1.本发明属于医药技术领域,涉及通过对天然产物hoec进行改构获得的抗炎化合物。
背景技术:
2.炎症性肠病(inflammatory bowel disease,ibd)是一种慢性非特异性的肠道炎症性疾病,有溃疡性结肠炎(ulcerative colitis,uc)和克罗恩病(crohn's disease,cd)两种临床亚型。ibd已被世界卫生组织列为难治性疾病之一。近年来,随着居民生活方式及饮食习惯的改变,ibd发病率呈上升趋势,成为继结直肠癌后危害肠道健康的又一重大威胁。ibd的主要临床表现为腹痛、腹泻等典型消化道症状,部分患者还可出现泌尿、呼吸及皮肤等消化系统外症状,病情加重时可出现发热、消瘦等全身体征。ibd具有易反复发作、迁延不愈,及病程较长的特点。多项临床调查结果显示,超过50%的uc患者为慢性复发型,这类患者诊断后1年、5年、10年的复发率分别高达30.2%、72.0%和88.4%。当病情迁延时,ibd患者可出现肠道穿孔、梗阻、腹腔脓肿,甚至是中毒性巨结肠、直结肠癌变等并发症,严重影响患者的生活质量,对患者造成极大的精神痛苦。
3.现代医学证实,在ibd条件下炎症的发生与花生四烯酸(arachidonic acid,aa)有着密切的关系。5-脂氧合酶(5-lipoxygenase,5-lox)是催化花生四烯酸(arachidonic acid,aa)代谢成白三烯(leukotrienes,lts)的关键酶。这些白三烯类物质是体内一类重要的炎症介质,参与多种炎症性疾病的病理过程。而且这些白三烯中的ltb4将白细胞吸引到炎症部位,促进它们与炎症和受损组织的粘附,从而扩增ibd中的炎症级联反应。
4.目前,5-lox抑制剂分为还原型抑制剂和非还原型抑制剂两类。还原型抑制剂如去甲二氢愈创木酸(ndga)、齐留通(zileuton)等,主要作用于5-lox活化部位的铁离子,把 3价活性状态的铁离子还原为 2价的非活性状态,干扰了铁离子催化循环,阻止5-lox的激活。体内外实验均证实其具有高效的5-lox抑制作用。还原型抑制剂属于非选择性抗氧化物,由于容易干扰其他活性自由基的合成和生物氧化还原系统,具有严重的副作用,从而限制了它们进一步在临床的研究和应用。
5.由于还原型抑制剂的多种毒性促使人们致力于寻找非还原型抑制剂。非还原型抑制剂是一类高选择性的5-lox抑制剂,与底物aa竞争结合5-lox。非还原型抑制剂中,甲氧基四氢吡喃衍生物zm230487是一种高效的lta4合成抑制剂,能明显降低急性炎性反应,但对于慢性炎症过程起效甚微;甲氧基四氢吡喃类与木脂素类的杂合体l-697198以及l-708780和l-739010在一系列动物实验中表现出极佳的生物利用度和半衰期,但后续的研究发现这些化合物的呋喃部分经代谢活化并作用于血浆和肝脏蛋白,呈现出多种毒性。
6.尽管上述这些5-lox抑制剂在对与5-lox酶有关疾病(包括ibd)的研究和实际防治中发挥着重要的作用,但它们却存在选择性较差(不是特异性的作用某一靶标)、毒副作用明显等问题,临床应用受到很大限制。因此亟需开发新的抗炎药物活性成分,以延长ibd缓解期时间,减少毒副作用,提高ibd患者生活质量。
7.天然产物由于其结构的多样性,已成为发现结构新颖、生物活性独特、具有特异性作用靶点的药物活性成分的重要来源。目前上市销售的药物小分子新化学实体中有61%可追溯到天然产物,例如青蒿素、吗啡、麻黄碱、东莨菪碱、利血平、青霉素、喜树碱、紫杉醇等。有研究报道(张杰.咖啡酸衍生物的合成及其抗炎活性研究.华东理工大学,2014.)了咖啡酸类天然化合物hoec的作用靶标为5-lox,其类似物hoeca(成药性提高)对ltb4的生成具有明显抑制活性。该研究中还获得了四种对由lps诱导的大鼠巨噬细胞raw264.7细胞no生成具有很强的抑制作用的咖啡酸酯衍生物,但并未能准确表明其抗炎活性优于hoec和hoeca这两个母体化合物。
技术实现要素:
8.本发明的目的在于提供一种抗炎化合物及其制备方法和用途,该抗炎化合物可用于治疗炎症性肠病等炎症性疾病。
9.为达到上述目的,本发明采用了以下技术方案:
10.一种抗炎化合物,该抗炎化合物为结构如式i所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0011][0012]
其中,r1、r2、r3分别为h、甲氧基、乙酰氧基、羟基、亚甲二氧基团中的任意一种,r4为h、羟基、卤素中的任意一种。
[0013]
优选的,所述抗炎化合物(式i)中,r3为h或甲氧基。
[0014]
优选的,所述抗炎化合物(式i)中,r4为h或为位于对位的羟基或卤素。
[0015]
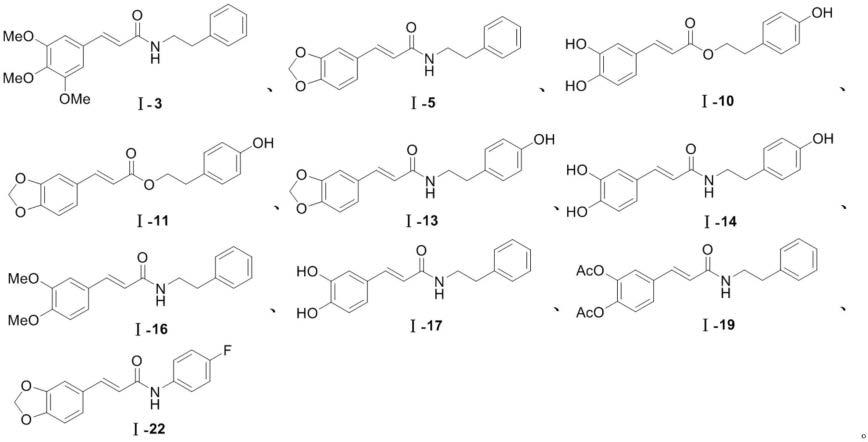
优选的,所述抗炎化合物为结构如下式(i-3、i-5、i-10、i-11、i-13、i-14、i-16、i-17、i-19或i-22)所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0016][0017]
一种抗炎化合物的制备方法,包括以下步骤:
[0018]
将肉桂酸衍生物与苯乙胺类化合物、取代苯或苯乙醇类化合物通过酰胺化反应或酯化反应连接,所述肉桂酸衍生物为采用吸电子基团或给电子基团分别对肉桂酸中苯环的对位和间位进行取代而得。
[0019]
优选的,所述抗炎化合物的制备方法具体包括以下步骤:将第一起始原料和第二起始原料在反应助剂(若需要则使用缩合剂、催化剂、碱性试剂)的作用下于溶剂中反应,并通过分子间脱水合成如以下式i所示的一系列肉桂酸的酯类衍生物或酰胺类衍生物:
[0020][0021]
所述第一起始原料为结构如下式所示的肉桂酸衍生物(反应前可以对苯环上的取代基进行必要的保护,以确保反应在远离苯环的羧基处进行):
[0022][0023]
所述第二起始原料的结构如下式所示:
[0024][0025]
其中,r1、r2、r3分别为h、甲氧基、乙酰氧基、羟基、亚甲二氧基中的任意一种,r4为h、羟基、卤素中的任意一种。
[0026]
优选的,所述缩合剂为n,n
′‑
二环己基碳二亚胺(dcc)、羰基二咪唑(cdi)、二异丙基碳二亚胺(dic)、1-(3-二甲胺基丙基)-3-乙基碳二亚胺(edc)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edci)、苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐(bop)中的一种或多种,催化剂为4-二甲氨基吡啶(dmap)、1-羟基苯并三氮唑(hobt)中的一
种或多种,碱性试剂为三乙胺(tea);溶剂为四氢呋喃、乙腈、苯、二氯甲烷(dcm)、n,n-二甲基甲酰胺、二甲亚砜(dmf)中的一种或多种。
[0027]
优选的,反应温度为25~30℃,反应时间为12~20h,羧酸(具体指第一起始原料)与胺或醇(具体指第二起始原料)的物质的量比为1:1~1:2,催化剂的用量控制在羧酸(具体指第一起始原料)物质的量的50%~110%,羧酸(具体指第一起始原料)与缩合剂的物质的量比为1:1~1:2,羧酸(具体指第一起始原料)与碱性试剂的物质的量比为1:1~1:7。
[0028]
优选的,所述抗炎化合物的制备方法还包括以下步骤:反应结束后进行萃取,然后洗涤(洗涤试剂从水、饱和食盐水、饱和nahco3溶液中选择一、两种)萃取所得有机相,然后依次经无水硫酸钠干燥、减压浓缩、硅胶柱层析,从而将反应体系中的目的产物分离出来。
[0029]
优选的,所述萃取的试剂为乙酸乙酯或二氯甲烷。
[0030]
优选的,所述柱层析的洗脱剂为二氯甲烷-乙酸乙酯混合溶剂,二氯甲烷:乙酸乙酯(ea)的体积比为5:1~1:1,或者为乙酸乙酯-石油醚混合溶剂,石油醚(pe):乙酸乙酯的体积比为5:1~1:1,或者为二氯甲烷-甲醇混合溶剂,二氯甲烷:甲醇的体积比为30:1~1:1。
[0031]
上述抗炎化合物可作为活性成分用于制备治疗炎症性疾病,尤其是治疗炎症性肠病的药物。
[0032]
优选的,所述抗炎化合物对lps诱导的炎症反应具有显著的抑制作用。
[0033]
优选的,所述药物采用能够使活性成分有效地到达体内(具体指血液循环系统)的剂型,具体选自片剂、胶囊剂、粉末、颗粒剂、糖浆、溶液、悬浮液、注射剂、酊剂、口服液、气雾剂、口含剂、冲剂、丸剂、散剂等常见剂型或纳米制剂等缓释剂型中的任意一种。
[0034]
优选的,所述药物中除了含有上述活性成分之外,还含有少量不影响活性成分有效性的次要成分和/或药学上可接受的载体,例如可以含有甜味剂以改善口味、抗氧化剂以防止氧化。
[0035]
优选的,所述药物中还含有制剂所必要的其他辅料。
[0036]
本发明的有益效果体现在:
[0037]
本发明以天然产物hoec、其类似物hoeac为先导化合物结构,经设计、合成以这两种化学结构为基本骨架的衍生物,并通过进行毒性、活性实验,获得了在lps刺激下对raw264.7细胞产生的炎性细胞因子具有更明显抑制作用的抗炎分子,其作为用于治疗炎症性疾病的药物的活性成分,有助于延长ibd缓解期时间。
[0038]
本发明通过缩合反应将肉桂酸衍生物进行酰胺化或酯化,形成了一系列肉桂酸的酯类衍生物或酰胺类衍生物,为筛选高效抗炎药物的活性成分提供了新的途径。
[0039]
进一步的,本发明通过考察肉桂酸苯环上引入的不同的吸电子基团和给电子基团、以及取代基在苯环上的位置,对目标分子抗炎活性作用的影响,发现肉桂酸苯环上引入甲氧基等给电子基团能够增强抗炎活性。
附图说明
[0040]
图1为肉桂酸衍生物的酰胺化或酯化路线。
[0041]
图2为化合物3的合成路线(a)、1hnmr图谱(b)及
13
cnmr图谱(c)。
[0042]
图3为化合物5的合成路线(a)、1hnmr图谱(b)及
13
cnmr图谱(c)。
[0043]
图4为化合物10的合成路线(a)、1hnmr图谱(b)及
13
cnmr图谱(c)。
[0044]
图5为化合物11的合成路线(a)、1hnmr图谱(b)及
13
cnmr图谱(c)。
[0045]
图6为化合物13的合成路线。
[0046]
图7为化合物14的合成路线。
[0047]
图8为化合物16的合成路线(a)及1hnmr图谱(b)。
[0048]
图9为化合物17的合成路线。
[0049]
图10为化合物19的合成路线。
[0050]
图11为化合物22的合成路线。
[0051]
图12为il-6的mrna表达结果,图中:lps组给予lps刺激,ctrl组不给予lps刺激,0/3/10/30表示14-25或14-28的浓度(μm)。
[0052]
图13为tnf-a的mrna表达结果。
[0053]
图14为ifn-β的mrna表达结果。
[0054]
图15为il-1β的mrna表达结果。
[0055]
图16为mip-2的mrna表达结果。
[0056]
图17为il-10的mrna表达结果。
具体实施方式
[0057]
下面结合附图和实施例对本发明作进一步详细说明。所述实施例仅用于解释本发明,而非对本发明的保护范围的限制。
[0058]
(一)候选抗炎化合物设计、合成策略
[0059]
本发明将天然产物hoec及其类似物hoeca(参见下式)作为母核结构,设计、合成一系列可能靶向5-lox酶,并具有较好抑制lps刺激raw264.7细胞产生的炎性细胞因子的肉桂酸酯类、酰胺类衍生物。
[0060][0061]
参见图1,各r基团用于通过引入的取代基形成功能结构。其中,r1、r2、r3上的相应取代(可分别用甲氧基、乙酰氧基、羟基、亚甲二氧基等进行取代)用以分别考察:a.在苯环中引入不同的吸电子基团和给电子基团对目标分子抗炎活性作用的影响;b.取代基在苯环上位置不同时对目标分子抗炎活性作用的影响。而r4上的相应取代(可分别用羟基、卤素等基团进行取代)用以考察苯环上取代基团和链的长短对目标分子抗炎活性作用的影响。
[0062]
(二)合成实例
[0063]
1.化合物3即i-3(84)的制备
[0064]
参见图2,在氮气保护下将3,4,5-三甲氧基肉桂酸(化合物1;238.24mg,1.0mmol,1.0eq)、edci(170.76mg,1.1mmol,1.1eq),及hobt(148.64mg,1.1mmol,1.1eq)溶于10ml二氯甲烷中,在0℃条件下,缓慢加入三乙胺(0.28ml,2.0mmol,2.0eq),然后缓慢加入含有2-苯乙胺(化合物2;0.14ml,1.1mmol,1.1eq)的无水二氯甲烷溶液,在0℃条件下搅拌30min之后,升温到室温反应过夜。tlc显示反应终点,反应结束之后,减压除去(0.09mpa、35℃)二氯
甲烷,加入20ml水稀释,并用乙酸乙酯(20ml
×
3)进行萃取,合并有机相并用水、饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃)后用ch2cl2:ea=4:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到87.4mg白色固体(即化合物3),产率25.6%。产物鉴定数据如下:1h nmr(500mhz,cdcl3)δ7.53(d,j=15.5hz,1h),7.33(t,j=7.4hz,2h),7.23(t,j=6.7hz,2h),6.70(s,2h),6.24(d,j=15.5hz,1h),5.67(t,j=5.5hz,1h),3.86(s,9h),3.73
–
3.57(m,2h),2.89(t,j=6.9hz,2h)。
13
c nmr(126mhz,cdcl3)δ165.92,153.51,141.17,139.68,138.99,130.49,128.94,128.83,126.69,120.04,105.05,61.08,56.25,40.87,35.77。
[0065]
2.化合物5即i-5(85)的制备
[0066]
参见图3,在氮气保护下将3,4-(亚甲二氧)肉桂酸(化合物4;192.17mg,1.0mmol,1.0eq)、edci(170.76mg,1.1mmol,1.1eq),及hobt(148.64mg,1.1mmol,1.1eq)溶于10ml二氯甲烷中,在0℃条件下,缓慢加入三乙胺(0.28ml,2.0mmol,2.0eq),然后缓慢加入含有2-苯乙胺(化合物2;0.14ml,1.1mmol,1.1eq)的无水二氯甲烷溶液,在0℃条件下搅拌30min之后,升温到室温反应过夜。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),加入20ml水稀释,并用乙酸乙酯(20ml
×
3)进行萃取,合并有机相并用水、饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用ch2cl2:ea=4:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃)得到176mg白色固体(即化合物5),产率60%。产物鉴定数据如下:1h nmr(500mhz,cdcl3)δ7.52(d,j=15.5hz,1h),7.32(t,j=7.4hz,2h),7.23(dd,j=14.4,7.2hz,3h),6.99
–
6.92(m,2h),6.78(d,j=7.9hz,1h),6.15(d,j=15.5hz,1h),5.97(s,2h),5.69(s,1h),3.65(dd,j=13.0,6.8hz,2h),2.88(t,j=6.9hz,2h)。
13
c nmr(126mhz,cdcl3)δ166.15,149.15,148.32,140.89,139.04,129.32,128.93,128.80,126.67,123.97,118.75,108.63,106.45,101.54,77.41,77.16,76.91,40.91,35.82。
[0067]
3.化合物10即
ⅰ‑
10(101)的制备
[0068]
参见图4,在氮气保护下将咖啡酸(化合物6;1.2g,6.7mmol,1.0eq)加入100ml的双颈瓶中,然后加入30ml无水二氯甲烷。在0℃条件下,缓慢加入三乙胺(6.13ml,44.1mmol,6.6eq),然后缓慢加入含有tbdmscl(4.5g,30mmol,4.5eq)的无水二氯甲烷溶液15ml,在0℃条件下搅拌反应30min之后,升温到室温反应过夜。tlc显示反应终点。反应结束之后,向反应体系中加入1mol
·
l-1
hcl 30ml,分离有机相,水相用二氯甲烷萃取(20ml
×
2),合并有机相,然后依次用饱和k2co3溶液、1mol
·
l-1
hcl溶液、饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用ch2cl2:ea=4:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到2.14g白色固体(即化合物7),产率78%。
[0069]
在氮气保护下将化合物7(408.2mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入15ml无水二氯甲烷。在搅拌的条件下,向反应体系中加入酪醇(化合物8;165.7mg,1.2mmol,1.2eq)和dmap(61mg,0.5mmol,0.5eq),搅拌5min之后,向反应体系中加入edci(306.7mg,1.6mmol,1.6eq),然后在室温条件下反应12h。tlc显示反应终点,反应结束之后,向反应体系中加入30ml水进行洗涤,分离有机相,水相用二氯甲烷萃取2次,合并有机相并用饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用pe:ea=3:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到397.2mg白色油状物,静置过
夜之后得到白色固体(即化合物9),产率75.1%。
[0070]
在氮气保护下将化合物9(235.4mg,0.38mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入10ml四氢呋喃。在0℃和搅拌的条件下,向反应体系中逐滴加入tbaf(1.0ml,1.0mmol,3eq),搅拌30min之后,向反应体系中加入40ml 1%的hcl溶液,水相用乙酸乙酯(20ml
×
3)萃取,合并有机相,用20ml饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用ch2cl2:ch3oh=30:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到79.5mg浅绿色粉末状固体(即化合物10),产率59.5%。产物鉴定数据如下:1h nmr(500mhz,meod)δ7.71(d,j=15.8hz,1h),7.27(d,j=8.4hz,1h),7.11(d,j=1.7hz,1h),7.05(t,j=8.5hz,1h),7.01(dd,j=8.2,1.7hz,1h),6.81(d,j=8.2hz,1h),6.45(d,j=15.8hz,1h),3.76(t,j=6.9hz,1h),2.83(t,j=6.9hz,1h)。
13
c nmr(126mhz,meod)δ167.84,150.78,150.01,148.61,146.90,138.00,130.94,127.55,123.37,122.61,116.55,115.31,114.23,64.13,39.57。
[0071]
4.化合物11即i-11(102)的制备
[0072]
参见图5,在氮气保护下将3,4-(亚甲二氧)肉桂酸(化合物4;192.17mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入15ml无水二氯甲烷。在搅拌的条件下,向反应体系中加入酪醇(化合物8;151.9mg,1.1mmol,1.1eq)和dmap(61mg,0.5mmol,0.5eq),此时反应液呈现为浑浊状态,补加1ml无水dmf,反应液变为澄清,搅拌25min之后,向反应体系中加入edci(306.7mg,1.6mmol,1.6eq),加入edci之后反应液变为嫩黄色,在室温条件下反应17h。tlc显示反应终点,反应结束之后,向反应体系中加入30ml水进行洗涤,分离有机相,水相用二氯甲烷萃取2次,合并有机相并用饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用pe:ea=2:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到221.7mg浅黄色油状物,静置过夜之后得到白色固体(即化合物11),产率71%。产物鉴定数据如下:1h nmr(500mhz,cdcl3)δ7.76(d,j=15.9hz,1h),7.26(dd,j=4.6,3.8hz,2h),7.15
–
6.99(m,4h),6.84(d,j=8.0hz,1h),6.44(d,j=15.9hz,1h),6.03(s,2h),3.85(t,j=6.5hz,2h),2.87(t,j=6.5hz,2h)。
13
c nmr(126mhz,cdcl3)δ165.89,150.13,149.55,148.58,146.43,136.16,130.13,128.73,125.06,121.84,115.21,108.77,106.72,101.79,63.71,38.72。
[0073]
5.化合物13即
ⅰ‑
13(107)的制备
[0074]
参见图6,在氮气保护下将3,4-(亚甲二氧)肉桂酸(化合物4;192.17mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入10ml无水dmf。将圆底烧瓶置于冰浴中,在搅拌的条件下,向反应体系中加入三乙胺(152.9μl,1.1mmol,1.1eq)。然后向反应体系中加入酪胺(化合物12;180mg,1.3mmol,1.3eq),随后缓慢加入含有bop(442.29mg,1.0mmol,1.0eq)的15ml二氯甲烷溶液,在0℃搅拌30min后,将反应液置于室温反应过夜(20h)。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),剩余物中加入50ml乙酸乙酯和25ml 1mol
·
l-1
hcl,分离出有机相,依次用饱和nahco3溶液(30ml)、饱和食盐水(20ml)洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用pe:ea=1:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到287.3mg白色固体(即化合物13),产率92.3%。产物鉴定数据如下:1h nmr(500mhz,meod)δ7.44(t,j=13.8hz,1h),7.08(d,j=1.6hz,1h),7.05(d,j=8.5hz,2h),7.01(dd,j=8.0,1.6hz,1h),6.82(dd,j=7.9,4.2hz,1h),6.75
–
6.65
(m,2h),6.40(d,j=15.7hz,1h),5.98(s,1h),3.46(t,j=7.4hz,2h),2.75(t,j=7.4hz,2h)。
13
c nmr(126mhz,meod)δ168.85,156.92,150.64,149.82,141.46,131.27,130.73,130.68,125.02,119.79,116.26,109.40,107.10,102.90,42.56,35.77。
[0075]
6.化合物14即
ⅰ‑
14(108)的制备
[0076]
参见图7,在氮气保护下将咖啡酸(化合物6;180.16mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入10ml无水dmf。将圆底烧瓶置于冰浴中,在搅拌的条件下,向反应体系中加入三乙胺(152.9μl,1.1mmol,1.1eq)。然后向反应体系中加入酪胺(化合物12;180mg,1.3mmol,1.3eq),随后缓慢加入含有bop(442.29mg,1.0mmol,1.0eq)的15ml二氯甲烷溶液,在0℃搅拌30min后,将反应液置于室温反应过夜(20h)。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),剩余物中加入50ml乙酸乙酯和25ml 1mol
·
l-1
hcl,分离出有机相,依次用饱和nahco3溶液(30ml)、饱和食盐水(20ml)洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用dcm:ch3oh=10:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到195.2mg无色固体(即化合物14),产率65.2%。产物鉴定数据如下:1h nmr(500mhz,meod)δ7.34(d,j=15.7hz,1h),7.02(d,j=8.4hz,2h),6.96(d,j=2.0hz,1h),6.86(dd,j=8.1,1.9hz,1h),6.73(dd,j=8.1,3.2hz,1h),6.71
–
6.66(m,2h),6.30(dd,j=15.7,2.4hz,1h),3.41(t,j=7.4hz,2h),2.71(t,j=7.4hz,2h)。
13
c nmr(126mhz,meod)δ169.30,156.79,148.67,146.63,142.16,131.36,130.74,128.31,122.12,118.40,116.48,116.25,115.07,42.53,35.76.
13
c nmr(126mhz,meod)δ142.20,130.77,122.16,118.41,116.50,116.28,115.10,42.57,35.79。
[0077]
7.化合物16即
ⅰ‑
16(83)的制备
[0078]
参见图8,在氮气保护下将3,4-二甲氧基肉桂酸(化合物15;208.21mg,1.0mmol,1.0eq)、edci(170.76mg,1.1mmol,0.2ml,1.1eq),及hobt(148.64mg,1.1mmol,1.1eq)溶于10ml无水二氯甲烷中,在0℃条件下,缓慢加入三乙胺(0.28ml,2.0mmol,2.0eq),然后缓慢加入含有2-苯乙胺(化合物2;0.14ml,1.1mmol,1.1eq)的无水二氯甲烷溶液5ml,在0℃条件下搅拌30min之后,升温到室温反应过夜。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),加入20ml水稀释,并用乙酸乙酯(20ml
×
3)进行萃取,合并有机相并用水、饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用ch2cl2:ea=4:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到130.1mg白色固体(即化合物16),产率42%。产物鉴定数据如下:1h nmr(400mhz,cdcl3)δ7.56(d,j=15.6hz,1h),7.37
–
7.31(m,2h),7.07(d,j=8.1hz,1h),7.01(s,1h),6.85(d,j=8.4hz,1h),6.18(d,j=15.5hz,1h),5.53(s,1h),3.90(s,4h),3.71
–
3.61(m,1h),2.89(t,j=6.8hz,1h)。
[0079]
8.化合物17即
ⅰ‑
17(109)的制备
[0080]
参见图9,在氮气保护下将咖啡酸(化合物6;180.16mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入10ml无水dmf。将圆底烧瓶置于冰浴中,在搅拌的条件下,向反应体系中加入三乙胺(152.9μl,1.1mmol,1.1eq)。然后向反应体系中加入2-苯乙胺(化合物2;180mg,1.3mmol,1.3eq),随后缓慢加入含有bop(442.29mg,1.0mmol,1.0eq)的15ml二氯甲烷溶液,在0℃搅拌30min后,将反应液置于室温反应过夜(20h)。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),剩余物中加入50ml乙酸乙酯和25ml 1mol
·
l-1
hcl,分离出有机相,依次用饱和nahco3溶液(30ml),饱和食盐水(20ml)洗涤,无水硫酸钠
干燥,有机相减压浓缩(0.09mpa、35℃),用dcm:ch3oh=10:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到170.6mg白色固体(即化合物17),产率60.2%。产物鉴定数据如下:1h nmr(500mhz,meod)δ7.39(dd,j=15.7,2.9hz,1h),7.31
–
7.25(m,2h),7.25
–
7.21(m,2h),7.21
–
7.16(m,1h),7.00(d,j=2.1hz,1h),6.90(dd,j=8.2,2.0hz,1h),6.76(dd,j=8.1,1.7hz,1h),6.33(dd,j=15.7,2.2hz,1h),3.50(t,j=7.4hz,2h),2.84(t,j=7.4hz,2h).
13
c nmr(126mhz,meod)δ169.30,148.75,146.70,142.22,140.54,129.80,129.50,128.29,127.36,122.10,118.33,116.45,115.05,42.25,36.63。
13
c nmr(126mhz,meod)δ142.24,129.82,129.52,127.38,122.12,118.35,116.47,115.06,42.27,36.65。
[0081]
9.化合物19即
ⅰ‑
19(113)的制备
[0082]
参见图10,在氮气保护下将3,4-二乙酰氧基肉桂酸(化合物18;264.3mg,1.0mmol,1.0eq)、edci(170.76mg,1.1mmol,0.2ml,1.1eq),及hobt(148.64mg,1.1mmol,1.1eq)溶于10ml无水二氯甲烷中,在0℃条件下,缓慢加入三乙胺(0.28ml,2.0mmol,2.0eq),然后缓慢加入含有2-苯乙胺(化合物2;0.14ml,1.1mmol,1.1eq)的无水二氯甲烷溶液5ml,在0℃条件下搅拌30min之后,升温到室温反应过夜。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),加入20ml水稀释,并用乙酸乙酯(20ml
×
3)进行萃取,合并有机相并用水、饱和食盐水洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用pe:ea=1:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到344.8mg浅黄色固体(即化合物19),产率94%。产物鉴定数据如下:1h nmr(500mhz,cdcl3)δ7.54(d,j=15.5hz,1h),7.35
–
7.29(m,4h),7.22(dd,j=9.3,8.1hz,3h),7.17(d,j=8.3hz,1h),6.23(d,j=15.5hz,1h),5.70(t,j=5.4hz,1h),3.64(dd,j=12.9,6.8hz,2h),2.88(t,j=6.9hz,2h),2.29(d,j=0.5hz,6h).
13
c nmr(126mhz,cdcl3)δ168.26,168.22,165.49,143.05,142.45,139.34,138.92,133.92,128.92,128.84,126.72,126.33,123.91,122.38,121.91,40.95,35.72,20.78,20.76。
13
c nmr(126mhz,cdcl3)δ139.23,128.82,128.73,126.61,126.23,123.81,122.28,121.80,40.84,35.61,20.67,20.65。
[0083]
10.化合物22即
ⅰ‑
22(123)的制备
[0084]
参见图11,在氮气保护下将3,4-(亚甲二氧)肉桂酸(化合物4;192.17mg,1.0mmol,1.0eq)加入到100ml圆底烧瓶中,然后加入4ml socl2(将羧基酰氯化,以增强反应的活性),及1滴dmf。回流反应6h之后,减压除去多余的socl2,然后向剩余物中加入5ml无水二氯甲烷,将反应瓶置于冰浴中,在搅拌的条件下,向反应体系中加入三乙胺(0.2ml,1.44mmol,1.44eq)。然后向反应体系中加入4-氟苯胺(化合物21;1.0eq,0.1ml),随后将反应液置于室温反应过夜(20h)。tlc显示反应终点,反应结束之后,减压除去二氯甲烷(0.09mpa、35℃),剩余物中加入50ml乙酸乙酯和25ml 1mol
·
l-1
hcl,分离出有机相,依次用饱和nahco3溶液(30ml)、饱和食盐水(20ml)洗涤,无水硫酸钠干燥,有机相减压浓缩(0.09mpa、35℃),用pe:ea=3:1的展开条件进行柱层析,对洗脱液减压浓缩(0.09mpa、35℃),得到246.3mg白色固体(即化合物22),产率86.4%。产物鉴定数据如下:1h nmr(400mhz,cdcl3)δ7.66(d,j=15.4hz,1h),7.57(s,2h),7.07
–
7.01(m,4h),6.82(d,j=7.8hz,1h),6.35(d,j=15.4hz,1h),6.01(s,2h)。
[0085]
(三)细胞毒性实验
[0086]
1.先导化合物hoeca(14-25)和hoec(14-28)的细胞毒性试验
[0087]
取生长良好的raw264.7细胞铺板,0.5ml/孔(24孔板),得到2
×
104个/孔的细胞悬液。分别向单独药物作用组和给予lps刺激(100ng/ml)的药物处理组每孔细胞中加入适量药物(hoeca或hoec)储备液(溶剂是dmso),使终浓度为5、15、50、70,及100μm。加入药物后,分别于6h、12h、24h、48h、72h观察并记录细胞的悬浮比例、细胞数目、细胞状态(是否圆润、透亮),以及触角的数目,从而判断药物所具有的毒性。结果表明hoeca在12h时,50μm浓度下有一定的细胞毒性,100μm浓度下有明显的细胞毒性。hoec(浓度是100μm)在0~48h无明显细胞毒性。
[0088]
2.药物储备液的配制
[0089]
将合成得到的化合物(化合物3、5、10、11、13、14、16、17、19和22)分别于dmso中溶解,得到40mm浓度母液,保存。在使用时取适量母液用高糖hyclone dmem培养基(ge,美国)稀释成4mm的溶液,备用。
[0090]
3.毒性观察实验
[0091]
取生长良好的raw264.7细胞,经胰酶-edta溶液消化,配制成1
×
105个/ml细胞悬液,加入24孔塑料细胞培养板(linbrou-s-a)孔内(0.5ml/孔)。分别向单独药物作用组和给予lps刺激的药物处理组每孔细胞中加入适量药物(化合物3、5、10、11、13、14、16、17、19、22)储备液,使终浓度为0μm、10μm、30μm和100μm。加入药物后,分别于6h、12h、24h、48h、72h观察并记录细胞的悬浮比例、细胞数目、细胞状态(是否圆润、透亮),以及触角的数目,从而判断药物所具有的毒性。结果表明:
①
对于单独药物作用组,各孔漂浮细胞增多,0μm、10μm和30μm孔约占3%,100μm孔约占5%;细胞数目0μm孔和10μm孔差异不大,30μm孔细胞数目稍减少,100μm孔细胞数目减少最为明显,细胞皱缩明显,细胞间隙增大;
②
给予lps刺激的药物处理组,0μm、10μm和30μm孔细胞漂浮情况约占3%,100μm孔约占4%,细胞数目0μm、10μm和30μm孔无明显差异,100μm孔略少,细胞破碎(细胞壁皱缩,胞质不见,细胞核破碎不完整),触角变多,受刺激分化的细胞比例约占95%,因细胞破碎颗粒度变化不明显。实验结果说明,药物(化合物3、5、10、11、13、14、16、17、19和22)在100μm浓度时,具有一定的毒性(与先导化合物hoec相似),在30μm以下是安全的。
[0092]
(四)pcr实验
[0093]
1.实验设计
[0094]
取生长良好的raw264.7细胞铺板,0.5ml/孔(24孔板),得到2
×
105个/孔的细胞悬液。
[0095]
药物干预:分别在给予lps(100ng/ml)同时,用合成所得化合物不同浓度药液(10μm、30μm、100μm)处理raw264.7细胞。处理6h之后,提取各浓度下细胞的总rna,反转录,荧光定量pcr检测il-6、tnf-α、il-1β、il-10、ifn-β和mip-2的mrna水平的表达情况。
[0096]
2.细胞总rna提取
[0097]
到时间点后,先在显微镜下快速观察细胞状态及有无污染。然后根据rna极速抽提试剂盒的说明书步骤进行如下操作:首先吸弃细胞上清,加入0.5ml冷的pbs,悬空摇晃细胞培养板,洗去残留的培养基并吸弃pbs,再加入0.5ml细胞裂解液ra2,轻轻吹打后,将细胞裂解液吸至内套柱中,离心(4℃,12000rpm,1min)。弃掉外套柱中的液体,在内套柱加入0.5ml用无水乙醇4倍稀释后的wash buffer,离心(4℃,12000rpm,1min),倒掉外套柱中的液体;
[0121]
配制荧光定量pcr反应体系(20μl;试剂名称、试剂体积):sense primer和anti-sense primer(各5μm,预混合)1μl;ddh2o 5μl;premix ex taqtm 10μl;cdna模板4μl。
[0122]
将上述混合物混匀后加入到pcr毛细管中,离心(3000rpm,1min),设置反应程序后检测,得出样本目的基因的ct值,减去对应样本β-actin的ct值,得到差值。此差值再减去阴性对照孔的差值,用2
‑△△
ct公式计算样品目的基因扩增倍数,做图并分析。
[0123]
5.实验结果
[0124]
5.1 il-6的实验结果
[0125]
参见图12,结果表明84号、101号药物(化合物3、10)能够显著降低lps诱导的raw264.7细胞il-6mrna的表达。14-25(hoeca)给药孔(10μm、30μm)较不给药孔(0μm,即仅给予lps刺激)il-6mrna表达更高,说明14-25可能具有促炎作用,14-28(hoec)表达量无明显区别,提示14-28可能没有调控作用。
[0126]
5.2 tnf-a的实验结果
[0127]
参见图13,结果表明83号、84号药物(化合物16、3)能够显著降低lps诱导的raw264.7细胞tnf-α mrna的表达。而14-25和14-28能够促进lps诱导的raw264.7细胞tnf-αmrna的表达。
[0128]
5.3 ifn-β的实验结果
[0129]
参见图14,结果表明84号药物(化合物3)能够显著降低lps诱导的raw264.7细胞ifn-β mrna的表达。
[0130]
5.4 il-1β的实验结果
[0131]
参见图15,结果表明84号、101号药物(化合物3、10)能够显著降低lps诱导的raw264.7细胞il-1βmrna的表达。
[0132]
5.5 mip-2的实验结果
[0133]
参见图16,结果表明83号、84号药物(化合物16、3)能够显著降低lps诱导的raw264.7细胞mip-2mrna的表达。
[0134]
5.6 il-10的实验结果
[0135]
参见图17,结果表明84号药物(化合物3)能够显著降低lps诱导的raw264.7细胞il-10mrna的表达。14-25和14-28可能具有促进lps诱导的raw264.7细胞il-10mrna的表达的作用。
[0136]
以上实验结果表明,化合物3、10、16具有比先导化合物更强的抑制炎性细胞因子il-6、tnf-α等的作用,而相应的先导化合物却对il-6、tnf-α等具有促进表达的作用。因此将化合物3、10、16作为治疗炎症性肠病的药物活性成分进行了实验验证。
[0137]
(五)制剂实例
[0138]
1.抗炎药物片剂的制备
[0139]
将化合物3、5、10、11、13、14、16、17、19、22各2g分别与17.5g辅料(白湖精:乳糖=7:3,质量比)混合之后,加入95%的乙醇制粒,干燥,整粒(过筛),加入硬脂酸钠0.5g,混合均匀之后压片,得到每片重100mg,相应化合物的含量为10mg的片剂。
[0140]
2.抗炎药物粉针剂的制备
[0141]
将化合物3、5、10、11、13、14、16、17、19、22各1g分别与5g甘露醇溶解于170ml的注
射用水中,初次混匀之后,定容到200ml,过滤所得到的溶液,装入西林瓶中,每瓶1ml,冻干,密封、灭菌,得到每支含有5mg对应化合物的冻干粉针剂。
[0142]
3.抗炎药物胶囊剂的制备
[0143]
将化合物3、5、10、11、13、14、16、17、19、22各3g分别与27g辅料(白湖精:乳糖=7:3,质量比)混合之后,加入95%的乙醇制粒,干燥,整粒(过筛),装入胶囊中,每粒重约30mg,相应化合物的含量约为3mg。
[0144]
总之,本发明合成的化合物具有明显优于先导化合物的抗炎活性,尤其是83、84和101号药物的抗炎活性最为显著,对lps刺激raw264.7细胞产生的炎性细胞因子具有更明显的抑制作用,具备用于制备抗炎药物(特别是治疗炎症性肠病药物)的潜力。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。