1.本发明属于基因技术与计算机技术结合领域,具体涉及一种面向群体基因组索引表示与构建的方法及设备。
背景技术:
2.基因组测序成本伴随着测序手段的进步正在逐年下降,目前进行中的单项人类基因组计划的规模正在向十万、百万人量级迈进。这些大型基因组计划的不断推进和实施,伴随而来的是基因组测序数据爆炸性的增长,所产生数据规模和对应的数据分析规模也将达到pb-zb级。数据量和数据分析需求的爆炸性增长,也导致了一些问题愈发凸显并亟待解决。
3.其中,基因组大数据索引结构的设计与实施是下游数据分析的基础和核心支持,索引结构的功能与性能直接影响基因组序列比对、序列拼接和变异检测等数据分析速度与精度。目前,现有索引技术是面向单一物种的参考基因组,且多采用序列图模型索引结构,内存占用较大,限制了整合群体基因组的样本数量。现有方法无法对pb级群体基因组数据构建有效的索引结构,且无法支撑日益增长的基因组大数据分析,严重制约基因数据价值挖掘与产业转化。
技术实现要素:
4.本发明为了解决现有基因组索引结构构建的方法无法对pb级群体基因组数据构建有效的索引结构的问题。
5.一种面向群体基因组索引表示与构建的方法,包括以下步骤:
6.步骤一、数据收集与预处理:
7.获取人类参考基因组数据和制定的已知包含不同类型变异的变异数据,并对两种数据文件进行去除冗余数据等规范化操作预处理;
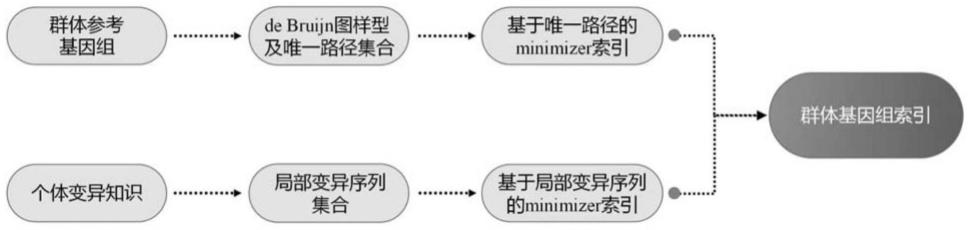
8.步骤二、参考基因组构建de bruijn图模型索引表示:
9.遍历完整的参考基因组,提取所有k-mer序列,同时记录每个k-mer对应的四元组(k-mer序列,入边碱基,出边碱基,偏移位置),基于每个k-mer对应的四元组,提取输入数据的不同k-mer序列以及k-mer序列之间的关联关系构建de bruijn图模型,de bruin图g=《v,e》是一个有向图模型,图中节点v是相互之间不同的长度为k的短序列片段k-mer,提取基因组上所有不同k-mer构成节点集合v={v1,v2,
…
,vm};对于两个节点vi和vj,如果vi与vj有(k-1)-mer的序列重叠,则存在有向边vi
→
vj;
10.每一个k-mer序列代表一个de bruijn图模型节点,根据节点的入边与出边数量情况确定唯一路径unipath,节点的入度和出度分别定义为节点的入边和出边的个数,且节点可能存在一个或多个入边和出边;图g中任意两个节点可形成的一条路径,如果路径的起始节点入度大于1且出度是1,结束节点出度大于1且入度是1,中间节点入度是1且出度是1,则该路径是一个唯一路径unipath;
11.利用生成的所有唯一路径;对于每一个路径,其上的节点关联了生成的k-mer序列位置集合;针对某路径,从头开始向后遍历每一个节点,获取当前节点和其后继节点的两个位置集合,找出两个集合的相近位置元素;由于每个位置集合是有序集合,遍历某集合元素并采用二分查找方法,找出所有相近位置并形成新位置集合;按照此方法,依次向后遍历路径上的每一个节点,每遍历一个新节点即执行以上位置查找和集合合并操作,直至遇到路径的结束节点,最后形成的位置集合即为当前唯一路径对应的位置集合;
12.按照此方法生成所有唯一路径对应的位置集合,并表示为二进制索引文件,表示为f-pos;
13.步骤三、局部变异序列索引表示与构建:
14.首先将参考基因组按照固定长度区间进行有重叠划分,每个划分的固定长度区间作为一个局部区域,每两个相邻局部区域有部分重叠,重叠区域的长度是划分区域长度的一半;
15.针对某个体的单体型,收集每个局部区域内的变异,并形成带有已知变异的局部个体基因组序列,即生成局部变异序列;重叠区域的长度是划分区域长度的一半;
16.通过以上方法生成变异序列索引文件,每一个变异序列索引文件对应作为一个alt string;
17.步骤四、针对“unipath”和“alt string”,基于minimizer的群体基因组索引表示与构建:
18.在针对“unipath”和“alt string”构建索引之前,分别将“unipath”图以及“alt string”列表转换为字符串列表;之后,针对字符串列表中的每一个字符串,依照minimizer选取准则,选取滑窗内哈希值最小的k-mer,作为局部的代表k-mer;这些局部最小k-mer被称为minimizer,将会被存储在一个统一的列表里;
19.所有字符串对应的minimizer列表,将会被合并成为一个完整的列表,该列表会被按照minimizer的哈希值的大小进行排序;之后,按照每个minimizer的哈希值的高k位构建miminizer的二级索引,k为二级索引参数;
20.二级索引将minimizer的总索引分割成为b=2
^k
个小区间,当假设minimizer的总数目为m个的时候,每个小区间平均包含m/b个minimizer;构建好的minimizer,每个占据96bit的存储空间,其中minimizer的哈希值的后q位占据30位存储空间;minimizer在原始“unipath”或者“alt string”列表上的位置,占据65bit的空间。
21.进一步地,在遍历完整的参考基因组提取所有k-mer序列时,需要对k-mer序列进行排序和去重操作,具体过程包括以下步骤:
22.根据k-mer的前f个bp的序列内容作为文件名,bp表示碱基总量,将k-mer序列对应的四元组写入对应临时文件;
23.然后,将一个文件内所有k-mer序列导入内存,对当前内存中的文件里的k-mer执行快速排序,使文件内k-mer序列对应的四元组有序,再输出到硬盘形成对应的有序文件;
24.快速排序:从头遍历当前文件内k-mer集合,对相邻的相同k-mer序列进行合并,并对k-mer对应的偏移位置结合进行合并;
25.对所有文件依次导入内存并进行快速排序,生成所有不同k-mer序列并保证k-mer对应的位置集合有序。
26.进一步地,根据节点的入边与出边数量情况确定唯一路径unipath的过程包括以下步骤:
27.首先根据节点的入边与出边数量情况,将节点分为如下几种类型:
28.1)
‘
x’型节点:多个入边且多个出边;
29.2)
‘
fy’型节点:多个入边且一个出边;
30.3)
‘
ry’型节点:一个入边且多个出边;
31.4)
‘
l’型节点:一个入边且一个出边;
32.唯一路径的起始和终止节点的构成方式有以下几种可能性:
33.(1)起始和终止节点是同一个
‘
x’型节点;
34.(2)起始节点是
‘
fy’型节点且结束节点是
‘
ry’型节点;
35.(3)起始节点是
‘
x’型节点的后驱节点或
‘
fy’型节点的后驱节点且结束节点
‘
x’型节点的前驱节点或
‘
ry’型节点的前驱节点;
36.然后,遍历图中所有节点,如果当前节点是唯一路径的起始节点,则执行向后延伸操作;向后延伸操作:通过当前节点的出边信息计算得出其后驱节点,如果后驱节点是
‘
l’型节点,则继续向后延伸,按照此方法依次循环,直至遇到结束节点;通过以上方法可生成图模型的所有唯一路径且保证路径中各节点不重复;
37.通过以上方法生成所有的唯一路径索引文件,每一个唯一路径索引文件对应作为一个unipath。
38.进一步地,步骤四中选取滑窗内哈希值最小的k-mer的具体过程包括以下步骤:
39.minimizer的选取有两个关键参数,分别是windows size以及k-mer长度;假设待构建索引的序列为l,k-mer长度为l_k,则滑窗选取时,将会有(l-l_k 1)个k-mer被选取出来;假设windows size是w,则从编号1~5这5个k-mer中,选取哈希值最小的一个min_k-mer1;之后,从2~6这5个中,选择最小的一个min_k-mer2;之后是3~7并依次类推;相邻的不同minimizer选取窗口选取出来的min_k-mer可能是同一个,此时每个重复的仅仅记录一次;min_k-mer被称为minimizer。
40.进一步地,所述哈希值的后q位中的q=minimizer_len*2-k,其中minimizer_len表示minimizer长度。
41.进一步地,针对“unipath”进行基于minimizer的群体基因组索引表示与构建时,unipath图模型中的minimizer的存储数据结构如下:
42.构建的unpath序列的minimizer索引,除了30bit的minimizer存储空间,还有65bit的位置区;
43.所述位置区为:其中32bit用于存储该minimizer所属的unitig的id,32bit存储该minimizer所在unitig的位置,1bit用于存储该minimizer相对于unitig的方向。
44.进一步地,针对“alt string”进行基于minimizer的群体基因组索引表示与构建时,alt string的minimizer的存储数据结构如下:
[0045]“alt string”的minimizer的每一项存储成为96bit的数据结构,所有的minimizer共同构成数据表;属于“alt string”的minimizer,其坐标以如下方法唯一标注:
[0046]
每个minimizer包含65位的位置区,其中,
[0047]
(a)25bit用于存储该minimizer所属的window block的id;
[0048]
(b)19bit存储该minimizer所在haplotype的id;
[0049]
(c)22bit用来存储在haplotype上的位置;
[0050]
(d)1bit用于存储该minimizer相对于haplotype上的方向。
[0051]
进一步地,针对“alt string”进行基于minimizer的群体基因组索引表示与构建的过程中,仅对相互靠近的每个window block中间的甜区变异构建索引,所述甜区为每个window block的从25%*l到75%l的一段区域。
[0052]
进一步地,所述方法还包括minimizer查找的过程,minimizer查找的过程包括以下步骤:
[0053]
首先,使用minimizer生成算法生成read的minimizer列表;之后针对其中的某个minimizer,获取其哈希值的前k位,定位到minimizer索引的一个小区间内,该小区间平均包含m/b个minimizer;之后,在该小区间内,使用剩余的q位作为key,进行二分查找;
[0054]
找到minimizer在索引中的具体位置之后,将通过其65位的位置区,解析出来read的minimizer在“unipath”图或者“alt string”列表中的位置;如果查找的是“unipath”图的minimizer索引,则解析为相应的unitig的id,unitig上的位置以及minimizer相对于unitig的方向;如果查找的是“alt string”列表的minimizer索引,则解析成为:window block的id,haplotype的id,haplotype上的位置,以及该minimizer相对于haplotype上的方向,即minimizer在wb结构上的坐标。
[0055]
一种面向群体基因组索引表示与构建设备,所述设备包括处理器和存储器,所述存储器中存储有至少一条指令,所述至少一条指令由处理器加载并执行以实现所述的一种面向群体基因组索引表示与构建方法。
[0056]
有益效果:
[0057]
本发明提出一种面向群体基因组索引表示与构建的方法,解决了现有基因组索引结构的性能瓶颈问题。本发明可以组织和表示pb级大规模群体基因组数据,并支持快速序列比对与序列拼接等数据分析任务。同时,有效整合大规模群体基因组序列信息,解决由单一基因组引起的系统性偏差问题。本发明充分利用和组织群体基因组的重复序列,构建出基于单一路径表示方法的图模型,实现有限内存空间下对大规模群体基因组构建图模型,有效突破索引群体基因组数量规模瓶颈,大幅提升群体基因组索引结构的应用价值。
附图说明
[0058]
图1为面向群体基因组索引表示与构建的方法示意图。
[0059]
图2为窗口块的划分示意图。
[0060]
图3为窗口块内单体型压缩示意图。
具体实施方式
[0061]
具体实施方式一:结合图1说明本实施方式,
[0062]
本实施方式为一种面向群体基因组索引表示与构建的方法,包括以下步骤:
[0063]
步骤一、数据收集与预处理:
[0064]
首先获取最新版本的人类参考基因组数据和制定的已知包含不同类型变异的变异数据,并对两种数据文件进行去除冗余数据等规范化操作预处理。
[0065]
步骤二、参考基因组构建de bruijn图模型索引表示:
[0066]
de bruijn图模型是表示和组织基因组和测序数据的重要数据结构,广泛应用于众多基因组序列分析任务。de bruijn图模型可有效组织参考基因组的重复序列。de bruin图g=《v,e》是一个有向图模型,图中节点v是相互之间不同的长度为k的短序列片段k-mer,提取基因组上所有不同k-mer构成节点集合v={v1,v2,
…
,vm}。对于两个节点vi和vj,如果vi与vj有(k-1)-mer的序列重叠,则存在有向边vi
→
vj。节点的入度和出度分别定义为节点的入边和出边的个数,且节点可能存在一个或多个入边和出边。图g中任意两个节点可形成的一条路径,如果路径的起始节点入度大于1且出度是1,结束节点出度大于1且入度是1,中间节点入度是1且出度是1,则该路径是一个唯一路径。因此,构建de bruijn图模型需要提取输入数据的不同k-mer序列以及k-mer序列之间的关联关系(图模型中边)。
[0067]
步骤二一、生成所有不同k-mer序列:
[0068]
遍历完整的参考基因组,提取所有k-mer序列,同时记录每个k-mer对应的四元组(k-mer序列,入边碱基,出边碱基,偏移位置)。由于基因组序列数据量大,k-mer序列数量多,无法将所有k-mer序列同时放入内存中进行排序和去重等操作,因此本发明采用分块排序的方法实现所有k-mer序列有序。
[0069]
分块排序:根据k-mer的前f(默认值为6)个bp(bp表示碱基总量)的序列内容(字串形成的序列)作为文件名,并将k-mer序列对应的四元组写入对应临时文件。通过该方法,每一个k-mer序列被写入对应的文件中,同时,保证文件之间k-mer集合有序(按照字典序)。此时,文件内的k-mer序列时无序的。
[0070]
然后,将一个文件内所有k-mer序列导入内存,对当前内存中的文件里的k-mer执行快速排序,使文件内k-mer序列对应的四元组有序,再输出到硬盘形成对应的有序文件。快速排序:从头遍历当前文件内k-mer集合,对相邻的相同k-mer序列进行合并,并对k-mer对应的偏移位置结合进行合并。
[0071]
对所有文件依次导入内存并进行快速排序,生成所有不同k-mer序列并保证k-mer对应的位置集合有序。由于是将f个bp作为文件名,使得文件之间是有序的,所以当所有的k-mer序列文件都分别排序后,所有k-mer也都有序。
[0072]
步骤二二、生成图模型的唯一路径:
[0073]
每一个k-mer序列代表一个de bruijn图模型节点,根据节点的入边与出边数量情况,将节点分为如下几种类型:
[0074]
1)
‘
x’型节点:多个入边且多个出边;
[0075]
2)
‘
fy’型节点:多个入边且一个出边;
[0076]
3)
‘
ry’型节点:一个入边且多个出边;
[0077]
4)
‘
l’型节点:一个入边且一个出边。
[0078]
唯一路径的起始和终止节点的构成方式有以下几种可能性:
[0079]
(1)起始和终止节点是同一个
‘
x’型节点;
[0080]
(2)起始节点是
‘
fy’型节点且结束节点是
‘
ry’型节点;
[0081]
(3)起始节点是
‘
x’型节点的后驱节点或
‘
fy’型节点的后驱节点且结束节点
‘
x’型节点的前驱节点或
‘
ry’型节点的前驱节点。
[0082]
遍历图中所有节点,如果当前节点是唯一路径的起始节点,则执行向后延伸操作。
向后延伸操作:通过当前节点的出边信息计算得出其后驱节点,如果后驱节点是
‘
l’型节点,则继续向后延伸,按照此方法依次循环,直至遇到结束节点。通过以上方法可生成图模型的所有唯一路径且保证路径中各节点不重复。
[0083]
通过以上方法生成所有的唯一路径索引文件,每一个唯一路径索引文件对应作为一个unipath,表示为f-unipath。
[0084]
步骤二三、生成唯一路径的位置集合索引文件:
[0085]
利用步骤二二生成的所有唯一路径。对于每一个路径,其上的节点关联了步骤二一生成的k-mer序列位置集合。针对某路径,从头开始向后遍历每一个节点,获取当前节点和其后继节点的两个位置集合,找出两个集合的相近位置元素(位置差小于某阈值,默认值设置为50bp)。由于每个位置集合是有序集合,遍历某集合元素并采用二分查找方法,找出所有相近位置并形成新位置集合。按照此方法,依次向后遍历路径上的每一个节点,每遍历一个新节点即执行以上位置查找和集合合并操作,直至遇到路径的结束节点,最后形成的位置集合即为当前唯一路径对应的位置集合。
[0086]
按照此方法生成所有唯一路径对应的位置集合,并表示为二进制索引文件,表示为f-pos。该索引文件有效支持基于图模型路径信息的基因组序列比对,是基于群体基因组索引结构的数据分析方法的基础。
[0087]
步骤三、局部变异序列索引表示与构建:
[0088]
参考基因组代表某物种的群体遗传特征。而基因组变异信息是不同于参考基因组的代表个体独有的遗传特征,其包括错配、插入、删除、倒置和转座等变异类型。故完整的群体基因组信息包括参考基因组和不同个体的变异信息。对于某区域内的多个变异,不同个体的变异组合相互独立,且同一个体的单倍体上的变异组合也相互独立。
[0089]
首先将参考基因组按照固定长度区间进行有重叠划分,每个划分的固定长度区间作为一个局部区域,每两个相邻局部区域(默认长度300bp)有部分重叠(默认长度150bp),重叠区域的长度是划分区域长度的一半。
[0090]
针对某个体的单体型,收集每个局部区域内的变异,并形成带有已知变异的局部个体基因组序列,即生成局部变异序列。同时,重叠区域的长度是划分区域长度的一半,支持并保证任意种子一定会落在基因组上的某一局部区域。不考虑某区域内没有已知变异的情况。
[0091]
通过以上方法生成变异序列索引文件,每一个变异序列索引文件对应作为一个alt string,表示为f-alt。
[0092]
步骤四、基于minimizer的群体基因组索引表示与构建:
[0093]
步骤四一、minimizer索引构建:
[0094]
我们面向“unipath”图结构,以及“alt string”列表分别构建索引。在minimizer索引的构建层面,这两个不同的索引拥有相似的构造方法与构造过程。整体上,minimizer索引构造程序以字符串列表作为输入,以构建好的索引作为输出。
[0095]
在针对“unipath”以及“alt string”构建索引之前,构造程序分别将“unipath”图以及“alt string”列表转换为字符串列表。之后,针对字符串列表中的每一个字符串,依照minimizer选取准则,选取滑窗内哈希值最小的k-mer,作为局部的代表k-mer;这些局部最小k-mer被称为minimizer,将会被存储在一个统一的列表里。
[0096]
选取滑窗内哈希值最小的k-mer的具体过程:minimizer的选取有两个关键参数,分别是windows size以及k-mer长度;假设待构建索引的序列为l,k-mer长度为l_k,则滑窗选取时,将会有(l-l_k 1)个k-mer被选取出来。假设windows size是w(通常选取默认值5),则从编号1~5这5个k-mer中,选取哈希值最小的一个min_k-mer1;之后,从2~6这5个中,选择最小的一个min_k-mer2;之后是3~7并依次类推。相邻的不同minimizer选取窗口选取出来的min_k-mer可能是同一个(例如1~5与2~6的最小值是一样的,都是3),此时每个重复的仅仅记录一次。该k-mer被称为minimizer。
[0097]
所有字符串对应的minimizer列表,将会被合并成为一个完整的列表,该列表会被按照minimizer的哈希值的大小进行排序(通常从小到大)。之后,按照每个minimizer的哈希值的高k位(k为二级索引参数,通常为26)构建miminizer的二级索引。
[0098]
二级索引将minimizer的总索引分割成为b=2
^k
(通常为64m)个小区间,当假设minimizer的总数目为m个的时候,每个小区间平均包含m/b个minimizer(假设m的值为4g个,则每个小区间平均包含64个minimizer)。构建好的minimizer,每个占据96bit的存储空间,其中minimizer的哈希值的后q(q=minimizer_len*2-k,minimizer_len表示minimizer长度,根据minimizer长度的不同,通常为10~30bit)位占据30位存储空间;minimizer在原始“unipath”,或者“alt string”列表上的位置,占据65bit的空间;外加1bit的无用的冗余空间。
[0099]
unipath图模型中的minimizer的存储数据结构:
[0100]
构建的unpath序列的minimizer索引,除了30bit的minimizer存储空间,还有65bit的位置区。位置区是如下定义的:其中32bit用于存储该minimizer所属的unitig的id;32bit存储该minimizer所在unitig的位置,1bit用于存储该minimizer相对于unitig的方向。
[0101]
局部变异序列(alt string)的minimizer的存储数据结构:
[0102]
与unipath图模型中的minimizer的存储数据结构相似,“alt string”的minimizer的每一项,也会存储成为96bit的数据结构,所有的minimizer共同构成数据表。
[0103]
属于“alt string”的minimizer,其坐标以如下方法唯一标注:每个minimizer包含65位的位置区,其中,
[0104]
(a)25bit用于存储该minimizer所属的window block的id(最多支持32m个window block个空间);
[0105]
(b)19bit存储该minimizer所在haplotype的id(每个window block最多包含0.5m个不同的haplotype);
[0106]
(c)22bit用来存储在haplotype上的位置(最多支持长度为4m的haplotype);
[0107]
(d)1bit用于存储该minimizer相对于haplotype上的方向(正向或者逆反方向)。
[0108]
为了充分说明alt string的含义,这俩地对window block等进行补充说明:
[0109]
解释1:window block。
[0110]
为了方便群体参考基因组的压缩,我们将线性参考基因组(即通常所说的人类参考基因组,包含22条常染色体与2条性染色体的碱基数据,由国际机构发布与更新。这是一个与泛基因组相对的概念)按照固定的长度划分成块。每个块的长度不低于待处理的二代测序序列的长度(记为l)的二倍,块之间有长度为l的交叠。划分的规则固定,且我们可以通
过块的id,获取其对应的参考基因组区间,并通过参考基因组坐标(或者区间),确定其所属的块id,这样的块称为windows block,以下简称wb,它有一个固定参数l,我们依赖已知的参数l以及线性参考基因组完成wb系统的划分以及构建,如图2所示。
[0111]
(a)假设单端测序数据的长度为l(个碱基),则对于参考基因组(例如grch38)的每个染色体,进行固定长度的区间划分。每个区间长度为2l,同时,相邻区间之间有l个碱基的交叠。第一个区间的范围是1到2l,第二个区间的范围是l 1到3l,第n个区间的范围是(n-1)l 1到nl,每一个如此的区间被称为窗口块(window block,缩写为wb)。以l=150为例,1号染色体的wb区间分别是[1,300];[151,450];[301,600]
……
(如图2)。
[0112]
解释2:haplotype以及hap_seq。
[0113]
(b)构造面向边合成边测序(即二代测序方法,ngs)方法的泛基因组(此处特指包含了变异数据的基因组)数据结构。假设人群变异数据库(大规模)共有h个样本,并有2h个单体型(即haplotype。人是双倍体生物,有两套染色体,分别来自父亲与母亲。单体型是其中的一套染色体。因此h个样本有2h个单体型)。则在每一个窗口块内,不同的单体型具有其特定的局部碱基序列(长度接近2l,或随着变异的引入而波动,以下简称hap_seq)。2h种单体型共有2h条局部碱基序列。
[0114]
(c)不同的单体型在同一个窗口块内的局部碱基序列,可能一致,也可能不一致。在同一个窗口块内,对于完全一致的hap_seq进行去重,同时删除与线性参考基因组的hap_seq完全一样(的hap_seq),则剩余互异的局部碱基序列,这些局部单体型序列每个都至少包含一个变异,如图3所示。
[0115]
解释3:alt string。
[0116]
alt string是hap_seq中包含了变异的局部区域。它被如下方式定义:在hap_seq中,发生变异的碱基以及其前方p个碱基(一般情况下为28个碱)以及其后方的28个碱基,将被标记为受变异影响碱基。hap_seq中所有的受变异影响碱基共同构成alt string。例如碱基8与10分别发生了snp,则alt string为hap_seq的1到38的子串。例如碱基8与100分别发生了snp,则alt string为hap_seq的1到36(共计36个碱基)以及72到128(共计57个碱基)这两个子串。
[0117]
解释4:minimizer列表。
[0118]
每个alt string有minimizer列表;每个hap_seq有alt string列表;每个wb有hap_seq列表,整个泛基因组结构有多个wb。整个泛基因组结构的所有minimizer按照先wb,后hap_seq的顺序合并称为完整的单一列表。
[0119]
局部变异序列(alt string)的minimizer的去重:
[0120]
当window block的长度为2l(通常值为300bp)时,每个相邻window block之间存在l/2的交叠区域(通常为150bp)。此时,每一个变异都会同时出现在相邻的两个window block内。该重复导致了minimizer数量翻倍。为了节约存储空间,我们仅仅对相互靠近的每个window block中间位置的变异(以及其周围的区域)构建索引,该区域被称为甜区(sweet region),代表了每个window block的从25%*l到75%l的一段区域(通常为window block内的从75bp到225bp的一段区域)。这样,既保证了所有的变异以及周边区域都被索引到,同时也保证了它们仅仅被索引一次。
[0121]
步骤四二、minimizer查找:
[0122]
我们使用哈希方案以及二分查找方案混合的方式对于read的minimizer在索引中的位置进行查找。有赖于二级索引的建立,该方案兼顾了哈希查找的速度,同时避免了空间冗余以及哈希冲突。
[0123]
具体的查找方法:
[0124]
首先,使用minimizer生成算法生成read的minimizer列表;之后针对其中的某个minimizer,获取其哈希值的前k位(k为二级索引参数,通常为26),定位到minimizer索引的一个小区间内,该小区间平均包含m/b个minimizer(通常平均值不超过64)。之后,在该小区间内,使用剩余的q(q=minimizer_len*2-k,通常为10~30bit)位作为key,进行二分查找。
[0125]
有赖于二级索引的建立,对于minimizer的二分查找过程,被局限在一个局部的连续内存区间内。该方法可以极大减少内存页置换错误(pages fault or miss),并减少二分查找次数,从而提升索引速度。
[0126]
找到minimizer在索引中的具体位置(一段连续区间)之后,将通过其65位的位置区,解析出来read的minimizer在“unipath”图或者“alt string”列表中的位置。如果查找的是“unipath”图的minimizer索引,则解析为相应的unitig的id,unitig上的位置(pos)以及minimizer相对于unitig的方向。如果查找的是“alt string”列表的minimizer索引,则解析成为:window block的id,haplotype的id,haplotype上的位置,以及该minimizer相对于haplotype上的方向,即minimizer在wb结构上的坐标。
[0127]
具体实施方式二:
[0128]
本实施方式为一种面向群体基因组索引表示与构建设备,所述设备包括处理器和存储器,应当理解,包括本发明描述的任何包括处理器和存储器的设备,设备还可以包括其他通过信号或指令进行显示、交互、处理、控制等以及其他功能的单元、模块;
[0129]
应当理解,包括本发明描述的任何方法对应的可以被提供为计算机程序产品、软件或计算机化方法,其可以包括其上存储有指令的非暂时性机器可读介质,所述指令可以用于编程计算机系统,或其他电子装置。存储介质可以包括但不限于磁存储介质,光存储介质;磁光存储介质包括:只读存储器rom、随机存取存储器ram、可擦除可编程存储器(例如,eprom和eeprom)以及闪存层;或者适合于存储电子指令的其他类型的介质。
[0130]
所述存储器中存储有至少一条指令,所述至少一条指令由处理器加载并执行以实现所述的一种面向群体基因组索引表示与构建方法。
[0131]
本发明的上述算例仅为详细地说明本发明的计算模型和计算流程,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。