1.本发明涉及基因检测技术领域,具体为一种基于序列比对骨架的基因组变异检测方法。
背景技术:
2.变异检测是许多基因组学研究的基础,它能够检测被测个体和参考基因组之间的核苷酸差异,从而发现基因组变异与重要表型和疾病之间的关联。在生物信息学工具的帮助下,快速发展的高通量测序技术能够以高分辨率检测各种类型的基因组变异,如单核苷酸变异(snvs)、短插入/删除(indels)和结构变异(svs)。特别是,最先进的单分子实时测序(smrt测序)技术能够产生长且高质量的测序数据,优异的长度和碱基质量能够实现个体基因组的高灵敏度和高准确性变异检测。然而,变异检测的计算成本仍然很高,例如,最先进的方法从原始测序数据到变异检测结果通常需要数百个cpu小时,这对大规模的基因组研究带来了巨大的挑战。因此,面对群体基因组计划,精准、快速的变异检测方法亟待开发。
3.近年来,人们提出了许多的变异检测方法,主要包括基于序列比对的变异检测和基于序列组装的变异检测方法。基于比对的方法首先将测序片段与参考基因组进行比对,通过分析它们之间的差异来实现变异检测。对于长度较短变异(即snvs和indels)的检测,从相对较小基因组区域的比对片段中提取snvs和indels的特征,并使用贝叶斯统计、卷积神经网络(cnn)和递归神经网络(rnn)等方法来分析这些特征以检测潜在变异。基于组装的方法首先进行基因组的从头组装,并将产生的共识序列与参考基因组进行比对,然后从比对信息中发现变异。然而,基因组的从头组装非常耗时,并且需要大量的内存支持。
4.因此,变异检测作为许多基因组学研究的基础,需要在保持高准确性、高敏感性的同时,开发更加高效的变异检测算法,以满足大规模基因组研究中的需求。
技术实现要素:
5.本发明的目的是:针对现有技术中变异检测效率低的问题,提出一种基于序列比对骨架的基因组变异检测方法。
6.本发明为了解决上述技术问题采取的技术方案是:
7.一种基于序列比对骨架的基因组变异检测方法,包括以下步骤:
8.步骤一:以参考基因组与基因组测序片段作为输入,采用基于de bruijn图索引结构查询定位算法和动态规划的最优路径查询算法生成比对骨架,并对骨架缝隙进行填充;
9.步骤二:提取骨架缝隙填充过程中出现的碱基错误匹配、插入与删除,并将其作为潜在的变异位点信息,将潜在的变异位点信息以sindel格式输出到外部存储,之后,通过调用内部脚本,将sindel文件中所有潜在的变异位点信息按照参考位置的顺序进行排序;
10.步骤三:基于已排序的sindel文件,并提取sindel中近似匹配块的比对片段和参考基因组之间的差异,并通过该差异识别候选的snv和indel位点;
11.步骤四:利用候选的snv和indel位点进行snv和indel检测,并对检测结果进行基
因型判定。
12.进一步的,所述步骤一中生成比对骨架的具体步骤为:
13.采用基于de bruijn图的参考基因组索引查询测序片段与参考基因组上的完全匹配块,并延伸至下一个碱基错误匹配,生成非固定长度的近似匹配块,以所有的近似匹配块为顶点,顶点之间在测序片段与参考序列上是共线性关系的存在一条边,以此构建有向无环图;
14.然后基于有向无环图,并使用稀疏动态规划算法构造3~5条可能的比对骨架并计算骨架中每个顶点的得分及前驱顶点,选择得分最大的顶点作为终止顶点,根据计算得到的前驱顶点依次进行回溯,直到达到起始顶点,将这个回溯过程中得到路径作为一条骨架输出;然后依次选取次优得分的顶点来进行回溯,构造路径,直到所有路径都被构建,完成原始骨架的识别;
15.得分最大的顶点输出的骨架表示为:其中k代表骨架的优先级,k值越小优先级越高,tk表示骨架中包含的顶点数量;
16.根据得到的骨架,以顶点的得分作为优先级从高到低依次选择待连接骨架,针对待连接骨架判断当前最高优先级的骨架与其他骨架之间的连接关系,当满足连接条件时进行连接,并更新待连接骨架,具体为:当两个骨架比对到同一染色体且满足下式时,则骨架为待连接骨架的拆分骨架,将二者进行连接,
[0017][0018]
其中,rs、re、gs、ge分别代表测序序列和参考序列的起始和结束位置,tgap代表连接骨架之间最大允许gap长度,abs代表取绝对值;
[0019]
当时,代表pi在待连接骨架p0的左侧,更新p0为:
[0020][0021]
当时,代表pi在待连接骨架p0的右侧,更新p0为:
[0022][0023]
直至所有骨架都被连接,得到多个连接骨架。
[0024]
进一步的,所述步骤一中生成比对骨架的步骤还包括:
[0025]
对连接骨架进行过滤的步骤,若连接骨架中包含碱基的总长度小于100bp,则将其过滤。
[0026]
进一步的,所述对骨架缝隙进行填充的具体步骤为:
[0027]
针对连接骨架,对骨架中任意两个相邻顶点之间的空白区域进行填充,具体步骤为:
[0028]
步骤一一:以顶点的右端点和的左端点为起点,采用landau-vishkin算法分
别向右和向左延伸,延伸时最大允许编辑错误为3,使用延伸到的右端终点更新顶点使用延伸到的左端终点更新顶点
[0029]
步骤一二:以更新后的顶点的右端点为起点,的左端点为终点,取该起点和终点的中间区域,并使用smith-waterman算法进行端到端的比对,完成空白区域的填充;
[0030]
步骤一三:对骨架中最左侧顶点与最右侧顶点的未比对区域,使用smith-waterman进行固定一端的延伸,当存在5个连续的碱基无法正确匹配时停止延伸;
[0031]
当所有空白区域被填充或延伸完毕后,提取过程中出现的碱基错误匹配、插入与删除,并将其作为潜在的变异位点信息,之后将潜在的变异位点信息以sindel格式输出到外部存储,最后,通过调用内部脚本,将sindel文件中所有潜在的变异位点信息按照参考位置的顺序进行排序。
[0032]
进一步的,所述识别候选的snv和indel位点的具体步骤为:
[0033]
首先将参考基因组进行窗口划分,得到多个窗口,每一个窗口为一条染色体上的一段区域,区域长度为20000000bp,同时保证每两段区域之间都没有交叠;
[0034]
针对每一个窗口,遍历参考基因组,并计算每个基因组位点上各种非参考碱基的频率;
[0035]
如果至少有一种非参考碱基的频率高于阈值时,则将其作为候选位点,并将候选位点分类为高置信度或低置信度位点。
[0036]
进一步的,所述每个基因组位点上各种非参考碱基的频率表示为:
[0037]
p
cv
=n
ab
/n
ts
[0038]
其中,n
ab
表示sindel文件中携带此种非参考碱基的信号的数量,n
ts
代表sindel文件中跨越此位置的所有信号数量。
[0039]
进一步的,所述计算每个基因组位点上各种非参考碱基的频率后还包括:
[0040]
计算所有非参考碱基的平均碱基质量,若质量低于20,则将其过滤。
[0041]
进一步的,所述将候选位点分类为高置信度或低置信度位点的具体步骤为:
[0042]
当一个候选位点不存在多个非参考碱基,并且满足下式时,则被认为是高置信度的位点:
[0043][0044]
其中,n
ts
代表sindel文件中跨越此位置的所有信号数量,p
cv
代表非参考碱基的频率,t
ts
、t
hc
为用户指定的参数;
[0045]
否则,将该位点判定为低置信度的位点。
[0046]
进一步的,所述步骤四的具体步骤为:
[0047]
对于高置信度的候选位点,统计支持参考碱基与非参考碱基的测序片段数目,并通过二项式模型估计各种基因型的概率:
[0048][0049]
其中,ε表示碱基被错误地估计的概率,sr
ref
表示携带参考碱基的信号的数量,sr
alt
表示携带非参考碱基的信号的数量,l(0/0)表示没有变异,l(0/1)代表杂合变异,l(1/1)代表纯合变异;
[0050]
对于低置信度的候选位点:
[0051]
步骤四一:设置一个长度为20bp的窗口,在参考基因组上滑动,当窗口内包含至少两个低置信度候选位点时,将其合并成一个低置信度区域,并继续滑动窗口,使其与上一窗口没有交叠,当窗口内存在其他低置信度候选位点时继续合并,直至窗口内不再包含低置信度候选位点,然后将每个低置信度区域上下游50bp的区域加入到对应的低置信度区域中,得到候选区域;
[0052]
步骤四二:提取所有覆盖候选区域的比对片段,然后使用基于simd的偏序比对算法abpoa对覆盖候选区域的比对片段进行多序列比对,生成至少一个比对片段的共识序列;
[0053]
步骤四三:调用比对算法ksw2将生成的共识序列与候选区域的参考序列进行比对,并从具体的比对信息中识别出非参考碱基,并记录支持参考和非参考碱基的比对片段数量;
[0054]
判断比对片段的碱基质量能都否使用,当比对片段的碱基质量不能使用时,使用与高置信度候选位点相同的二项式模型计算基因型;否则,将所有比对片段与步骤四二中的共识序列进行比对,得到比对分数,并使用贝叶斯模型计算各种基因型的概率。
[0055]
进一步的,所述步骤四二中生成至少一个比对片段的共识序列的具体步骤为:
[0056]
当候选区域内存在杂合变异时,产生两条共识序列,一条包含基因组变异,另一条不包含基因组变异;当候选区域不存在变异或存在纯合变异时,会产生一条共识序列。
[0057]
本发明的有益效果是:
[0058]
本技术提出的基于比对骨架和共识序列重构的变异检测方法,通过稀疏动态规划构建测序片段比对骨架,采用landau-vishkin和smith-waterman算法对骨架gap区域进行填充;据不同候选变异位点的序列和分布特征将其划分为高置信度候选变异位点和低置信度候选变异位点,并分别使用两种不同的策略进行变异检测和基因型推断。
[0059]
1)序列比对阶段,本技术较当前主流比对方法比对效率提升7倍以上;
[0060]
2)变异检测阶段,本技术较当前主流变异检测方法检测效率提升20-70倍。
附图说明
[0061]
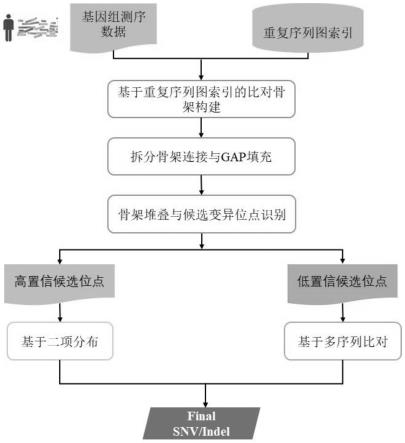
图1为本技术的整体流程图。
具体实施方式
[0062]
需要特别说明的是,在不冲突的情况下,本技术公开的各个实施方式之间可以相互组合。
[0063]
具体实施方式一:参照图1具体说明本实施方式,本实施方式所述的一种基于序列比对骨架的基因组变异检测方法,包括以下步骤:
[0064]
步骤一:以参考基因组与基因组测序片段作为输入,采用基于de bruijn图索引结构查询定位算法和动态规划的最优路径查询算法生成比对骨架,并对骨架缝隙进行填充;
[0065]
步骤二:提取骨架缝隙填充过程中出现的碱基错误匹配、插入与删除,并将其作为潜在的变异位点信息,将潜在的变异位点信息以sindel格式输出到外部存储,之后,通过调用内部脚本,将sindel文件中所有潜在的变异位点信息按照参考位置的顺序进行排序;
[0066]
步骤三:基于已排序的sindel文件,并提取sindel中近似匹配块的比对片段和参考基因组之间的差异,并通过该差异识别候选的snv和indel位点;
[0067]
步骤四:利用候选的snv和indel位点进行snv和indel检测,并对检测结果进行基因型判定。
[0068]
下面进行详细的说明:
[0069]
比对骨架的生成
[0070]
在比对骨架的生成中,以参考基因组与基因组测序片段作为输入,采用基于de bruijn图索引结构查询定位和动态规划的最优路径查询算法来生成比对骨架,并将骨架信息写入外部存储以便后续的使用。
[0071]
1.1骨架构建
[0072]
首先,本方法采用基于de bruijn图的参考基因组索引快速查询测序片段与参考基因组上的完全匹配块(exact match),并延伸至下一个碱基错配,生成非固定长度的近似匹配(anchor)。以所有的anchor为顶点,顶点之间在测序片段与参考序列上是共线性关系的存在一条边,构建有向无环图。
[0073]
然后根据顶点的位置关系,首先构造3~5条可能的比对骨架,然后使用稀疏动态规划算法来计算每个顶点的得分及其前驱节点(节点即顶点),选择得分最大的节点作为终止节点,根据计算得到的前驱节点信息依次进行回溯,直到达到起点,将这个回溯过程中得到路径作为一条骨架输出;然后依次选取次优得分的节点来进行回溯,构造路径,直到所有路径都被构建,完成原始骨架的识别,以此来表示序列比对的信息。
[0074]
算法中对节点按照其拓扑顺序进行计算,因此在计算一个节点时,其所有前驱节点一定已被计算过,能够有效减小稀疏动态规划过程中的计算量。设在稀疏动态规划中共构建了k条比对骨架,最优比对骨架可以表示为其中k代表骨架的优先级,k值越小优先级越高,tk表示骨架中包含的顶点数量。
[0075]
1.2拆分骨架连接
[0076]
完整的测序序列比对骨架会因为边的连接约束而被拆分,形成多段骨架,因此需要将拆分骨架进行连接,从而得到完整的比对骨架,然后通过填充骨架中的gap区域实现变异的识别。
[0077]
在拆分骨架链接中,首先根据优先级从高到低依次选择待连接骨架,判断当前最高优先级的骨架与其他骨架之间的连接关系,当满足连接条件时进行连接,并更新待连接骨架。具体来说,首先选择待连接骨架遍历其他骨架遍历其他骨架当两个骨架比对到同一染色体且满足下式时,代表pi是待连接骨
架p0的拆分骨架,进行连接,
[0078][0079]
其中rs,re,gs,ge分别代表测序序列和参考序列的起始和结束位置,tgap代表连接骨架之间最大允许gap长度。当时,代表pi在待连接骨架p0的左侧,更新p0为:
[0080][0081]
相反的,当时,代表pi在待连接骨架p0的右侧,更新p0为:
[0082][0083]
按照这个流程对骨架pi进行连接,当所有骨架都被连接完毕后,记连接后的骨架为p0,集合t={i|pi∈p0}为构成p0的所有拆分骨架的优先级列表,并根据优先级依次选取目标骨架,直至没有满足上式的可合并骨架。在完成连接后对假阳性骨架进行过滤,若骨架pi中包含碱基的总长度小于100bp,则将其过滤。
[0084]
1.3骨架缝隙填充
[0085]
由于基因组上可能存在的变异,以及在测序中存在不可避免的系统性测序错误,连接后的比对骨架中依然会存在一些无法匹配的空白区域(gap),需要使用更精确的方法对gap区域进行填充。进行碱基级别的空白区域填充通常使用smith-waterman算法以及landau-vishkin算法,考虑二者的优缺点,smith-waterman算法有着较高的时间复杂度,landau-vishkin算法对于单碱基错误率较低的数据有着较低的时间复杂度,但它无法识别长度大于最大允许编辑错误的indels。因此,为了同时考虑检测的准确性与效率性能,本研究采用smith-waterman与landau-vishkin相结合的算法进行空白区域的填充。
[0086]
对1.2中已连接的比对骨架对骨架中任意两个相邻节点对骨架中任意两个相邻节点之间的空白区域进行填充,具体步骤为:
[0087]
(1)分别以节点的右端点和的左端点为起点,采用landau-vishkin算法分别向右和向左延伸,延伸时最大允许编辑错误默认取为3,使用延伸到的终点更新节点
[0088]
(2)以更新后的节点的右端点为起点,的左端点为终点,取这一段局部区域使用smith-waterman算法进行端到端的比对,完成空白区域的填充。
[0089]
对于节点左侧与右侧的未比对区域,同样使用上述方法,但将其中smith-waterman进行的端到端的比对改为固定一端的比对,当存在5个连续的碱基无法正确匹配时停止延伸。
[0090]
当所有空白区域被填充或延伸完毕后,提取过程中出现的碱基错误匹配、插入与删除,作为潜在的变异位点信息,并将这些信息以sindel格式输出到外部存储。最后,通过
调用内部脚本,将sindel文件中的所有信号分别按照参考位置的顺序进行排序,为下文的变异检测提供基础。
[0091]
snv/indel检测
[0092]
snv与indel候选区域识别
[0093]
snv与indel候选区域识别中使用已排序的sindel格式文件作为输入,推断出高度可能的snv和indel候选位点。所有近似匹配中的非参考等位基因以及它们的位置都被记录在了sindel文件中,因此该文件可以被看作比对骨架中包含的snv/indel信号的堆积,通过收集近似匹配的比对片段部分和参考基因组之间的差异,以识别候选的snv和indel位点。具体来说,按照以下三步进行候选变异区域的识别:
[0094]
(1)无交叠窗口划分
[0095]
为了提高算法的运行效率,首先将全基因组按照一定大小的区域进行划分,得到多个窗口,每一个窗口为一条染色体上的一段区域,其长度默认为20,000,000bp,同时保证每两段区域之间都没有交叠。对于每一个窗口,提取输入sindel文件中所有落在该窗口内的信号信息,进行变异信号的检测。
[0096]
(2)变异信号检测
[0097]
遍历参考基因组,并计算每个基因组位点上各种非参考等位基因的频率,如下所示:
[0098]
p
cv
=n
ab
/n
ts
[0099]
其中n
ab
表示sindel文件中携带此种非参考等位基因的信号的数量,n
ts
代表sindel文件中跨越此位置的所有信号数量。如果至少有一种碱基的频率高于阈值,则该位置被识别为候选变异位点。当存在多个频率高于阈值的非参考等位基因时,记录所有的这些非参考等位基因作为变异信息。
[0100]
为了减少由于测序错误导致的假阳性候选区域,需要进行进一步的过滤,计算所有非参考等位基因的平均碱基质量,若质量低于20,则认为当前位置测序质量过低,将其过滤。
[0101]
(3)候选变异位点分类
[0102]
被识别的候选位点被进一步分类为高置信度或低置信度位点。当一个候选位点不存在多个非参考等位基因,并且满足下式,则被认为是高置信度的位点:
[0103][0104]
其中,n
ts
代表sindel文件中跨越此位置的所有信号数量,p
cv
代表非参考等位基因的频率,t
ts
、t
hc
为用户指定的参数。否则,将该位点判定为低置信度的位点。
[0105]
snv与indel检测与基因型判定
[0106]
在进行snv与indel检测时,对高置信度与低置信度位点采取不同的策略:对于一个高置信度的候选位点,因为它是一个单等位基因位点,因此将其非参考等位基因作为变异等位基因,统计支持参考基因组与变异序列的测序片段数目,并通过二项式模型估计各种基因型的概率:
[0107][0108]
其中,ε表示等位基因被错误地估计的概率,sr
ref
表示携带参考等位基因的信号的数量,sr
alt
表示携带非参考等位基因的信号的数量。
[0109]
对于低置信度的候选位点,它包含多个等位基因,因此使用以下四个步骤实现对它的检测:
[0110]
首先,设置一个长度默认为20bp的窗口,在参考基因组上滑动,当窗口内包含至少两个低置信度候选位点时,将其合并以形成一个低置信度区域,并继续滑动窗口,使其与上一窗口没有交叠,当窗口内存在其他低置信度候选位点时继续合并,直至窗口内不再包含低置信度候选位点,然后将整个低置信度区域上下游50bp的区域加入到候选区域来进行进一步的扩展。
[0111]
根据候选区域的坐标,提取所有跨越候选区域的比对片段。然后使用一种基于simd的偏序比对算法abpoa对局部片段进行多序列比对,来快速地生成一个或多个比对片段的共识序列,一般来说,当候选区域内存在杂合变异时,会产生两条共识序列,一条包含基因组变异,另一条不包含基因组变异;反之,当候选区域不存在变异或存在纯合变异时,会产生一条共识序列。
[0112]
调用比对算法ksw2将生成的共识序列与候选区域的参考序列进行比对,并从具体的比对信息中识别出非参考等位基因。此外,分别记录支持参考和非参考等位基因的比对片段数量,以便后续对基因型的计算。
[0113]
对于不同情况使用不同的方法来计算基因型。当比对片段的碱基质量不能使用时,就使用与高置信度候选位点相同的二项式模型;否则,对所有比对片段、参考等位基因和非参考等位基因进行比对,计算出各种等位基因的概率,然后使用贝叶斯模型来计算各种基因型的概率。
[0114]
需要注意的是,具体实施方式仅仅是对本发明技术方案的解释和说明,不能以此限定权利保护范围。凡根据本发明权利要求书和说明书所做的仅仅是局部改变的,仍应落入本发明的保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。