1.本发明涉及化合物检测技术领域,具体而言,涉及一种河鲀毒素的检测样品的制备方法和检测方法、应用。
背景技术:
2.河鲀毒素(tetrodotoxin,ttx)是天然生物碱类神经毒素,因其最早从河鲀中分离而得名,是目前已知毒性最强的非蛋白类生物毒素之一,其毒性强度为氰化钠的1250倍,麻醉作用为可卡因的16万倍。河鲀毒素毒性作用快,且尚无特效解毒剂,这也导致河鲀毒素中毒者死亡率极高。部分河鲀体内含有剧毒的河鲀毒素,最少仅需2mg即可致死体重为50kg左右的成年人。
3.由于河鲀毒素在极低浓度就可以对人体或动物体产生毒害作用,因此亟需开发一种精准检测河鲀毒素的方法,为保障水产品安全提供理论基础。
4.鉴于此,特提出本发明。
技术实现要素:
5.本发明的目的在于提供一种河鲀毒素的检测样品的制备方法和检测方法、应用。
6.本发明是这样实现的:
7.第一方面,本发明提供了一种河鲀毒素的检测样品的制备方法,包括将样品置于乙酸甲醇溶液中提取,提取后固液分离,取上清液作为样品液;其中,提取和固液分离过程中的温度均低于10℃。
8.在可选的实施方式中,样品与乙酸甲醇溶液的质量体积比为0.1~0.5g:0.15~1.5ml。
9.优选地,乙酸甲醇溶液中乙酸的含量为0.5~1.5%。
10.优选地,样品包括鱼肉、鱼糜、鱼皮和鱼内脏中的至少一种。
11.在可选的实施方式中,提取包括将样品和乙酸甲醇溶液的混合物进行研磨和超声。
12.优选地,样品和乙酸甲醇溶液的混合物的制备包括向样品中加入乙酸甲醇溶液和钢珠涡旋20~40s。
13.优选地,研磨包括将混合物置于研磨机中研磨,研磨时间为2~6min。
14.优选地,超声为冰水浴超声,水浴温度为0~5℃,超声时间10~20min;
15.优选地,提取次数为2~5次。
16.在可选的实施方式中,固液分离为离心;离心的温度为2~6℃,转速为10000~15000rpm,离心时间10~20min。
17.在可选的实施方式中,固液分离后还包括对底部沉淀进行二次提取和二次固液分离。
18.优选地,二次提取过程中加入的乙酸甲醇溶液的体积为提取过程中加入的乙酸甲
醇溶液的体积的1/3~1/5。
19.在可选的实施方式中,还包括将固液分离获得的上清液制成粉末,再对粉末进行复溶,制得样品液。
20.优选地,粉末的制备包括用氮气吹干固液分离获得的上清液。
21.优选地,复溶的溶剂为乙腈、水和甲酸的复合溶液;更优选地,乙腈和水的体积比为0.8~1.2:0.8~1.2;更优选地,复溶的溶剂中,甲酸的浓度为0.001~0.2%。
22.优选地,复溶的溶剂体积为50~200μl。
23.优选地,复溶包括将粉末与复溶溶剂的混合液涡旋后,置于冰水浴中超声,再固液分离,取上清液为样品液。
24.优选地,复溶的涡旋时间为20~40s,超声时间2~8min,固液分离采用离心的方式分离,离心温度2~6℃,离心转速10000~15000rpm,离心时间10~20min。
25.第二方面,本发明提供了一种河鲀毒素的检测方法包括采用超高液相色谱-串联质谱法检测如前述实施方式任一项所述的制备方法制得的河鲀毒素的检测样品。
26.在可选的实施方式中,包括采用超高液相色谱-串联质谱法检测河鲀毒素标准品,获得河鲀毒素浓度和峰面积的标准曲线;采用超高液相色谱-串联质谱法检测样品液,通过样品液的峰面积和标准曲线获得样品液中河鲀毒素的浓度。
27.在可选的实施方式中,超高液相色谱的色谱条件包括:色谱仪为agilent1290infinity ii series(agilent technologies)超高效液相色谱仪,色谱柱为waters acquity uplc beh amide(100
×
2.1mm,1.7μm,waters),柱温35~45℃,样品盘温度2~6℃,流动相a为5mm乙酸铵和0.1%甲酸水溶液,流动相b为乙腈,进样量5~20μl。
28.在可选的实施方式中,串联质谱的质谱条件包括:质谱仪为三重四极杆质谱仪,离子源为iondrive turbo v esi,气帘气30~40psi,离子喷雾电压为4500~5500v,温度为350~450℃,离子源气体1为50~70psi,离子源气体2为50~70psi。
29.第三方面,本发明提供了一种如前述实施方式任一项的制备方法制得的河鲀毒素的检测样品或如前述实施方式任一项的检测方法在食品、药品或农产品领域的应用。
30.本发明具有以下有益效果:
31.本发明提供了一种河鲀毒素的检测样品的制备方法和检测方法、应用,通过采用超高液相色谱-串联质谱法检测技术能够精确检测出样品中的河鲀毒素,配合河鲀毒素的提取方法,可以在更低的样品加入量下,精确检测出河鲀毒素的含量,该方法检测精度高,检测灵敏,稳定性好,为食品、药品和农产品中的河鲀毒素安全应用提供依据。
附图说明
32.为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
33.图1为本发明实施例1提供的河鲀毒素标准品溶液的高效液相色谱图;
34.图2为本发明实施例1提供的河鲀毒素标准品溶液的标准曲线图;
35.图3为本发明对比例2提供的河鲀毒素在样品中的高效液相色谱图。
具体实施方式
36.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
37.第一方面,本发明提供了一种河鲀毒素的检测样品的制备方法,包括如下步骤:
38.样品液的制备方法包括将样品置于乙酸甲醇溶液中提取,提取后固液分离,取上清液作为样品液;其中,提取和固液分离过程中的温度均低于10℃。通过在较低温度下提取和固液分离不仅可以较好地保持河鲀毒素的活性,同时还可以提高河鲀毒素的溶出度,有利于河鲀毒素的检出。
39.在可选的实施方式中,样品与乙酸甲醇溶液的质量体积比为0.1~0.5g:0.15~1.5ml。本发明的提取方法可以使用较少的样品精确地检测出河鲀毒素的含量,实现了河鲀毒素的微量检测。
40.优选地,乙酸甲醇溶液中乙酸的含量为0.5~1.5%。
41.优选地,样品来源于河鲀、螺类、蓝环章鱼、圆尾鲎中的任一种,优选为河鲀;优选地,河鲀包括暗纹东方鲀、晕环东方鲀、弓斑东方鲀、铅点东方鲀、兔头鲀和横纹东方鲀中的任一种;更优选地,样品包括鱼肉、鱼糜、鱼皮和鱼内脏中的至少一种。
42.在可选的实施方式中,提取包括将样品和乙酸甲醇溶液的混合物进行研磨和超声。
43.优选地,样品和乙酸甲醇溶液的混合物的制备包括向样品中加入乙酸甲醇溶液和钢珠涡旋20~40s。
44.优选地,研磨包括将混合物置于研磨机中研磨,研磨时间为2~6min。由于样品在加入时可以是块状的组织,也可以是更细小的肉糜,因此对样品进行研磨可以使大块样品破碎,使样品中的河鲀毒素更好地溶出。
45.优选地,超声为冰水浴超声,水浴温度为0~5℃,超声时间10~20min;
46.优选地,提取次数为2~5次。多次提取的方法可以是研磨和超声过程交替进行,例如当提取次数为2次时,先将混合物放入研磨机中研磨破碎,然后进行水浴超声,超声结束后再次进行研磨破碎,然后再次超声,使河鲀毒素溶出。
47.在可选的实施方式中,固液分离采用离心的方式分离,离心的温度为2~6℃,转速为10000~15000rpm,离心时间10~20min。
48.在可选的实施方式中,为了保证样品中尽可能多的河鲀毒素溶出,固液分离后还包括对底部沉淀进行二次提取和二次固液分离。
49.优选地,二次提取过程中加入的乙酸甲醇溶液的体积为提取过程中加入的乙酸甲醇溶液的体积的1/3~1/5。由于在第一次提取的时候样品会吸收部分乙酸甲醇进入样品的组织中暂存,同时,第一次提取的过程中已经溶出了大量河鲀毒素,因此在二次提取的过程中可以适当减少乙酸甲醇的添加量。
50.优选地,二次提取和二次固液分离的参数除另有说明外均与第一次提取的参数相同。
51.在可选的实施方式中,为了获得更佳的检测结果,能够明显且直观地观察到河鲀
毒素的色谱峰,还包括将固液分离获得的上清液制成粉末,再对粉末进行复溶,制得样品液。
52.优选地,粉末的制备包括用氮气吹干固液分离获得的上清液。
53.优选地,复溶的溶剂为乙腈、水和甲酸的复合溶液;更优选地,乙腈和水的体积比为0.8~1.2:0.8~1.2;更优选地,复溶的溶剂中,甲酸的浓度为0.001~0.2%。复溶的溶剂既可以较佳地溶解河鲀毒素,同时与检测过程中使用的流动相溶液相似,河鲀毒素的检出结果较好。
54.优选地,复溶的溶剂体积为50~200μl。
55.优选地,复溶包括将粉末与复溶溶剂的混合液涡旋后,置于冰水浴中超声,再固液分离,取上清液为样品液。
56.优选地,复溶的涡旋时间为20~40s,超声时间2~8min,固液分离采用离心的方式分离,离心温度2~6℃,离心转速10000~15000rpm,离心时间10~20min。
57.第二方面,本发明提供了一种河鲀毒素的检测方法,包括如下步骤:
58.1)、配制河鲀毒素标准品溶液
59.称取河鲀毒素标准品于10ml容量瓶中,配制成浓度为10mg/l的标准品储备液。分别量取不同体积的标准品储备液于10ml容量瓶中,依次稀释获得一系列浓度梯度的标准品溶液。
60.在可选的实施方式中,标准品溶液的浓度梯度为62.5ng/l、125ng/l、250ng/l、500ng/l、1000ng/l、2000ng/l、4000ng/l和8000ng/l。
61.2)、绘制标准曲线
62.采用超高液相色谱-串联质谱法依次检测河鲀毒素标准品溶液,获得河鲀毒素的峰面积,将峰面积与标准品溶液的浓度对应,绘制标准曲线。
63.在可选的实施方式中,超高液相色谱的色谱条件包括:色谱仪为agilent1290infinity ii series(agilent technologies)超高效液相色谱仪,色谱柱为waters acquity uplc beh amide(100
×
2.1mm,1.7μm,waters),柱温35~45℃,样品盘温度2~6℃,流动相a为5mm乙酸铵和0.1%甲酸水溶液,流动相b为乙腈,进样量5~20μl。
64.在可选的实施方式中,串联质谱的质谱条件包括:质谱仪为三重四极杆质谱仪,离子源为iondrive turbo v esi,气帘气30~40psi,离子喷雾电压为4500~5500v,温度为350~450℃,离子源气体1为50~70psi,离子源气体2为50~70psi。
65.3)、检测样品液中河鲀毒素的含量
66.采用超高液相色谱-串联质谱法检测样品液,通过样品液的峰面积和标准曲线获得样品液中河鲀毒素的浓度。
67.其中,采用超高液相色谱-串联质谱法检测样品液的色谱条件和质谱条件与2)步骤中检测标准品溶液的条件相同。
68.第三方面,本发明提供了一种如前述实施方式任一项的制备方法制得的河鲀毒素的检测样品或如前述实施方式任一项的检测方法在食品、药品或农产品领域的应用。
69.以下结合实施例对本发明的特征和性能作进一步的详细描述。
70.实施例1
71.本实施例提供了一种河鲀毒素的检测方法,包括如下步骤:
72.s1、制备样品液
73.称取0.25g样品于ep管中,加入钢珠和1ml乙酸甲醇溶液(乙酸含量1%),涡旋30s,混合均匀获得混合物。其中,样品为暗纹东方鲀的肠道组织。
74.第一次提取:将样品和乙酸甲醇溶液的混合物置于研磨机中研磨4min,再置于冰水浴中超声,水浴温度为0℃,超声时间15min,超声结束后重复上述研磨和超声过程3遍。
75.离心:将ep管置于离心机中离心,离心温度4℃,离心转速13000rpm,离心时间15min,将上清液转移至另一ep管中。
76.二次提取和二次离心:向ep管内的底部沉淀中再加入250μl的乙酸甲醇(乙酸含量1%),涡旋30s后冰水浴超声15min;4℃下1300rpm离心15min。合并两次上清液,并用氮气吹干获得粉末。
77.复溶:向粉末中加入100μl复溶溶液,涡旋30s后冰水浴超声5min;4℃下1300rpm离心15min,取80μl上清液作为样品液备用。其中,复溶溶剂为乙腈、水和甲酸的混合溶液,乙腈和水的体积比为1:1,甲酸的浓度为0.1%。
78.s2、配制河鲀毒素标准品溶液
79.称取河鲀毒素标准品于10ml容量瓶中,加入乙酸甲醇溶液(乙酸含量1%)配制成浓度为10mmol/l的标准品储备液。分别量取不同体积的标准品储备液于10ml容量瓶中,依次稀释获得一系列浓度梯度的标准品溶液。
80.在本实施例中,标准品溶液的浓度梯度为1ng/l、2.5ng/l、5ng/l、12.5ng/l、25ng/l、50ng/l、125ng/l、250ng/l、500ng/l、1000ng/l、2000ng/l、4000ng/l和8000ng/l。
81.s3、绘制标准曲线
82.采用超高液相色谱-串联质谱法依次检测河鲀毒素标准品溶液,获得标准品溶液的色谱图,其中,50ng/l的标准品溶液的色谱图如图1所示,通过计算各标准品溶液的色谱图中河鲀毒素的峰面积,将峰面积与标准品溶液的浓度对应,绘制标准曲线,如图2所示。
83.超高液相色谱的色谱条件包括:色谱仪为agilent 1290infinity ii series(agilent technologies)超高效液相色谱仪,色谱柱为waters acquity uplc beh amide(100
×
2.1mm,1.7μm,waters),柱温40℃,样品盘温度4℃,流动相a为5mm乙酸铵和0.1%甲酸水溶液,流动相b为乙腈,进样量10μl。
84.串联质谱的质谱条件包括:质谱仪为三重四极杆质谱仪,离子源为iondrive turbo v esi,气帘气35psi,离子喷雾电压为5000v,温度为400℃,离子源气体1为60psi,离子源气体2为60psi。
85.s4、检测样品液中河鲀毒素的含量
86.采用超高液相色谱-串联质谱法检测样品液,色谱条件和质谱条件与s3步骤中检测标准品溶液的条件相同,获得河鲀毒素的峰面积,通过标准曲线计算出样品液中河鲀毒素的浓度为656.33ng/kg。
87.实施例2
88.本实施例提供了一种河鲀毒素的检测方法,包括如下步骤:
89.s1、制备样品液
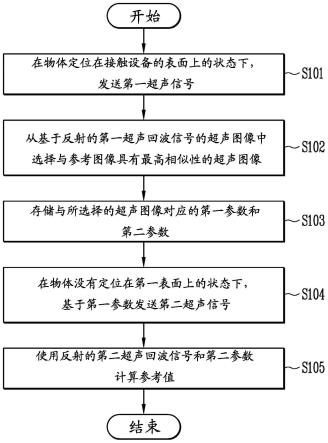
90.称取0.5g样品于ep管中,加入钢珠和1ml乙酸甲醇溶液(乙酸含量1%),涡旋30s,混合均匀获得混合物。其中,样品为暗纹东方鲀的肠道组织。
91.第一次提取:将样品和乙酸甲醇溶液的混合物置于研磨机中研磨4min,再置于冰水浴中超声,水浴温度为0℃,超声时间15min,超声结束后重复上述研磨和超声过程3遍。
92.离心:将ep管置于离心机中离心,离心温度4℃,离心转速13000rpm,离心时间15min,将上清液转移至另一ep管中。
93.二次提取和二次离心:向ep管内的底部沉淀中再加入250μl的乙酸甲醇(乙酸含量1%),涡旋30s后冰水浴超声15min;4℃下1300rpm离心15min。合并两次上清液,并用氮气吹干获得粉末。
94.复溶:向粉末中加入100μl复溶溶液,涡旋30s后冰水浴超声5min;4℃下1300rpm离心15min,取80μl上清液作为样品液备用。其中,复溶溶剂为乙腈、水和甲酸的混合溶液,乙腈和水的体积比为1:1,甲酸的浓度为0.1%。
95.标准曲线为实施例1中获得的标准曲线。样品液的检测方法与实施例1相同。
96.实施例3
97.本实施例提供了一种河鲀毒素的检测方法,其中,样品和标准曲线的绘制方法与实施例1相同,超高液相色谱-串联质谱法的检测条件不同,具体如下:
98.超高液相色谱的色谱条件包括:色谱仪为agilent 1290infinity ii series(agilent technologies)超高效液相色谱仪,色谱柱为waters acquity uplc beh amide(100
×
2.1mm,1.7μm,waters),柱温40℃,样品盘温度6℃,流动相a为5mm乙酸铵和0.1%甲酸水溶液,流动相b为乙腈,进样量10μl。
99.串联质谱的质谱条件包括:质谱仪为三重四极杆质谱仪,离子源为iondrive turbo v esi,气帘气40psi,离子喷雾电压为5000v,温度为400℃,离子源气体1为60psi,离子源气体2为60psi。
100.对比例1
101.本对比例提供了一种河鲀毒素的检测方法,其步骤与实施例1相同,区别仅在于:提取溶剂为1ml乙酸乙腈溶液(乙酸含量1%),加标回收实验结果显示乙腈对河鲀毒素的提取效果不如甲醇。
102.对比例2
103.本对比例提供了一种河鲀毒素的检测方法,其步骤与实施例1相同,区别仅在于:色谱柱为waters acquity uplc beh c18(100
×
2.1mm,1.7μm,waters),得到如图3所示色谱结果。由图3可知,实验结果显示c18色谱柱无法有效分离鱼肉样品中的河鲀毒素,分离效果远差于amide色谱柱。
104.对比例3
105.本对比例提供了一种河鲀毒素的检测方法,其步骤与实施例1相同,区别仅在于:离子源为iondrive turbo v esi-(负离子模式),实验结果显示河鲀毒素在负离子模式下无法电离。
106.对比例4
107.本对比例提供了一种河鲀毒素的检测方法,其步骤与实施例1相同,区别仅在于:两次提取的水浴温度均为60℃。
108.试验例1
109.采用实施例1的方法,分别对暗纹东方鲀的心脏、血清、卵巢、肌肉、肝、胆等13个不
同部位进行检测,检验不同暗纹东方鲀样品中的河鲀毒素含量,得到如表1所示结果。
110.表1暗纹东方鲀组织样品中河鲀毒素的含量
111.组织浓度(ng/kg)组织浓度(ng/kg)组织浓度(ng/kg)肠656.33肌肉746.08鳃612.82肠757.54卵巢674.76鳃835.98胆706.93脾162.47心脏680.69胆389.96脾545.65心脏179.11肝94.61鳔176.87血清129.87肝45.36鳔130.69眼睛787.26精巢664.22皮肤330.23眼睛871.61肌肉1129.71皮肤762.72
ꢀꢀ
112.由表1可知,河鲀毒素广泛存在于鲀鱼的身体各个部位,针对同一种鲀鱼,其不同部位的样品中,河鲀毒素的含量差异较大;相同部位但不同样品,例如不是同一条鲀鱼,其河鲀毒素的含量差异也较为明显。
113.试验例2
114.以加标浓度为100ng/l的实施例1的样品和50ng/l浓度的标准品作为qc样本进行重复性实验,分别采用实施例1和对比例1~3的方法检测qc样本中的河鲀毒素,计算其加标回收率和标准相对偏差rsd值,得到如表2所示结果。
115.表2河鲀毒素的检测方法的精确度
[0116][0117][0118]
由表2可知,采用本发明实施例1的方法,可以准确可靠地检测出样品中河鲀毒素的含量。而对比例1由于乙腈对河鲀毒素提取效果弱于甲醇,导致其加标回收率低,rsd值高,因此该方法对河鲀毒素的检测精度较低,难以实现低含量的河鲀毒素含量检测。
[0119]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。