胎死亡的基因(例如毒素)。例如,这可以通过用如白喉毒素a(dta)等毒素诱导程序性细胞死亡(凋亡),通过表达如胱天蛋白酶3等胱天蛋白酶基因或突变组成型活性突变形式的胱天蛋白酶3,或通过表达bmp4
‑
头蛋白的抑制剂蛋白来实现。dta的核苷酸序列在seq id no:92中示出。dta的氨基酸序列在seq id no:93中示出。
275.在一个实施例中,鸡casp3 cds(wt)的核苷酸序列在seq id no:94中示出。鸡casp3cds(wt)的氨基酸序列在seq id no:95中示出。
276.在一个实施例中,鸡casp3的组成型活性(突变)形式的核苷酸序列在seq id no:96中示出。鸡casp3蛋白的组成型活性(突变)形式的氨基酸序列在seq id no:97中示出。
277.在一个实施例中,头蛋白的核苷酸序列在seq id no:98中示出。头蛋白的氨基酸序列在seq id no:99中示出。
278.某些实施例中,“致死诱导盒”基于破坏基本信号传导通路的基因。越早诱导死亡,
胚胎死亡就越早。在某些实施例中,基本信号传导通路涉及骨形态发生蛋白(bmp)。在某些实施例中,bmp是bmp4。在某些实施例中,致死促进蛋白是bmp4的拮抗剂。在某些实施例中,bmp4拮抗剂是头蛋白。在某些实施例中,在bmp通路有活性并且需要的发育阶段诱导致死促进蛋白头蛋白的表达。在某些实施例中,bmp通路在囊胚形成、原肠胚形成、神经形成或器官发生期间有活性或进行诱导。在某些实施例中,光遗传学系统在产卵(阶段x
‑
xiii eg&k)后不久激活。在某些实施例中,光遗传学系统在阶段x激活以诱导致死促进蛋白。在某些实施例中,光遗传学系统在阶段xi激活。在某些实施例中,光遗传学系统在阶段xii激活。在某些实施例中,光遗传学系统在阶段xiii激活。在某些实施例中,光遗传学系统在开始于受精直到孵化的胚胎发育期间的任何给定时间激活。在某些实施例中,致死在从受精到孵化的21天期间诱导。在某些实施例中,致死在受精后1天诱导。在某些实施例中,致死在受精后2天诱导。在某些实施例中,致死在受精后3天诱导。在某些实施例中,致死在受精后4天诱导。在某些实施例中,致死在受精后5天诱导。在某些实施例中,致死在受精后6天诱导。在某些实施例中,致死在受精后7天诱导。在某些实施例中,致死在受精后8天诱导。在某些实施例中,致死在受精后9天诱导。在某些实施例中,致死在受精后10天诱导。在某些实施例中,致死在受精后11天诱导。在某些实施例中,致死在受精后12天诱导。在某些实施例中,致死在受精后13天诱导。在某些实施例中,致死在受精后14天诱导。在某些实施例中,致死在受精后15天诱导。在某些实施例中,致死在受精后16天诱导。在某些实施例中,致死在受精后17天诱导。在某些实施例中,致死在受精后18天诱导。在某些实施例中,致死在受精后19天诱导。在某些实施例中,致死在受精后20天诱导。在某些实施例中,致死在受精后21天诱导。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多30小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多31小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多32小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多33小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多34小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多35小时的培育后激活。在某些实施例中,光遗传学系统在鸡胚胎发育期间在至多36小时的培育后激活。在某些实施例中,光遗传学系统在鸟体内激活。在某些实施例中,光遗传学系统在围绕胚胎的蛋壳形成之前激活。在某些实施例中,光遗传学系统在围绕胚胎的蛋壳形成之前在鸟体内激活。在某些实施例中,光遗传学系统通过将胚胎与诱导子直接接触激活。在某些实施例中,将诱导子插入到蛋中。在某些实施例中,首先至少部分地打开蛋壳,并且然后将诱导子直接施用于胚胎。
279.为了验证在鸡细胞中对细胞死亡的诱导,将dta和两种形式的胱天蛋白酶3克隆到具有其后是ires gfp的用于证实转染效率的pgk(磷酸甘油酸激酶)或cagg启动子的表达载体中。在鸡胚胎细胞中,pgk启动子比pcagg更弱且更慢。
280.在一个实施例中,产生的载体是:pgk
‑
dta
‑
ires
‑
gfp、pcagg
‑
dta
‑
ires
‑
gfp、pgk
‑
casp3
‑
ires
‑
gfp、pcagg
‑
casp3
‑
ires
‑
gfp、pgk
‑
mcasp3
‑
ires
‑
gfp、pcagg
‑
mcasp3
‑
ires
‑
gfp。使用了不具有致死诱导基因的表达载体作为对照(pgk
‑
ires
‑
gfp和pcagg
‑
ires
‑
gfp)。
281.通过电穿孔将表达载体转染到pgc并且将细胞培育24小时、48小时和72小时,之后使用流式细胞术进行分析,所述流式细胞术通过绿色荧光蛋白(gfp)和使用碘化丙啶(pi)染色鉴定的死细胞检测阳性表达细胞。对gfp进行编码的核苷酸序列在seq id no:115中列
出。
282.图24a
‑
24b中呈现的结果证明了死细胞占总gfp阳性细胞的比例。
283.在一个实施例中,构建了将上述三个元件合并到可以整合到z染色体的单个活性单元中的靶向载体。如图25所示,该单元侧接有5'同源臂和3'同源臂。启动子(pcagg)驱动光遗传学系统的由ires序列分离的两个单元的表达。光遗传学系统其后是聚腺苷酸化序列(pa)。光学基因盒侧接有两个loxp位点。光遗传学系统可以基于cibn
‑
cry2、magnet或如上所述的任何其它系统。在第二loxp位点之后是致死诱导编码序列,之后是第二聚腺苷酸化序列。由于此致死诱导编码序列没有启动子,因此其在应用蓝光照射之后才有活性,所述蓝光照射导致光学基因二聚化和cre酶激活,所述酶去除了侧接在loxp位点之间的光学基因盒。此切除导致将致死编码序列直接置于驱动其表达的pcagg启动子的下游。此策略受益于较短的设计,因为所述设计含有单个启动子并且不需要“stop”序列。
284.在一个实施例中,pcagg启动子的核苷酸序列在seq id no:100中示出。pgk启动子的核苷酸序列在seq id no:109中示出。pcmv启动子的核苷酸序列在seq id no:110中示出。phsyn启动子的核苷酸序列在seq id no:111中示出。pefl
‑
a启动子的核苷酸序列在seq id no:112中示出。
285.在一个实施例中,光学基因1
‑
nls
‑
cry2
‑
δ535
‑
l348f
‑
cren(aa 19
‑
104)的核苷酸序列在seq id no:101中示出。光学基因2
‑
cibn(aa1
‑
170)
‑
nls
‑
cre
‑
c(aa106
‑
343)的核苷酸序列在seq id no:102中示出。cre重组酶的核苷酸序列在seq id no:113中示出。cren(aa 19
‑
104)的核苷酸序列在seq id no:117中示出。crec(aa 106
‑
343)的核苷酸序列在seq id no:118中示出。
286.在一个实施例中,ires序列的核苷酸序列在seq id no:103中示出。聚腺苷酸化位点序列(兔β珠蛋白)的核苷酸序列在seq id no:104中示出。5'同源臂(lha)的核苷酸序列在seq id no:105中示出。3'同源臂(rha)的核苷酸序列在seq id no:106中示出。
287.在一个实施例中,关于magnet系统,以下序列是可以代替cry2
‑
cibn系统使用的替代光学基因。例如,cren(aa18
‑
59)_n
‑
mag_nls的核苷酸序列在seq id no:107中示出。nls_p
‑
mag_cre
‑
c(aa60
‑
237)的核苷酸序列在seq id no:108中示出。
288.在一个实施例中,如下执行将经修饰的pgc移植到胚胎中并且产生将进行筛选以供种系传递的嵌合体鸡和潜在创始者载体。在生成靶向载体之后,源自雌性胚胎和雄性胚胎两者的pgc系经历与px330
‑
一体化crispr
‑
cas9
‑
gfp编码质粒和靶向载体进行共电穿孔。crispr质粒在染色体z的设计的位点处产生dna双链断裂,由此促进靶向载体的同源重组。crispr质粒对与gfp框内融合的cas9进行编码,由此允许鉴定成功转染。转染后24
‑
72小时,将阳性单个细胞facs分选为96孔板中的单个细胞并且生长以形成纯pgc集落,所述集落通过pcr和southern印记进行筛选以鉴定经历适当同源重组的集落。
289.如下生成嵌合体鸡。嵌合体鸡是将经遗传修饰的pgc转换为功能配子的手段。为了生成嵌合体,将阶段14
‑
16h&h胚胎注射到血流(例如心脏)中,其中具有>3000个pgc(通常至多8000
‑
10000个)。这些细胞与内源pgc一起定殖于胚胎性腺,由此其被称为嵌合体。种系传递的效率,即将经遗传修饰的pgc转换为功能pgc的能力,取决于几个因素,包含性腺中的内源pgc与注射的pgc之间的比率。为了减少内源pgc的量,用600
‑
800拉德对新鲜产下的受精的蛋进行γ辐照。在某些实施例中,辐照包括600
‑
800拉德的辐照。在某些实施例中,辐照包
括400
‑
1000拉德的辐照。在某些实施例中,辐照包括200
‑
1200拉德的辐照。
290.认为pgc对γ辐照比体细胞更具易感性,或者在辐照后,pgc不如体细胞更有能力再生。因此,辐照后内源pgc的总量减少。辐照后,通常将蛋温育到胚胎达到足以进行pgc注射的阶段14
‑
16h&h。作为辐照效率的指标,胚胎达到阶段14
‑
16h&h所需的培育时间在约10小时内增加,并且多达70%的经辐照的胚胎未能正常发育并且因此不用于pgc注射。不同鸡品系、蛋壳类型、稠度和颜色可能需要不同辐照条件,由此能量的量保证了校准。注射后,将蛋密封并且温育直到孵化出嵌合体雏鸡。然后将雏鸡饲养到性成熟,并且在雄性中收集精子以使用半定量pcr或实时pcr分析种系传递。
291.实例3
292.本实例呈现了含有附加元件的靶向载体,所述元件在本文中被称为“安全锁定”元件。此元件如此通过默认基本上锁定光遗传学致死系统,所述系统为非活性的。仅通过与“安全非锁定”品系杂交,光遗传学致死系统将会变得有活性。此元件通过更好地保护品种使整个系统受益,因为为了被激活,需要使用另外的“非锁定”品系。这也确保了在整个生产过程中,不需要使经历hr的细胞免受光,因为光遗传学系统基本上是非活性的。
293.本实例公开了八种“一体式”靶向载体,并且在体外和体内两者证明了其如所设计的工作。数据还示出magnet光遗传学系统在鸡胚胎中工作并且能够激活致死诱导机制。magnet系统已在上文进行了描述。此外,数据示出,作为胚胎致死诱导基因的头蛋白阻止胚胎从产卵(来自新鲜产下的蛋)开始发育。
294.本文公开的八个靶向载体涵盖2个光遗传学系统(magnet和cibn
‑
cry2)和4个致死诱导基因(dta、头蛋白、cacasp3和mcherry,其用于控制和验证过程)的8种组合选项。为了进行说明,mcherry如此不诱导致死并且用于证实其它元件的动作。
295.如上所讨论的,本文所述的实验的目的是将致死诱导盒引入到雄性或雌性来源的pgc的z染色体中。最终目标是获得在z染色体上携带基因组插入的母鸡。此染色体将仅分离到下一代雄性胚胎,所述雄性胚胎在蓝光诱导下将激活致死诱导盒,由此雄性胚胎将在胚胎发生的早期阶段死亡。因此,由于性别染色体分离,雌性产蛋母鸡不会获得经修饰的遗传材料,因为其从雄性公鸡侧得到wt z染色体并且从母鸡妈妈侧得到wt w染色体(参见图1)。在此实例中,靶向载体包括如下所述的4个元件(参见图26中的实例)。
296.如图26所示,第一元件是“同源臂”。在一个实施例中,两个约1.5kb同源臂位于载体的5'端部和3'端部两者上。这被设计成将同源重组(hr)导引到位于z染色体上的hint1z基因座下游。选择了hr的此位点,因为hint1z基因是在pgc和以及整个胚盘(这些是新鲜产下的雏鸡胚胎)中转录的。z染色体上所有其它经开放转录的区域也是出于此目的的潜在良好候选区。
297.第二元件是确保在去除stop盒之前光遗传学致死机制为非活性的“安全锁定”机制。stop盒侧接有两个frt位点,并且其位于pcagg启动子与光学基因之间,由此防止光学基因表达。在与如下所述的flp表达品系杂交时,去除stop元件,从而允许光学基因转录并且以光依赖性方式变得有活性(图26a,活性“非锁定”状态)。在此实例中,“安全锁定”元件包括gfp和其后是聚腺苷酸化位点的编码序列。将防止下游元件转录的任何其它序列也可以用作“安全锁定”元件。
298.第三元件是“光遗传学诱导型元件”。如上所述,光遗传学系统基于在某些光波长
下会被激发并改变构象以与第二特异性蛋白质二聚化的蛋白质。上文已经讨论了光遗传学系统的实例。
299.在此实例中,测试和使用了两个替代性光遗传学系统。第一个是magnet系统,其中两种光遗传学蛋白质——带正电的p
‑
mag和带负电的n
‑
mag——在蓝光照射下二聚化。对位点特异性重组酶mag进行编码的序列在seq id no:114和seq id no:65中列示。这些蛋白质中的每种蛋白质与cre重组酶的非活性部分融合,并且在两个光学基因二聚化时,会产生活性形式的cre(参见《自然化学生物学》第12卷,1059
–
1064 2016)。在此情况下,自切割肽p2a(图26中的“连接”)的序列位于两个光学基因之间。
300.所使用的第二光遗传学系统是改进的cibn(crec)
‑
cry2(cren)的系统。在此情况下,修饰包含在截短的cry2基因中插入l348f突变,所述基因含有仅前535个氨基酸
‑
cry2
‑
δ535
‑
l348f,(参见《自然化学生物学》25第12卷,第425
–
430页(2016))。在一个实施例中,两个光学基因之间的接头是ires序列(图26中的“连接”)。
301.第四元件是“致死诱导盒”。在一个实施例中,致死诱导盒包括通过细胞死亡或如bmp4等胚胎发生的早期阶段所需的分子信号传导通路的严重干预的手段来促进早期胚胎死亡的基因。如上所述,致死诱导基因的实例包含但不限于如白喉毒素a(dta)等毒素、如胱天蛋白酶3或突变组成型活性突变形式的胱天蛋白酶3等胱天蛋白酶基因或bmp4
‑
头蛋白的抑制剂蛋白。
302.下表列出了这八个靶向载体的特征。在一个实施例中,将这些载体克隆到pjet1.2穿梭载体质粒中。
[0303][0304][0305]
在hek293细胞中在体外(图27)并且在鸡胚胎中在卵内(图28
‑
29)中评估靶向载体(tv)中的元件的活性。对于体外验证,用单独的tv4(图27a),用pcagg
‑
cre(seq id no:128)(图27b),或用pcagg
‑
flpo(seq id no:129)质粒对hek293细胞进行转染。在两种处理下完成后者:一种是保持在黑暗中(图27c),并且另一种是在转染后24小时暴露于蓝光持续15秒。在照射之后,将细胞进一步培育24小时(图27d)。
[0306]
当单独表达时,tv4像所有其它tv一样,表达gfp,由此指示“安全锁定”状态的活性(图27a)。当与pcagg
‑
cre质粒共表达时,一起去除“安全锁定”和光学基因盒(图26b;图27b),所述去除由gfp的表达的丧失和mcherry表达的开始(作为致死诱导基因的替代物)指示。此结果证实了在切除“安全锁定”和光学基因元件时,致死诱导元件变得有活性。当tv4与pcagg
‑
flpo质粒共表达时,去除了仅“安全锁定”元件,如由gfp的表达的丧失所指示的(图26a,图27c
‑
d),并且光遗传学系统以光依赖性方式变得有活性。当将细胞保持在黑暗中时,未激活光遗传学系统并且mcherry未表达,这指示致死盒未表达(图27c)。然而,在相同
的条件下,当对细胞进行照射时,光遗传学系统变得有活性并被切除(参见图26a)以允许致死元件进行表达。这由如图27d所示的mcherry的表达指示。
[0307]
为了体内证明tv元件的活性,将鸡胚胎与质粒一起注射到神经管中,并如上所述进行电穿孔。在图28中,白线表示用于定向目的的神经管和肢芽的背中线。对四个处理组进行了测试:1.单独的tv4的表达(图28a),2.tv4和作为阳性对照的pcagg
‑
cre质粒的共电穿孔(图28b),3.tv4和保持在黑暗中的pcagg
‑
flpo质粒的共电穿孔(图28c),以及4.在tv4和pcagg
‑
flpo质粒共电穿孔之后暴露于蓝光持续15秒,并且进一步培育12小时(图28d)。
[0308]
当单独电穿孔时,tv4表达gfp以指示默认的非活性“安全锁定”状态,并且mcherry未表达(图28a)。当与pcagg
‑
cre质粒共表达时,去除“安全锁定”和光学基因盒(图26b;图28b),因此无gfp表达,但mcherry进行了表达。这用作用于激活致死诱导元件的阳性对照。通过将tv4与pcagg
‑
flpo质粒共表达,去除了仅frt侧接的“安全锁定”元件,因此无gfp表达(图28c
‑
28d),但是光遗传学系统以光依赖性方式被激活。当将细胞保持在黑暗中时无gfp表达,这指示光遗传学系统处于其活性状态,但是mcherry未表达,即致死盒是非活性的(图28c)。然而,当电穿孔后12小时对胚胎进行照射时,光遗传学系统变得有活性并被切除以允许致死元件进行表达。这由如图28d所示的mcherry的表达指示。
[0309]
为了证明致死诱导基因头蛋白以光依赖性方式的活性,将含有作为致死诱导元件的头蛋白的编码序列的tv1与pcagg
‑
flpo质粒共电穿孔到已培育36小时的鸡胚胎的喙神经管(中脑和后脑的轴向水平)。在此阶段,神经嵴细胞以bmp4依赖的方式从背侧神经管分层。因此,预测通过使头蛋白在背侧神经管中异位表达来抑制bmp4信号传导通路可抑制神经嵴细胞分层。为了使神经嵴细胞可视化,使用了抗hnk
‑
1抗体以对神经嵴标记物hnk
‑
1进行染色。
[0310]
图29示出了在神经管中用tv1(seq id no:120)、pcagg
‑
flpo(seq id no:129)和pcagg
‑
ires
‑
gfp(seq id no:59)质粒进行电穿孔的胚胎。添加后者作为阳性对照以允许监测经转染的细胞。转染后二十四小时,在整个实验中保持在黑暗中的胚胎中(图29a,上排,背侧视图,下排,右侧视图),神经嵴细胞从背侧神经管分层并法向迁移。hnk
‑
1染色揭示了右侧神经褶和左侧神经褶两者(产生迁移性神经嵴细胞的神经管的最背侧方面)上以及迁移性神经嵴细胞(图29a,箭头)上的背管中的双侧对称表达模式。然而,在电穿孔后暴露于光12小时的胚胎中,神经嵴细胞未能从管分层和迁移,并且hnk
‑
1染色揭示了右侧神经褶——在电穿孔的侧(图29b,箭头)的表达显著减少。因此,这些结果指示在从tv1中去除“安全锁定”元件时,由magnet光遗传学系统以光依赖性方式调节头蛋白的表达和活性。
[0311]
为了证明头蛋白能够在产蛋时就能停止胚胎发育,在胚盘胚胎阶段,用外源来源的头蛋白对胚盘进行卵内处理(图30)。为此,将pcagg
‑
头蛋白
‑
ires
‑
gfp(seq id no:130)或作为阴性对照的pcagg
‑
ires
‑
gfp(seq id no:59)质粒转染到hek293细胞中。为了验证头蛋白在经转染的细胞中的表达,利用抗头蛋白抗体(艾碧康公司,ab16054,预测大小为约24kda)和抗α
‑
tubulin
‑
hrp抗体(艾碧康公司,ab40742,预测大小为约55kda)作为加载对照通过western印记对从经转染的细胞中提取的总蛋白进行分析。图30a示出了虽然经阴性对照转染的细胞(con)中无头蛋白表达,但用pcagg
‑
头蛋白
‑
ires
‑
gfp(nog)转染的细胞产生了头蛋白。左泳道中呈现了经预染色的蛋白序列梯(赛默科技公司(thermo scientific),pageruler#26617)。将来自对照和头蛋白表达细胞的条件培养基注射到新鲜产下的受精的
蛋中,随后分别如图30b
‑
c和图30d
‑
e所示将所述受精的蛋温育24或54小时。图30b
‑
c示出了经处理的胚胎的高分辨率反式显微镜(hrem)3d重建的模型。虽然经对照处理的胚胎(图30b)继续正常发育并经历正常原肠胚形成,如通过原始条纹的形成(用箭头标出)所指示的,但经头蛋白处理的胚胎未能形成原肠胚且无明显的原始条纹,这指示胚胎发生过程基本上停止(图30c)。当使胚胎进一步发育持续54小时的培育(图30d
‑
e)时,经对照处理的胚胎正常发育,经历正常的神经形成过程并形成跳动的心脏(图30d),而经头蛋白处理的胚胎形成没有明显特性的细胞团(图30e,箭头)。
[0312]
总体来说,上述结果示出,图26中呈现的分子作用模式策略如所预测的工作,包含“安全锁定”元件和以光依赖性方式激活致死诱导元件的magnet光遗传学系统。另外,通过用头蛋白处理新鲜产下的蛋中的胚盘来抑制bmp信号传导通路终止了胚胎发育的进程。
[0313]
在z染色体上创建携带整合的靶向载体的纯的pgcs系
[0314]
在一个实施例中,为了生成具有靶向整合到z染色体的pgc,使用了用于递送crispr/cas9的核糖核蛋白(rnp)系统。rnp系统的使用尤其得益于高效率、快速的dna切割和从经转染的细胞中快速清除rnp复合物。rnp复合物包含重组cas9或高保真cas9核酸酶和crrna:tracrrna复合物的混合物。tracrrna可商购获得(idt),并且可以将crrna定制成具有对应于靶向的基因组dna上的期望的切割位点的特定20个核苷酸序列。在一个实施例中,本文公开的seq id 66和seq id 68用于合成两个crrna寡核苷酸(参见图8)。
[0315]
上文已经描述了pgc培养基和系衍生。为了对pgc进行电穿孔,在akodmem中对5
×
105个细胞进行洗涤,然后转移到含有0.7μg的靶向载体质粒(在本实例中为tv1)、1μmcas9电穿孔增强剂(idt)和最终浓度为1.5μm重组cas9或高保真cas9核酸酶(分别为s.p.cas9核酸酶或s.p.hifi cas9核酸酶v3;idt)的rnp复合物和1.8μmcrrna:tracrrna(sgrna复合物,定制crispr
‑
cas9 crrna和tracrrna;idt)中。
[0316]
通过将rna寡核苷酸加热到95℃1分钟并且冷却到室温,制备了crrna:tracrrna复合物。通过向crrna:tracrrna复合物添加重组cas9或高保真cas9核酸酶蛋白,并且在室温下温育10
‑
20分钟来制备rnp复合物(cas9蛋白 crrna:tracrrna)混合物。普遍认为,高保真cas9应该导致较少脱靶。
[0317]
对于电穿孔,将含有rnp复合物、电穿孔增强子试剂和靶向载体质粒的10μl缓冲液“r”添加到pgc团粒中,并使用neon电穿孔仪(英杰公司)立即用3个1000v的脉冲对混合物进行电穿孔,每个脉冲持续13毫秒。随后,在48孔板中将细胞立即接种在含有1μm scr7吡嗪(西格玛公司的sml1546)的pgc培养基中。1
‑
4小时后更换培养基,并且允许经转染的细胞恢复7
‑
10天。电穿孔后48小时添加scr7吡嗪。通过facs分选单独分离经转染的细胞。对于facs分选,轻轻完成细胞移液,并在pgc培养基中对细胞进行分选。用facs索尼分选仪(索尼公司(sony))将单个gfp阳性细胞分选到u形96孔板中。使经分选的细胞生长2
‑
3周以形成纯的集落。从这些集落中提取总基因组dna以供分析,将冷冻保存的且阳性的集落注射到替代宿主胚胎中。上文已经描述了用于验证靶向载体的正确整合的pcr和southern印记分析(参见例如图15
‑
16)。上文也已经描述了产生替代嵌合体雏鸡的方法。
[0318]
图31a示出了用tv1进行hr的纯雌性pgc系和所表达的gfp。将来自此集落的细胞注射到移植后培育5天的替代宿主胚胎中。图31b示出了pgc注射之后5天胚胎的腹侧视图。在此阶段,pgc定殖于生殖脊,所述生殖脊是性腺的原基(图31b,箭头)。将其它注射的胚胎温
育至孵化,并且在孵化后第10天处死雌性雏鸡以对卵巢进行分析。图31c示出了含有大量gfp阳性pgc的卵巢(用线描绘)。这些结果指示在具有tv1的z染色体上经历hr的pgc成功地定殖于性腺。
[0319]
创建表达cre或flpo的“非锁定”品系.
[0320]
表达靶向载体tv1
‑
8的转基因鸡品系在其基因组中将具有“安全锁定”元件,因此光遗传学致死系统将是非活性的。为了去除侧接有frt位点的“安全锁定”元件(参见图26a),这些品系需要与flpo表达的鸡品系杂交。为了生成此品系,创建了含有flpo酶和其后的ires
‑
gfp的靶向载体(tv
‑
flpo
‑
ires
‑
gfp,seq id no:131)。作为阳性对照和以光独立性方式激活致死诱导盒的手段,可以使用cre表达的品种。在此情况下,去除了侧接有loxp位点的“安全锁定”和光遗传学元件,以激活致死诱导元件(参见图26b)。为了生成此品系,创建了含有cre酶和其后的p2a
‑
gfp的靶向载体(tv
‑
cre
‑
p2a
‑
gfp,seq id no:132)。这两个靶向载体共享相同的5'和3'同源臂以及报告基因gfp,并且被设计成整合到上述针对tv1
‑
8的z染色体中。
[0321]
已使用了上述靶向载体(tv
‑
flpo
‑
ires
‑
gfp和tv
‑
cre
‑
p2a
‑
gfp)来生成注射到替代嵌合体胚胎中的pgc。上文已经描述了pgc的转染、facs分析、hr整合的验证、胚胎注射等的方法。
[0322]
在另一个实施例中,有使用重组cre或flpo蛋白来去除“安全锁定”元件的选项。在一个实施例中,可以对细胞或胚胎应用对重组蛋白进行编码的序列,所述序列含有作为细胞穿透肽的tat肽的序列,所述序列其后是核定位序列(nls),所述nls进而其后是对cre或flpo酶进行编码的序列。这些重组蛋白在经培养的细胞中有效,并且其可商购获得。据信,这些重组蛋白可以注射到胚胎中或对成年进行注射,并且在无需生成cre/flp转基因鸡和杂交的情况下潜在地激活系统。鸡胚胎通过蛋壳上的开口的可获得性允许直接注射到血流中或胚胎附近。因此,在一个实施例中,上述重组蛋白可以直接注射到胚胎中以去除“安全锁定”元件。
[0323]
尽管已结合特定实施例描述了某些实施例,但很明显,许多替代方案、修改以及变化对本领域的技术人员应是显而易见的。因此,意图涵盖落入所附权利要求书的精神和广泛范围内的所有此类替代方案、修改以及变化。
[0324]
本说明书中所提及的所有公开、专利以及专利申请在此通过引用以其整体并入到本说明书中,达到如同每一个单独的公开、专利或专利申请被专门地并且单独地指示通过引用并入本文的相同的程度。此外,本技术中对任何参考的引用或识别不应理解为承认此参考可作为本公开的现有技术获得。在使用章节标题的意义上,其不应被解释为必要地限制。
[0325]
虽然本文已展示并描述了方面或实施例的某些特征,但本领域的普通技术人员现在将想到许多修改、取代、变化和等效形式。因此,应当理解的是,随附的权利要求书的意图是涵盖落入本文提供的方面或实施例的真实精神内的所有此类修饰和变化。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。