1.本发明属于医药技术领域,具体涉及一种立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法。
背景技术:
2.立他司特(lifitegrast) 是由sar code bioscience设计开发的用于治疗干眼病的药物,其是一种新的细胞间黏附因子的抑制剂,可以通过阻断细胞间黏附分子和整合素蛋白淋巴细胞功能相关抗原之间的结合起效。
3.2016年7月,美国fda正式批准了5%立他司特滴眼剂(商品名xiidratm)的申请。立他司特是fda批准的第一个可以改善和治疗干眼症症状的新药,在不久的将来其临床应用会更广泛。5,7-二氯-1,2,3,4-四氢异喹啉作为立他司特的一种重要中间体,该中间体的检测分析尤为重要,其结构式如下:高效液相色谱(hplc)于20世纪70年代获得迅猛发展,成为一种常规高效分离分析技术,约有80%的化合物可以用不同模式的hplc进行分离分析。
4.根据《中国药典》记载,分离度(简称为r)也叫分辨率,分离度表示相邻两峰的分离程度,r越大,表明相邻两组分分离越好。一般说当r<1时,两峰有部分重叠;当r=1.0时,分离度可达98%;当r=1.5时,分离度可达99.7%。通常用r=1.5作为相邻两组分已完全分离的标志。
5.中国专利cn 111060629 a公开一种利非司特有关物质的检测方法,具体步骤如下:a、取利非司特,加乙腈水溶液溶解,得供试品溶液;b、取利非司特对照品和杂质对照品,加乙腈水溶液溶解,得分离度溶液;c、采用高效液相色谱法分别对分离度溶液,供试品溶液、对照溶液进行检测。
6.中国专利cn 114689733 a公开一种立他司特或其制剂有关物质检测方法,采用高效液相色谱法以十八烷基硅烷键合硅胶为填充剂;以磷酸、磷酸水溶液、有机溶剂,或上述混合液为流动相,梯度洗脱;柱温20-60℃;检测波长200-240nm;流速0.5-2.0ml/min。
7.上述专利仅是对立他司特中有关物质进行检测,而对于中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法至今未有报道。
8.目前,亟需研制开发一种立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法,对产品的生产和质量控制具有重要的意义。
技术实现要素:
9.本发明的目的是提供一种立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法,具有专属性好、灵敏度高、耐用性好、操作简单的特点。
10.本发明所述的立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法是采用高效液相色谱法对立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉进行含量测定,其中,色谱条件包括流动相,流动相是以质量浓度为0.05%的磷酸水溶液为流动相a,以乙腈为流动相b。
11.所述的色谱条件还包括洗脱方式,洗脱方式为梯度洗脱,梯度洗脱的洗脱程序如下:。
12.所述的色谱条件还包括色谱柱,色谱柱为c18,优选为ymc-pack ods-aq 4.6*250mm,s-3μm,12nm。
13.所述的色谱条件还包括检测器,检测器为紫外检测器,优选为thermo scientific 紫外检测器;检测器的波长为210-230nm,优选为215nm。
14.所述的色谱条件还包括柱温,柱温为25-30℃,优选为25℃。
15.所述的色谱条件还包括流速,流速为0.8-1.5ml/min,优选为1.0ml/min。
16.所述的色谱条件还包括进样体积,进样体积为10-30μl,优选为20μl。
17.本发明所述的立他司特中间体5,7-二氯-1,2,3,4-四氢异喹啉的检测方法是配制供试品溶液和标准溶液,通过标准溶液绘制标准曲线,将空白溶液和供试品溶液注入高效液相色谱仪中,记录色谱图,采用外标法计算得到供试品溶液中5,7-二氯-1,2,3,4-四氢异喹啉的含量。
18.所述的空白溶液为50%乙腈水溶液。
19.所述的供试品溶液的制备方法是将5,7-二氯-1,2,3,4-四氢异喹啉样品用空白溶液进行溶解定容,配制成浓度为0.2-0.25mg/ml的供试品溶液。
20.所述的标准溶液的制备方法是称取5,7-二氯-1,2,3,4-四氢异喹啉标准品200mg,置于100ml量瓶中,用空白溶液溶解并稀释定容至刻度作为标准溶液。
21.所述的标准曲线的绘制方法是将标准溶液逐级稀释制成浓度分别为0.04、0.05、0.07、0.1、0.2、0.25mg/ml的5,7-二氯-1,2,3,4-四氢异喹啉溶液,将不同浓度的5,7-二氯-1,2,3,4-四氢异喹啉溶液分别注入高效液相色谱仪中,分别计算同一保留时间下的峰面积,以5,7-二氯-1,2,3,4-四氢异喹啉溶液的不同浓度x作为横坐标,以其峰面积y为纵坐标,绘制出标准曲线。
22.本发明的有益效果如下:本发明具有专属性好、灵敏度高、耐用性好的特点。本发明使用的梯度洗脱程序可以改善峰形,提高柱效应,减少分离时间,使强保留成分不易残留在柱上,保持了柱子的良好性能;同时采用乙腈-磷酸水溶液作为流动相,其中,磷酸一方面可以通过调节酸碱性来修饰峰型,另一方面可以作为离子抑制剂利于分离物质。本发明非常适合对立他司特中间
体5,7-二氯-1,2,3,4-四氢异喹啉的纯度检测进行质量控制。
附图说明
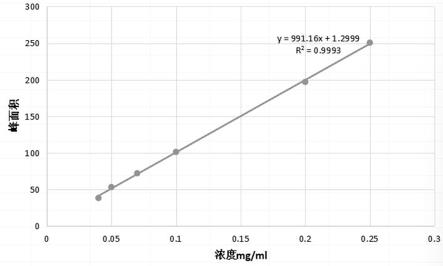
23.图1是实施例1中的标准曲线图。
24.图2是实施例1中的空白溶液色谱图。
25.图3是实施例1中的供试品溶液色谱图。
26.图4是方法1中的供试品溶液色谱图。
27.图5是方法2中的供试品溶液色谱图。
28.图6是方法3中的供试品溶液色谱图。
29.图7是方法4中的供试品溶液色谱图。
30.图8是方法5中的供试品溶液色谱图。
31.图9是方法6中的供试品溶液色谱图。
具体实施方式
32.以下结合实施例对本发明做进一步描述。
33.实施例1(1)空白溶液:50%乙腈水溶液。
34.(2)供试品溶液:5,7-二氯-1,2,3,4-四氢异喹啉样品用空白溶液进行溶解定容,配制成浓度为0.22mg/ml供试品溶液。
35.(3)标准溶液:精密称取5,7-二氯-1,2,3,4-四氢异喹啉标准品200mg,置于100ml量瓶中,用空白溶液溶解并稀释定容至刻度作为标准溶液。
36.(4)标准曲线的绘制:将标准溶液逐级稀释制成浓度分别为0.04、0.05、0.07、0.1、0.2、0.25mg/ml的5,7-二氯-1,2,3,4-四氢异喹啉溶液,将不同浓度的5,7-二氯-1,2,3,4-四氢异喹啉溶液分别注入高效液相色谱仪中,分别计算同一保留时间下的峰面积。以5,7-二氯-1,2,3,4-四氢异喹啉溶液的不同浓度x为横坐标,以其峰面积y为纵坐标,做出标准曲线方程(即关系函数) y=991.16x 1.2999,r2=0.9993,如图1所示,结果表明医药中间体5,7-二氯-1,2,3,4-四氢异喹啉在0.04-0.25mg/ml范围内,峰面积和浓度成以上线性关系。
37.(5)将空白溶液和供试品溶液注入高效液相色谱仪中,记录色谱图,空白溶液色谱结果见图2,供试品溶液色谱结果见图3,采用外标法计算得到供试品溶液中5,7-二氯-1,2,3,4-四氢异喹啉的含量为0.216mg/ml,按照面积归一化法进行积分得到供试品的纯度为99.41%,单杂小于1.0%。
38.色谱条件如下:色谱柱:ymc-pack ods-aq 4.6*250mm,s-3μm,12nm检测器:紫外检测器流动相:a:质量浓度为0.05%的磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm柱温:25℃流速:1.0ml/min进样体积:20μl
洗脱方式:梯度洗脱梯度洗脱的洗脱程序见表1。
39.表1 实施例1的梯度洗脱的洗脱程序实施例2供试品溶液:5,7-二氯-1,2,3,4-四氢异喹啉样品用空白溶液进行溶解定容,配制成浓度为0.2mg/ml供试品溶液。
40.色谱条件如下:色谱柱:ymc-pack ods-aq 4.6*250mm,s-3μm,12nm检测器:紫外检测器流动相:a:质量浓度为0.05%的磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为210nm柱温:28℃流速:0.8ml/min进样体积:30μl洗脱方式:梯度洗脱其它步骤同实施例1。
41.经计算得到供试品溶液中5,7-二氯-1,2,3,4-四氢异喹啉的含量为0.197mg/ml,供试品的纯度为99.42%,单杂小于1.0%。
42.实施例3供试品溶液:5,7-二氯-1,2,3,4-四氢异喹啉样品用空白溶液进行溶解定容,配制成浓度为0.25mg/ml供试品溶液。
43.色谱条件如下:色谱柱:ymc-pack ods-aq 4.6*250mm,s-3μm,12nm检测器:紫外检测器流动相:a:质量浓度为0.05%的磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为230nm柱温:30℃流速:1.5ml/min进样体积:10μl洗脱方式:梯度洗脱其它步骤同实施例1。
44.经计算得到供试品溶液中5,7-二氯-1,2,3,4-四氢异喹啉的含量为0.246mg/ml,供试品的纯度为99.40%,单杂小于1.0%。
45.方法验证如下:精密度考察:取实施例1中的0.2mg/ml供试品溶液6份进行连续6次进样,精密度检测结果见表2。
46.表2 精密度检测结果6次进样的峰面积相对标准偏差(rsd)为0.19%,结果表明,本发明hplc法检测5,7-二氯-1,2,3,4-四氢异喹啉样品的精密度高、重现性好。
47.本发明的研究过程如下:除色谱条件外,其它的5,7-二氯-1,2,3,4-四氢异喹啉的检测步骤同实施例1。
48.对5,7-二氯-1,2,3,4-四氢异喹啉的检测条件进行筛选。
49.首先筛选流动相体系,对比甲醇与乙腈,乙腈的洗脱能力较强,并且乙腈的黏度较甲醇偏小,所以选用乙腈作为流动相的有机体系。为了保持色谱峰峰型的良好以及相对保留时间的稳定,所以选择在流动相的水相体系中加入一定的缓冲试剂,故选用0.05%的磷酸水溶液作为流动相体系。采用0.05%的磷酸水溶液和乙腈作为流动相进行试验,分别尝试等度洗脱以及不同的梯度洗脱方式进行洗脱试验。
50.(1)方法1:现初步使用以下色谱条件进行分析试验。
51.色谱柱:eclipse pluse 4.6*50mm,s-3.5μm,12nm检测器:紫外检测器流动相:0.05%磷酸水溶液:乙腈(体积比)=9:1检测器的波长为215nm柱温:25℃流速:1.0ml/min进样体积:20μl洗脱方式:等度洗脱运行时间:30min该方法检测得到的谱图见图4。
52.从图4中可知,通过面积归一化法所得供试品纯度仅有66.66%,整体的基线不是特别稳定,主峰的峰型不好。
53.(2)方法2:等度洗脱的方法不能满足5,7-二氯-1,2,3,4-四氢异喹啉产品的检测,为了得到5,7-二氯-1,2,3,4-四氢异喹啉产品更好的分离度,决定采用梯度洗脱的方式。
54.色谱柱:eclipse pluse 4.6*50mm,s-3.5μm,12nm检测器:紫外检测器流动相:a:0.05%磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm
柱温:25℃流速:1.0ml/min进样体积:20μl洗脱方式:梯度洗脱,梯度洗脱的洗脱程序见表3。
55.表3 方法2的梯度洗脱的洗脱程序该方法检测得到的谱图见图5。
56.结果分析:从图5可知,通过面积归一化法所得供试品纯度为98.00%,出现主峰杂质与主峰的分离度不好,基线不够稳定,仍需继续改善。
57.(5)方法3:为了考察温度对色谱峰的影响进行了柱温的研究,通过适当提高柱温,看是否影响色谱峰的分离度等因素。主要色谱条件参数如下:色谱柱:eclipse pluse 4.6*50mm,s-3.5μm,12nm检测器:紫外检测器流动相:a:0.05%磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm柱温:35℃流速:1.0ml/min进样体积:20μl洗脱方式:梯度洗脱,梯度洗脱的洗脱程序同方法2的表3。
58.该方法检测得到的谱图见图6。
59.结果分析:从图6与图5进行比较可以看出,图6中通过面积归一化法所得供试品纯度为94.60%,基线稳定性变差,主峰及其周围杂质峰的分离度有所改善,所以提高柱温对色谱条件的优化没有更加明显的优势。
60.(3)方法4:为了得到更好的检测数据进行了色谱柱的更换,来寻求方法的最优化。
61.色谱柱:vp-ods 4.6*150mm,s-5μm,12nm检测器:紫外检测器流动相:a:0.05%磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm柱温:25℃流速:1.0ml/min进样体积:20μl洗脱方式:梯度洗脱,梯度洗脱的洗脱程序同方法2的表3。
62.该方法检测得到的谱图见图7。
63.结果分析:从图7可知,通过面积归一化法所得供试品纯度为85.97%,可以看出主峰周围存在杂质峰的包裹,主峰杂质与主峰的分离度并未得到改善,基线仍旧不稳定。
64.(5)方法5:更换另外一种色谱柱,其色谱条件如下:色谱柱:ymc-pack ods-aq 4.6*250mm,s-3μm,12nm检测器:紫外检测器流动相:a:0.05%磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm柱温:25℃流速:1.0ml/min进样体积:20μl洗脱方式:梯度洗脱,梯度洗脱的洗脱程序同方法2的表3。
65.该方法检测得到的谱图见图8。
66.结果分析:从图8可知,通过面积归一化法所得供试品纯度为99.24%,可以看出更换色谱柱后,发现该主峰与杂质已得到分开,但看的出整体的基线不是特别稳定,仍需继续改善。
67.(6)方法6:为了得到更加优化的色谱条件,在上述条件的基础之上进行略微的调整,更改流动相的梯度洗脱情况,保持原本具有优势的色谱条件。液相色谱条件如下:色谱柱:ymc-pack ds-aq 4.6*250mm,s-3μm,12nm检测器:紫外检测器流动相:a:0.05%磷酸水溶液
ꢀꢀꢀ
b:乙腈检测器的波长为215nm柱温:25℃流速:1.0ml/min进样体积:20μl洗脱方式:梯度洗脱梯度洗脱的洗脱程序见表4。
68.表4 方法6的梯度洗脱的洗脱程序该方法检测得到的谱图见图9。
69.结果分析:从图9可知,通过面积归一化法所得供试品纯度为99.42%,该色谱条件的分离度、专属性及重现性等均达到要求。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。