1.本发明涉及一种用于血浆中莠去津的检测方法。
背景技术:
2.中国是农业大国,农药制剂对农业发展具有关键性作用,其主要可分为杀虫剂、杀菌剂、除草剂等,而莠去津就是一种使用普遍的三嗪类除草剂。1958年由瑞士geigy公司研制研发,由于其具有除草效果好、价格低廉等特点被广泛应用。我国从20世纪70年代初开始使用,随着农业的发展莠去津的使用面积不断扩大。
3.研究发现,莠去津低剂量长期接触会对哺乳动物的生殖系统、免疫系统、神经系统、内分泌系统产生影响。莠去津慢性动物试验中动物出现血生化指标异常和乳腺肿瘤。人群长期接触莠去津农药可能会产生潜在的健康损害,特别是职业接触人群。为了更好地了解莠去津的健康风险,有关莠去津经口或经皮后农药在动物体中的药代/毒代动力学研究尤为重要,亟待一种快捷精准的、可用于药代/毒代动力学的莠去津检测方法。
4.目前莠去津的检测方法的开发主要集中在环境样品中的残留量上,包括土壤、水质以及农产品等。现有技术中,主要的检测方法为色谱分析法,但现行的莠去津检测方法存在分析灵敏度不够、处理过程相对复杂及仪器分析时间长等不足。例如,气相色谱-质谱联用法(gc-ms)前处理过程较为复杂,一般需要固相顶空微萃取,时间较长。另外,高效液相色谱法(hplc-uv)由于检测器的原因导致分析时间长且灵敏度不够。
技术实现要素:
5.本发明要解决的技术问题在于现有技术中对于莠去津的检测方法存在分析灵敏度不够、处理过程相对复杂及仪器分析时间长等,而提供了一种用于血浆中莠去津的检测方法。本发明的检测方法能够快速、简便的测定莠去津在经口给药或经皮给药后在血浆中的浓度,测试准确率高,稳定性好,回收率高,线性范围大,从而能够满足莠去津在动物体中的药代/毒代动力学的分析需求。
6.本发明通过以下技术方案解决上述技术问题。
7.本发明提供了一种用于血浆中莠去津的检测方法,其步骤包括:
8.将所述血浆经蛋白沉淀法处理得到的上清液用液相色谱-质谱联用分析方法检测,即可;
9.所述液相色谱的检测条件为:
10.流动相包括流动相a和流动相b;
11.流动相a为甲酸与水的混合液,其中,所述甲酸占所述流动相a总体积的0.01%~0.5%;
12.流动相b为甲酸与乙腈的混合液,其中,所述甲酸占所述流动相b的总体积的0.01%~0.5%;
13.采用梯度洗脱,以所述流动相的总体积为100%计;在0min,所述流动相a的体积为
65%;在0~2min,所述流动相a的体积由65%递减至10%;在2~2.5min,所述流动相a的体积为10%;在2.5~2.51min,所述流动相a的体积由10%递增至65%;在2.51~3min,所述流动相a的体积为65%。
14.本发明中,可采用内标法进行血浆中莠去津的检测。
15.其中,所述内标法使用的内标物较佳地为莠去津-d5。
16.其中,较佳地,将内标物溶于甲醇中。
17.一优选实施方式中,将内标物莠去津-d5溶解于适量甲醇中,配制成合适浓度的内标储备液,经乙腈稀释得到不同浓度的内标工作液。
18.一优选实施方式中,所述的内标工作液的浓度为500ng/ml。
19.本发明中采用的蛋白沉淀法具有简单、快捷的优点。所述蛋白沉淀法的操作和条件可为本领域常规,较佳地包括以下步骤:向所述血浆中(例如考察基质效应样品及回收率样品)或空白基质中(即空白血浆)中加入沉淀剂,涡旋、离心,吸取上清液,加入稀释剂,再次涡旋、离心,取上清液进样检测。而对于标准曲线、质控以及其他考察项样品在其中添加内标工作液后,再加入沉淀剂,涡旋、离心,吸取上清液,加入稀释剂,再次涡旋、离心,取上清进样检测。
20.其中,所述沉淀剂较佳地为乙腈。
21.其中,所述沉淀剂与所述血浆的体积比较佳地为(10~20):1,更佳地为18:1。
22.其中,较佳地,所述稀释剂为乙腈和水的混合液,其中,所述乙腈占所述稀释剂总体积的10%~50%;更佳地,所述乙腈占所述稀释剂总体积的30%。
23.其中,较佳地,所述离心的温度为4℃。
24.其中,较佳地,所述离心的转速为4000rpm。
25.其中,较佳地,所述离心的时间为10min。
26.本发明中,采用液相色谱质谱联用分析方法,通过对于所述液相色谱检测条件中例如流动相配比和洗脱方式的筛选,大大缩短了检测所需时间。较佳地,lc-ms/ms仪器的型号为ab sciex triple quad 6500plus。
27.本发明中,所述液相色谱的色谱柱的固定相较佳地为十八烷基键合硅胶。
28.其中,所述液相色谱的色谱柱的型号较佳地为infinitylabporoshell 120sb-c18。
29.其中,较佳地,所述液相色谱的色谱柱的内径为2.1mm,长度为50mm。
30.其中,所述液相色谱的色谱柱的孔径为较佳地2.7μm。
31.本发明中,所述液相色谱的运行时间较佳地为3min。
32.本发明中,较佳地,所述流动相a为甲酸与水的混合液,其中,所述甲酸占所述流动相a的总体积的0.05%。
33.本发明中,较佳地,所述流动相b为甲酸与乙腈的混合液,其中,所述甲酸占所述流动相b的总体积的0.05%。
34.本发明中,所述液相色谱的进样量较佳地为1~10μl,更佳地为5μl,所需进样量较少。
35.本发明中,所述液相色谱的柱温较佳地为35℃~45℃,更佳地为40℃。
36.本发明中,所述液相色谱的流速较佳地为0.4ml/min。
37.本发明中,所述质谱的操作和检测条件可为本领域常规。
38.其中,所述质谱的离子源较佳地为电喷雾电离esi。
39.其中,所述质谱的离子化模式较佳地为正离子化模式。
40.其中,较佳地,所述质谱采用多反应监测模式(multiple reaction monitoring,mrm)。
41.其中,所述质谱的仪器型号较佳地为ab sciex triple quad
tm 4500。
42.较佳地,所述多反应监测模式中,莠去津的母离子和子离子对的质荷比分别为216.0和174.0;采用内标法进行血浆中莠去津的检测,当内标物为莠去津-d5时,莠去津-d5的母离子和子离子对的质荷比分别为220.9和179.2;扫描间隔时间为100ms。
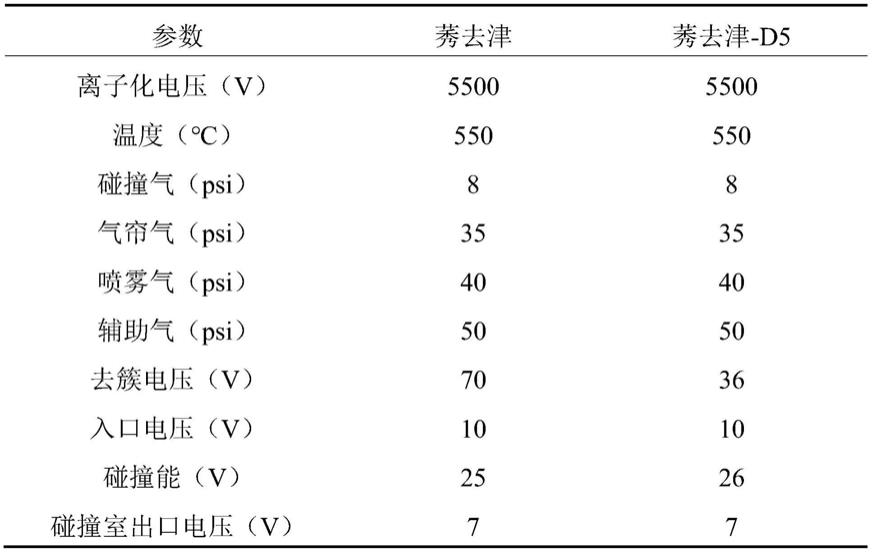
43.较佳地,所述多反应监测模式中,仪器参数如下:
[0044][0045]
本发明中,可将经所述液相色谱-质谱联用分析方法的检测结果与莠去津标准曲线对照,得知所述血浆中莠去津的浓度。所述莠去津标准曲线可将不同浓度的莠去津标准品进行液相色谱-质谱联用分析方法测试,拟合得到。本领域技术人员知晓,可通过配制多个浓度梯度的若干莠去津标准品,进行液相色谱-质谱联用分析方法测试,提取多反应监测模式色谱图,以标准样品与内标峰面积比值为纵坐标,设置权重为1/x2,忽略原点,拟合莠去津线性标准曲线。
[0046]
一优选实施方式中,将莠去津标准品溶解于适量甲醇中,配制成适合浓度标准品储备液,经甲醇和水的混合溶剂稀释得到多个浓度梯度的多个标准品工作液;其中,所述混合溶剂较佳地为30%~70%甲醇水溶液,例如为50%甲醇水溶液。
[0047]
一优选实施方式中,所述标准品工作液的浓度梯度依次为40ng/ml、80ng/ml、400ng/ml、2000ng/ml、4000ng/ml、16000ng/ml、32000ng/ml和40000ng/ml;将所述标准品工作液中分别加入空白血浆,得到标准样品,所述标准样品的浓度分别为2、4、20、100、200、800、1600和2000ng/ml。
[0048]
一优选实施方式中,将莠去津标准品溶解于适量甲醇中,配制成适合浓度质控储
备液,经甲醇和水的混合溶剂稀释得到多个浓度梯度的多个质控工作液;其中,所述混合溶剂较佳地为30%~70%甲醇水溶液,例如为50%甲醇水溶液。
[0049]
一优选实施方式中,所述质控工作液的浓度梯度依次为40ng/ml、120ng/ml、2400ng/ml和30000ng/ml;将所述质控工作液中分别加入空白血浆,得到质控样品,所述质控样品的浓度分别为2、6、120和1500ng/ml。
[0050]
本发明的有益效果:
[0051]
本发明的方法系统地根据中国药典和nmpa非临床药代动力学研究技术指导原则全面的验证了考察项。本方法所采取前处理方法为蛋白沉淀法,该方法与常用的萃取的方法流程相比,更为简单、用时短且易操作;同时,本发明的方法所需的样本进样量小,所需的分析时间较短;本发明开发的处理方式使标准曲线线性精准,保证了检测的准确性和可靠性;因此本发明能够保证在较短的分析时间里获得更准确、稳定、高精密度、高灵敏度的检测数据,为血浆中莠去津药代动力学的分析奠定了夯实的基础。
附图说明
[0052]
图1为空白基质在莠去津离子通道时的图谱。
[0053]
图2为空白基质在内标离子通道时的图谱。
[0054]
图3为莠去津定量下限(2.000ng/ml)的图谱。
[0055]
图4为定量下限样品中内标(25ng/ml)的图谱。
[0056]
图5为莠去津标准曲线图。
具体实施方式
[0057]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0058]
以下实施例中,“fa”是指甲酸;“h2o”是指水,“acn”是指乙腈;“ipa”是指异丙醇;“meoh”是指甲醇。
[0059]
实施例1
[0060]
1、试剂的配制:
[0061]
1.1流动相a(mpa):0.05%fa in h2o
[0062]
取500μl fa加入1000ml h2o中,混匀。室温保存有效期1周。
[0063]
1.2流动相b(mpb):0.05%fa in acn
[0064]
取500μl fa加入1000mlacn中,混匀。室温保存有效期1周。
[0065]
1.3强洗溶液(snw):meoh:acn:ipa:h2o=1:1:1:1,v/v/v/v
[0066]
取500mlmeoh,500ml acn,500ml ipa,500ml h2o,混匀。室温保存有效期1个月。
[0067]
1.4弱洗溶液(wnw):meoh:h2o=1:1,v/v
[0068]
取500ml meoh,500ml h2o,混匀。室温保存有效期1个月。
[0069]
1.5沉淀剂&内标稀释液:acn
[0070]
取1000mlacn。室温保存有效期1个月。
[0071]
1.6稀释液1(diluent1):meoh:h2o=1:1,v/v
[0072]
取100mlmeoh,100mlh2o,混匀。室温保存有效期1个月。
[0073]
1.7稀释液2(diluent2):acn:h2o=3:7,v/v
[0074]
取300ml acn,700ml h2o,混匀。室温保存有效期1个月。
[0075]
2、标准曲线(以下简称标曲)和质控样品的配制:
[0076]
储备液和工作液均储存于超低温冰箱(-70~-90℃)中。
[0077]
2.1标曲储备液的配制
[0078]
精密称取适量莠去津于透明样品瓶中,加入适量meoh溶解、摇匀,配制成浓度为1.000mg/ml的标曲储备液。(其中,所需甲醇体积根据莠去津折算因子计算后所得。质量折算因子由市售的标准品的纯度计算得到。莠去津的质量折算因子为0.947。)
[0079]
2.2标曲&sst工作液的配制(稀释液:meoh:h2o=1:1,v/v)
[0080]
使用标曲储备液,依照下表1配制,得到标曲&sst工作液:
[0081]
表1莠去津标曲&sst工作液配制
[0082][0083]
2.3标准曲线&sst样品的配制
[0084]
使用标曲&sst工作液,依照下表2配制,得到标准曲线&sst样品:
[0085]
表2莠去津标准曲线&sst样品的配制
[0086][0087]
标准曲线样品储存于超低温冰箱(-70~-90℃)中。
[0088]
2.4质控储备液的配制
[0089]
精密称取适量莠去津于透明样品瓶中,加入适量meoh液溶解、摇匀,配制成浓度为1.000mg/ml的质控储备液。(其中,所需甲醇体积根据莠去津折算因子计算后所得。莠去津的质量折算因子为0.947。)
[0090]
2.5质控工作液的配制(稀释液:meoh:h2o=1:1,v/v)
[0091]
使用质控储备液,依照下表3配制,得到质控工作液:
[0092]
表3莠去津质控工作液配制
[0093][0094][0095]
2.6质控样品的配制
[0096]
使用质控工作液,依照下表4配制,得到质控样品:
[0097]
表4莠去津质控样品的配制
[0098][0099]
质控样品储存于超低温冰箱(-70~-90℃)中。
[0100]
2.7内标储备液的配制
[0101]
精密称取适量莠去津-d5于透明样品瓶中,加入适量meoh液溶解、摇匀,配制成浓度为1.000mg/ml的内标储备液。(其中,所需甲醇体积根据莠去津-d5折算因子计算后所得。莠去津-d5的质量折算因子为0.9955。)
[0102]
2.8内标工作液的配制(稀释液:acn)
[0103]
使用内标储备液,依照下表5配制,得到内标工作液:
[0104]
表5莠去津-d5内标工作液配制
[0105][0106][0107]
2.9基质效应、回收率测试用纯溶液配制
[0108]
基质效应样品、基质效应纯溶液样品和回收率样品,依照下表6配制得到。
[0109]
表6纯溶液配制
[0110][0111]
注:
①
稀释液1:meoh:h2o=1:1,v/v;
②
稀释液2:acn:h2o=3:7,v/v
[0112]
3、样品处理步骤
[0113]
在步骤2中的样品分别进行以下处理:
[0114]
3.1样品涡旋(如果样品需要融化,在室温融化后再涡旋)。
[0115]
3.2将样品分别按照以下程序进行处理:
[0116]
标曲:20μl std 20μl内标 360μl acn
→
100μl上清 300μl acn:h2o=3:7,v/v;
[0117]
质控:20μlqc 20μl内标 360μl acn
→
100μl上清 300μl acn:h2o=3:7,v/v;
[0118]
双空白db、carryover(db和carryover均为无待测物、无内标的空白血浆):20μl血浆 20μl acn 360μl acn
→
100μl上清 300μl acn:h2o=3:7,v/v;
[0119]
blank(blank为含内标的空白血浆):20μl血浆 20μl内标 360μl acn
→
100μl上清 300μl acn:h2o=3:7,v/v;
[0120]
uloq:20μl std8 20μl acn 360μl acn
→
100μl上清 300μl acn:h2o=3:7,v/v;
[0121]
基质效应样品:20μl血浆 20μl acn 360μl acn
→
100μl上清 100μl(lqc-is_n或hqc-is_n) 200μl acn:h2o=3:7,v/v;
[0122]
基质效应纯溶液样品:20μl h2o 20μl acn 360μl acn
→
100μl上清 100μl(lqc-is_n或hqc-is_n) 200μl acn:h2o=3:7,v/v;
[0123]
回收率样品:20μl qc 20μl acn 360μl acn
→
100μl上清 100μl(is_n) 200μl acn:h2o=3:7,v/v;
[0124]
回收率参比样品:20μl血浆 20μl acn 360μl acn
→
100μl上清 100μl(低浓度:lqc-is_n;中浓度:mqc-is_n;高浓度:hqc-is_n) 200μl acn:h2o=3:7,v/v;
[0125]
以上
“→”
代表:在4℃,4000rpm条件下离心10min。
[0126]
4、测试步骤
[0127]
样本进样,进行液相色谱串联质谱检测:
[0128]
其中,液相色谱的检测条件为:
[0129]
进样量:5μl
[0130]
色谱柱:infinitylabporoshell 120sb-c18,2.1
×
50mm,2.7-micron,agilent
[0131]
柱温:40℃
[0132]
流动相a:0.05%甲酸与水的混合液
[0133]
流动相b:0.05%甲酸与乙腈的混合液
[0134]
运行时间:3.0min
[0135]
采用梯度洗脱,洗脱梯度如表7:
[0136]
表7流动相梯度
[0137][0138]
液相色谱的洗针溶剂中,强洗溶剂为甲醇、乙腈、异丙醇和水的混合溶剂,甲醇、乙腈、异丙醇和水的体积比为1:1:1:1;弱洗溶剂为甲醇和水的混合溶剂,甲醇和水的体积比为1:1。
[0139]
液相色谱的洗针程序为:
[0140]
冲洗类型:仅外部;
[0141]
冲洗模式:抽吸前后,浸入时间:2s;
[0142]
冲洗泵方式:冲洗泵,然后停止,时间:2s;
[0143]
冲洗设置:冲洗速度:35μl/s;
[0144]
冲洗体积:1000μl;
[0145]
测量管路吹扫量:100μl。
[0146]
其中,质谱检测条件为:
[0147]
仪器型号:ab sciex triple quadtm 4500
[0148]
离子源:esi
[0149]
离子化模式:正离子
[0150]
mrm多反应监测离子对如表8所示:
[0151]
表8 mrm离子对
[0152][0153]
[0154]
仪器参数如表9所示:
[0155]
表9仪器参数
[0156][0157]
5、分析批接收标准和标准曲线回归方法
[0158]
5.1回归方法
[0159]
以分析物峰面积与内标峰面积比对标准曲线中分析物的理论浓度进行线性最小二乘法回归计算,以所得回归方程计算样品中分析物的实测浓度。
[0160]
样品中分析物的实测浓度由以下回归方程计算:
[0161]
y=ax b
[0162]
其中,y=分析物与内标峰面积比
[0163]
a=标准曲线之斜率
[0164]
x=分析物浓度(单位ng/ml)
[0165]
b=标准曲线之截距(权重因子为1/x2)
[0166]
5.2分析批接收标准
[0167]
1.标准曲线(如图5所示)各浓度点的回算值与标示值之间的偏差应在
±
15.0%范围内(定量下限处的偏差在
±
20.0%范围内)。
[0168]
2.至少75%的标准曲线样品,且每个浓度点至少50%的样品应符合接受标准。
[0169]
3.回归方程的相关系数(r2)必须大于等于0.98。
[0170]
4.当至少67%的质控样品结果(每个浓度至少50%)与它们相应标示值的偏差在
±
15.0%之内时,该分析批认为可接受。
[0171]
实施例2:方法学验证
[0172]
本验证考察项目各分析批测定情况,见附表10。
[0173]
1、系统适用性
[0174]
制备1份系统适用性样品(配制过程参见实施例1中表2的sst样品)并连续进样3次以上,以最后3次进样来评估系统适用性。批次测定结果见附表11,所有结果均符合中国药
典和nmpa非临床药代动力学研究技术指导原则,表明进样时检测仪器均处于正常运转状态。
[0175]
2、残留
[0176]
通过在定量上限样品(uloq样品,配制过程参见实施例1)进样后分析双空白样品(即空白血浆;其中无待测物,无内标)评价残留。每一批次的残留量考察结果见附表12,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则符合接受标准,表明以本方法测定血浆中莠去津的浓度时,高浓度样品进样后在系统中无残留,对于后续低浓度样品的测定无影响。
[0177]
3、选择性
[0178]
3.1基质选择性
[0179]
选择性是区分目标分析物和内标与基质的内源性组份的能力。通过考察6个不同个体血浆(不同个体sd大鼠的edta-k2血浆)分别进行不加内标的空白样品和lloq水平样品(配制过程参见实施例1)测定来评价基质选择性。
[0180]
其中,不加内标的空白样品为使用6个不同sd大鼠edta-k2血浆,处理方式同实施例1中db样品;
[0181]
6个lloq水平样品为使用6个不同sd大鼠edta-k2血浆,处理方式同实施例1中lloq样品。
[0182]
代表色谱图见附图(空白基质色谱图见附图1-2,定量下限色谱图见附图3-4),测定结果见附表13-1和附表13-2,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。结果表明,本方法测定血浆中莠去津浓度的选择性良好,血浆基质对测定无干扰。
[0183]
3.2待测物与内标间的干扰
[0184]
使用单个基质或混合基质(sd大鼠edta-k2血浆),分别平行制备、处理并分析三份仅含待测物且不加内标的定量上限浓度样品(uloq样品,配制过程参见实施例1)。待测物对内标的干扰结果见附表14,结果表明,待测物对内标无干扰。
[0185]
使用单个基质或混合基质(sd大鼠edta-k2血浆),分别平行制备、处理并分析三份仅含内标且不加待测物的样品(对应于实施例1中的blank样品),内标浓度与方法验证时所用内标浓度一致(均为500ng/ml)。内标对待测物的干扰结果见附表14,结果表明,内标对待测物无干扰。
[0186]
4、基质效应
[0187]
分别取6个不同个体(不同个体sd大鼠的edta-k2血浆)的空白基质,每个个体配制一份,经前处理后分别加入与处理后的低、高浓度质控样品浓度相当的标准品及内标纯溶液进行分析。如果需要,体积可以适当调整。无基质存在的与低、高浓度质控样品浓度相当的纯溶液,平行六份,进行分析。如果需要,体积可以适当调整。结果见附表15。莠去津浓度为6.000ng/ml时,经内标归一化的平均基质效应因子为1.00
±
0.02,相对标准偏差为2.0%;莠去津浓度为1500.000ng/ml时,经内标归一化的平均基质效应因子为1.00
±
0.01,相对标准偏差为1.0%,小于15.0%,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。
[0188]
5、批内/批间准确度及精密度
[0189]
取四个不同浓度的质控样品(莠去津浓度分别为2.000ng/ml、6.000ng/ml、120.000ng/ml和1500.000ng/ml),进行批内/批间准确度和精密度的考察。每个分析批每个浓度平行处理6份,需新鲜配制准确度精密度样品。批内准确度和精密度通过每个分析批的质控样品进行评价;批间准确度和精密度通过三个独立分析批(至少2天进行)中的质控样品进行评价。
[0190]
根据质控样品测定结果计算准确度及精密度,结果见附表16。结果表明,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。因此,质控样品的批内、批间精密度和准确度均符合生物样品接受标准。
[0191]
6、质控样品
[0192]
每个分析批至少包含低、中、高3个浓度的质控样品(每个浓度水平至少2份)。含准确度精密度的分析批,低、中和高浓度的准确度与精密度样品也作为质控样品。除准确度及精密度分析批的质控样品结果见附表17。结果表明,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。
[0193]
7、回收率
[0194]
7.1待测物的提取回收率
[0195]
提取回收率样品由低、中、高浓度水平的基质样品分别平行制备六份,不加入内标溶液。在提取后的上清中,加入含有合适浓度内标的纯溶液进行稀释。如果需要,体积可以适当调整。
[0196]
回收率参比样品:提取过程与上面描述相同,在提取后的上清中,加入含有合适浓度待测物和内标的纯溶液进行稀释。最终浓度与100%提取的理论提取回收率样品相同。
[0197]
对于蛋白沉淀的前处理方法,回收率参比样品外加待测物和内标纯溶液后,将改变基质中的溶剂组成,此时提取回收率样品需补充相应的溶剂。
[0198]
按以下公式计算待测物的提取回收率:
[0199][0200]
结果见附表18。结果表明,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。
[0201]
7.2内标的提取回收率
[0202]
由于使用的是稳定同位素标记内标,内标的提取回收率参见待测物的回收率数据,因此内标的提取回收率符合标准。
[0203]
8、稀释可靠性
[0204]
采用配制浓度高于uloq的dqc样品(含莠去津浓度为15000.000ng/ml)进行稀释可靠性考察。用空白大鼠血浆10倍稀释该样品,平行稀释6份,稀释10倍后莠去津的浓度为1500.000ng/ml。随行标准曲线样品对稀释后样品进行测定。结果见附表19。结果表明,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则。
[0205]
9、稳定性
[0206]
9.1标准品储备液/工作液稳定性
[0207]
为考察莠去津储备液在室温条件下(6小时至48小时)和-70~-90℃超低温冰箱(1个月,允许的时间误差范围:
±
7天)中的稳定性,与对照莠去津储备液比较。将储备液稀释
后,加入内标处理,平行操作6份。结果见附表20和附表21。结果表明,莠去津储备液可在室温条件下稳定至少48小时,在超低温冰箱(-70~-90℃)中稳定至少34天;。
[0208]
为考察莠去津工作液(考察用于配制lloq和uloq的工作液)在室温条件下(通常为6小时至48小时)和-70~-90℃超低温冰箱(1个月,允许的时间误差范围:
±
7天)中的稳定性,与对照莠去津工作液比较。将工作液稀释后,加入内标处理,平行操作6份。结果见表22和表23。结果表明,莠去津工作液可在室温条件下稳定至少27小时,在超低温冰箱(-70~-90℃)中稳定至少33天。
[0209]
9.2内标储备液/工作液稳定性
[0210]
由于使用了稳定同位素标记内标,内标的稳定性参见待测物的稳定性数据。
[0211]
9.3质控样品稳定性
[0212]
9.3.1短期稳定性
[0213]
分别将低、高浓度的质控样品放置于室温条件下(4小时至24小时)后进行分析,各浓度水平平行测定6份。测定结果见表24。结果表明,莠去津的血浆样品可在室温条件下稳定至少24小时。
[0214]
9.3.2冻融稳定性
[0215]
分别将低、高浓度的质控样品经过5次冻融(-70~-90℃/室温)循环(首次冷冻至少24小时,之后至少12小时)后进行分析。各浓度水平平行测定6份。测定结果见表25。结果表明,莠去津的sd大鼠edta-k2血浆样品可稳定至少5次(-70~-90℃/室温)冻融循环。
[0216]
9.3.3处理后样品的稳定性
[0217]
经处理后的样品(可采用某一个分析批已成功进样的低、高浓度水平准确度和精密度样品),放置在自动进样器中或与自动进样器相同温度的环境中一段时间后(通常为24小时至144小时)随行新鲜配制的标准曲线再进样分析。结果见表26。结果表明,处理后的莠去津的血浆样品在自动进样器中可稳定至少72小时。
[0218]
9.3.4长期稳定性
[0219]
分别将低、高浓度的质控样品储存于超低温冰箱(-70~-90℃)中(1个月,允许的时间误差范围:
±
7天)。各浓度水平平行测定6份。样品稳定性至少应涵盖从第一个样品采集至样品分析结束的时间段。如有需要,可以评估更长的周期。结果见表27。结果表明,莠去津的血浆样品可在超低温冰箱(-70~-90℃)中分别稳定至少31天。
[0220]
10、全血稳定性
[0221]
为考察样品在采集和处理过程中的稳定性,分别配制低、高两个浓度的全血质控样品。配制完毕后,在37℃水浴中孵育15分钟后,将低、高两个浓度的全血质控样品分别分为两部分。将一部分立即离心处理,制备得到零时间(t0)的血浆样品。另一部分于碎冰中放置2小时后离心,制备得到稳定性考察的血浆样品。每个时间点的血浆样品平行测定6份,一起进样分析。结果见表28。结果表明,莠去津的全血样品在碎冰中放置可稳定至少2小时。
[0222]
11、溶血效应
[0223]
使用模拟的溶血血浆样品(加入2%的溶血全血至未溶血的血浆中,视为严重溶血)制备低浓度和高浓度质控样品进行评估。每个浓度平行测定6份样品。以正常血浆配制的标准曲线样品测定上述溶血质控样品,并随行正常血浆配制的质控样品。结果见表29。结果表明,所有结果均符合中国药典和nmpa非临床药代动力学研究技术指导原则,溶血对本
方法定量测定血浆中莠去津无影响。
[0224]
12、分析批最大进样数
[0225]
为评估分析批最大进样数及方法的耐用性,应至少有一个验证分析批的样品数量接近于样品分析时预估的一个分析批中包括的样品数量。此分析批中可以包括更多的验证样品,重复进样质控样品或者添加内标的空白基质。本验证实验中最大进样针数为117针。
[0226]
综上,本方法中所考察的验证项均符合中国药典和nmpa非临床药代动力学研究技术指导原则,可以用于测定血浆中的莠去津的浓度。
[0227]
表10批量样品检测情况列表
[0228][0229][0230]
表11系统适用性
[0231][0232][0233]
表12批量样品检测过程的残留
[0234][0235][0236]
表13-1莠去津及atrazine-d5(内标)基质选择性(批次2)
[0237][0238]
表13-2莠去津选择性定量下限样品基质选择性
[0239][0240][0241]
表14待测物与内标间的干扰
[0242]
待测物莠去津对内标atrazine-d5的干扰
[0243][0244]
内标atrazine-d5对待测物莠去津的干扰
[0245][0246]
表15基质效应
[0247]
莠去津基质效应
[0248]
[0249][0250]
表16批内、批间样品检测的精密度和准确度
[0251]
[0252]
[0253][0254]
表17莠去津质控样品浓度数据汇总(除准确度精密度分析批外)
[0255]
[0256][0257]
表18待测物提取回收率
[0258]
莠去津提取回收率
[0259]
[0260][0261]
表19稀释可靠性
[0262]
莠去津稀释可靠性(10倍)
[0263]
[0264][0265]
表20储备液短期稳定性
[0266]
莠去津储备液短期稳定性:
[0267][0268]
表21工作液短期稳定性(uloq,lloq工作液)
[0269]
莠去津工作液短期稳定性(uloq,lloq工作液):
[0270][0271]
表22储备液长期稳定性
[0272]
莠去津储备液长期稳定性:
[0273]
[0274][0275]
表23工作液长期稳定性(uloq,lloq工作液)
[0276]
莠去津工作液长期稳定性(uloq,lloq工作液):
[0277]
[0278][0279]
表24质控样品短期稳定性
[0280]
莠去津质控样品短期稳定性
[0281]
试验条件:在室温条件下放置24小时
[0282][0283][0284]
表25质控样品冻融稳定性
[0285]
莠去津质控样品冻融稳定性
[0286]
试验条件:在超低温冷冻冰箱(-70~-90℃)冻存首次至少24小时,之后至少12小
时
[0287][0288]
表26处理后样品的稳定性
[0289]
莠去津处理后样品的稳定性
[0290]
试验条件:在自动进样器(4℃)中放置72小时后的稳定性
[0291]
[0292][0293]
表27质控样品长期稳定性
[0294]
莠去津质控样品长期稳定性
[0295]
试验条件:-70~-90℃条件下放置31天
[0296]
[0297][0298]
表28全血稳定性
[0299]
莠去津全血稳定性
[0300]
试验条件:全血样品在碎冰中放置2小时后的稳定性
[0301]
[0302][0303]
表29溶血效应
[0304]
莠去津溶血效应
[0305]
[0306]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。