1.本发明涉及材料领域,具体涉及碳化物及其制备方法。
背景技术:
2.相关技术采用碳热还原法制备金属或非金属碳化物。
3.以碳热还原法制备碳化硅(sic)为例。相关技术一般在1400℃以上 的常压高温环境中,在非氧化性气氛下,对二氧化硅和碳单质进行长时 间烧结来制备碳化硅,反应方程式如下:
4.sio2 3c
→
sic 2co
↑
5.现有技术的制备碳化物的方法具有反应温度高、反应时间长、碳化 硅产物比表面积低等缺点,这些不利因素阻碍了sic作为催化剂载体的 规模化工业应用。
技术实现要素:
6.鉴于现有技术存在的问题,本技术提出了一种创新的制备碳化物的 策略:在低压环境下通过碳热还原氧化物制备碳化物,即低压碳热还原 策略。
7.以碳热还原法制备sic为例。本技术通过对碳热还原反应进行热力 学和动力学分析发现:在碳热还原反应过程中,sio2被还原至sio气体 中间反应物分子需要克服非常高的热力学能量势垒(室温室压,~593.5 kj/mol),这一步是sic制备的决速步骤。本技术进一步发现:低压环 境对于碳化硅的制备是有利的,原因在于:
8.第一、低压环境有利于降低整体反应的吉布斯自由能,从而使得反 应发生的起始温度向低温区移动,降低了反应温度;
9.第二、低压环境可以加速sio气态中间反应物分子向碳基反应物的 扩散,进而提高sic的生成速率,加快了反应效率;
10.第三、鉴于上述第一、二点,低压环境使得碳热还原反应能够在较低 的温度下以较高的速度进行。这有助于避免sic晶粒因长时间暴露于高 温环境而熟化、团聚、烧结,进而有助于获得高比表面积的sic。
11.本技术实施例的实验结果表明:在低压环境下,利用焦耳加热设备 制备sic,反应温度可以减少至1200℃,反应时间可以减少至30秒,sic 产物的bet比表面积可以高达569.9m2/g。
12.本技术实施例的实验结果还表明,在低压环境下,利用真空加热炉 制备sic,在1300℃的反应温度下,在5分钟的反应时间,能够制备12.29 g的sic。
13.本技术实施例的实验结果还表明,低压碳热还原策略还能够制备多 种金属碳化物,例如nbc、mo2c、tac、wc等,甚至是高熵金属碳化 物vnbmotawc5,tivnbtawc5等。
14.在一些方面,本技术提供一种制备碳化物的方法,包括:
15.(1)提供第一前驱体,所述第一前驱体中含有:待碳化元素的氧化 物和碳质材料;
16.(2)加热第一前驱体,使待碳化元素的氧化物和碳质材料发生碳热 还原反应,生
成碳化物;
17.其中,在所述加热的至少部分阶段,将反应的环境的压强降低至p0帕以下,0<p0<10000。
18.在一些实施方案中,步骤(2)中,加热第一前驱体至1200℃以上。
19.在一些实施方案中,步骤(2)中,加热第一前驱体至1200℃-2000℃, 例如1200℃-1300℃、1300℃-1400℃、1400℃-1500℃、1500℃-1600℃、 1600℃-1700℃、1700℃-1800℃、1800℃-1900℃或1900℃-2000℃。
20.在一些实施方案中,步骤(2)中,加热的时间为10秒以上,例如 10秒-30秒,例如30秒-60秒,例如1分钟-5分钟,例如5分钟-10分 钟,例如10分钟-15分钟,例如15分钟-20分钟。
21.在一些实施方案中,0<p0<1000。
22.在一些实施方案中,0<p0<100。
23.在一些实施方案中,p0的取值为0.1以下、0.1-1、1-10、10-100、100
‑ꢀ
200、200-300、300-400、400-500、500-600、600-700、700-800、800-900 或900-1000。
24.在一些实施方案中,所述加热的至少部分阶段占整个加热时间的10% 以上,例如10%-20%、20%-30%、30%-40%、40%-50%、50%-60%、 60%-70%、70%-80%、80%-90%或90%-100%。
25.在一些实施方案中,所述第一前驱体为粉末混合物,所述粉末混合 物含有待碳化元素的氧化物颗粒和碳质材料颗粒。
26.在一些实施方案中,所述第一前驱体中,所述待碳化元素的氧化物 的部分或全部表面被所述碳质材料覆盖。基于此,碳质材料能够有效低 俘获加热过程中形成的中间产物分子,进而与之反应生成碳化物。
27.在一些实施方案中,所述第一前驱体中,所述待碳化元素的氧化物 的50%-100%(例如50%-60%、60%-70%、70%-80%、80%-90%、90%
‑ꢀ
100%)的表面被所述碳质材料覆盖。
28.在一些实施方案中,制备碳化物的方法还包括制备第一前驱体的步 骤,具体包括:
29.(1.1)提供第二前驱体,所述第二前驱体含有待碳化元素的氧化物 和含碳有机物,所述待碳化元素的氧化物的至少部分表面被所述含碳有 机物覆盖;
30.(1.2)在非氧化条件下烧结第二前驱体,使所述含碳有机物热解为 碳质材料。
31.在一些实施方案中,制备碳化物的方法还包括制备第二前驱体的步 骤,具体包括:将含有待碳化元素的氧化物与溶解由含碳有机物的溶液 充分混合,然后将混合物干燥。
32.在一些实施方案中,待碳化元素的氧化物为介孔材料。
33.在一些实施方案中,在一些实施方案中,待碳化元素的氧化物的bet 比表面积为100m2/g以上,例如100m2/g-200m2/g、200m2/g-300m2/g、 300m2/g-400m2/g、400m2/g-500m2/g、500m2/g-600m2/g、600m2/g-700 m2/g、700m2/g-800m2/g、800m2/g-900m2/g或900m2/g-1000m2/g。
34.在一些实施方案中,待碳化元素的氧化物为含硅元素的分子筛材料, 例如mcm-41
材料。
35.在一些实施方案中,步骤(2)中,采用以下一种或多种方式实施所 述加热:焦耳加热、电磁感应加热、电弧加热、电阻加热、激光加热、电 子束加热、等离子弧加热、燃料燃烧加热、炬焰加热、微波加热、射频辐 射加热或其组合。
36.在一些实施方案中,步骤(2)中,所述加热包括焦耳加热,所述焦 耳加热包括以下操作:
37.(i)将所述第一前驱体置于焦耳加热设备的发热体附近;
38.(ii)通过向所述发热体施加焦耳加热电流以执行所述加热。
39.在一些实施方案中,所述加热具有500℃/秒以上的升温速度,例如 800℃/秒的升温速度,例如1000℃/秒的升温速度。较快的升温速度对 于碳化物的制备有以下益处:(1)减少产物在高温场中的暴露时间, 抑制产物团聚烧结;(2)制备超小尺寸或者高比表面积的产物;(3) 缩短反应时间,降低能耗;(4)减少能量散失到环境中,提高设备和 操作人员安全性。
40.在一些实施方案中,所述碳化物选自:非金属碳化物、金属碳化物。
41.在一些实施方案中,所述碳化物是离子型碳化物、共价型碳化物 或间隙型碳化物。
42.在一些实施方案中,所述金属碳化物是过渡金属碳化物。
43.在一些实施方案中,所述金属碳化物是难熔金属的碳化物。
44.在一些实施方案中,难溶金属是指熔点在1000℃以上,例如1500℃ 以上,例如1800℃以上的金属。难溶金属例如选自ti、zr、v、cr、 nb、w、ta、mo、或其组合。
45.在一些实施方案中,所述碳化物为难熔材料(refractory material)。
46.在一些实施方案中,所述碳化物选自以下一种或多种:sic、nbc、 mo2c、tac、wc、vnbmotawc5、tivnbtawc5。
47.在一些实施方案中,待碳化元素的氧化物是一种或多种氧化物。
48.在一些方面,本技术提供一种碳化物,由上述任一项所述的方法 制备获得。
49.在一些实施方案中,所述碳化物包括sic,所述sic的bet比表 面积为100m2/g以上,例如100m2/g-600m2/g,例如100m2/g-200m2/g、 200m2/g-300m2/g、300m2/g-400m2/g、400m2/g-500m2/g、500m2/g-600 m2/g、600m2/g-700m2/g、700m2/g-800m2/g、800m2/g-900m2/g或900 m2/g-1000m2/g。
50.术语解释
51.本公开如果使用了以下的术语,它们可以具有如下的含义
52.本文所用术语“碳化物”是指碳和一种或多种比碳更正电的非碳元素 的化合物,非碳元素不包括氢。非碳元素可以选自金属元素或非金属元 素。金属元素可以是过渡金属元素。非碳元素可以选自硅或硼。可选地, 非碳元素可以选自硼,硅,钛,铌,铁,钨,钼,锆,铪,钒,钽,铬, 锰,锝,铼,锇,钴,镍,镧系元素,钪,钇,镧,锌,铝,铜,锗,镉, 铟,锡,铅或铊中的一种或多种。碳化物还可以包括任选的掺杂元素。碳 化物可以是二元或多元碳化物。碳化物与非碳元素的摩尔比可以为0.5
‑ꢀ
2:1。
53.术语“高熵碳化物”是指包含四种以上非碳元素的碳化物。
54.术语“低压”是指低于1个标准大气压的压力。例如105pa以下,例 如104pa以下,例
如103pa以下,例如102pa以下。
55.术语“碳热还原反应”是指在大约500℃以上的高温下,将碳用作还 原剂去制备碳化物。
56.术语“待碳化元素的氧化物”是指含有待碳化元素的氧化物。
57.术语“碳质材料”是指石墨形式的单质碳或无定形碳形式的单质碳。
58.术语“难熔”是指至少在1000℃不熔化、不软化、不分解。例如至少 在1500℃不熔化、不软化、不分解。例如至少在1800℃不熔化、不软化、 不分解。
59.术语“介孔”定义为孔径在2nm~50nm范围的孔。
60.有益效果
61.本公开一项或多项方案具有以下一项或多项有益效果:
62.(1)制备碳化物所需的加热温度较低;
63.(2)提高了碳热还原反应的反应速率,缩短了反应时间;
64.(3)制备的碳化物比表面积较大;
65.(4)制备的碳化物稳定性较高;
66.(5)从原料到产物的转化率较高;
67.(6)具备工业化宏量生产的理论和实验可行性。
附图说明
68.图1的(a)(b)(c)(d)分别示出mcm-41的x射线衍射 图谱,mcm-41的实物图及结构示意图,mcm-41的n2分子吸脱附 曲线,mcm-41的孔径分布曲线。
69.图2的(a)和(b)分别示出mcm-41的扫描电子显微镜图像及 对应的edx元素分布图。图2的(c)(d)(e)分别示出不同倍数 mcm-41的透射电子显微镜图像。图2的(f)示出mcm-41的x射线 光电子能谱。图2的(g)示出mcm-41的si 2p的x射线光电子能谱。 图2的(h)示出mcm-41的o1s的x射线光电子能谱。
70.图3的(a)示出mcm-41@c制备示意图,局域放大图为蔗糖分 子的结构示意图。图3的(b)示出mcm-41@c的扫描电子显微镜图。
71.图3的(c)示出n2分子吸脱附曲线,mcm-41@c的bet比表面积为 840.2m2/g。
72.图4的(a)示出了一些实施方式采用焦耳加热设备制备sic的示意图。 图4的(b)示出了一些实施方式中低压碳热还原制备sic过程的温度和压强 变化。图4的(c)示出了实施例3.1的低压碳热还原制备金属碳化物过程的 温度和压强变化。图4的(d)示出了实施例4的低压碳热还原制备高熵碳化 物过程的温度和压强变化。
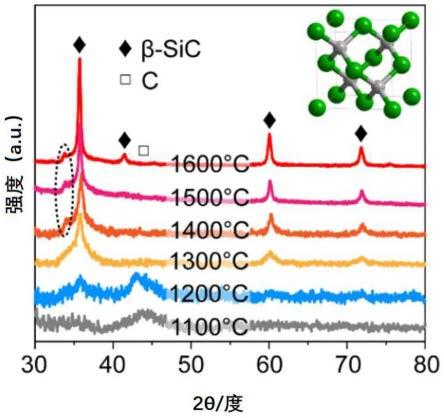
73.图5示出采用焦耳加热设备在不同加热温度制备的sic的x射线衍 射图谱。
74.图6的(a)示出采用焦耳加热设备制备的sic的透射电子显微镜 (tem)图像,图6的(b)示出sic的高分辨透射电子显微镜(hr
‑ꢀ
tem)图像。图6的(c)示出sic的选区电子衍射图像。
75.图7的(a)示出一些实施方式通过真空烧结炉制备的sic的x射 线衍射图,图7的(b)示出sic的n2分子的吸脱附曲线。图7的(c) (d)示出sic的透射电子显微镜图片。图7的(e)示出sic纳米晶的 晶格条纹间距测量。
76.图8的(a)示出金属氧化物nb2o5和制备的金属碳化物nbc的x 射线衍射光谱。图8的
(b)示出金属氧化物moo3和制备的金属碳化物 mo2c的x射线衍射光谱。图8的(c)示出金属氧化物nb2o5和制备的 金属碳化物nbc的x射线衍射光谱。图8的(d)示出金属氧化物nb2o5和制备的金属碳化物wc的x射线衍射光谱。
77.图9的(a)(b)(c)分别示出vnbmotawc5高熵碳化物的x 射线衍射图谱、高倍透射电子显微镜图像、高角环形暗场扫描透射电子 显微镜图像及对应的edx元素分布图。
78.图10的(a)tivnbtawc5高熵碳化物的x射线衍射图谱,插图为 其示意图。图10的(b)和(c)分别示出tivnbtawc5高熵碳化物的 透射电子显微镜图。图10的(d)示出10条tivnbtawc5高熵碳化物 的晶格条纹间距。
79.图11(a)(b)(c)(d)分别示出tivnbtawc5高熵碳化物的 透射电子显微镜图像。
80.图12的(a)示出高温水蒸气和空气混合处理sic粉末在不同时间 后的实物图;图12的(b)示出高温水蒸气和空气混合处理sic粉末在 不同时间后的x射线衍射图谱;图12的(c)示出真空烧结炉实物图及 对应的sic产物实物称重图。
81.图13的示出(a)sic的差热热重曲线。图13的(b)示出100小 时水汽高温热处理后的sic的n2分子的吸脱附曲线。图13的(c)示出 650℃水汽饱和空气流高温处理不同时间后的sic的x射线光电子能谱。
具体实施方式
82.下面将结合实施例对本公开的实施方案进行详细描述,但是本领 域技术人员将会理解,下列实施例仅用于说明本公开,而不应视为限 定本公开的范围。实施例中未注明具体条件者,按照常规条件或制造 商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通 过市购获得的常规产物。
83.1.化学试剂
84.mcm-41介孔硅酸盐分子筛(含1wt%的na2o)购买自天津南化 催化剂有限公司(nankai university catalyst co.,ltd.)。蔗糖(纯度, 99.5%)、碳粉(纯度,99.9%)、二氧化钒(纯度,99%)、五氧化二 铌(纯度,99.99%)、三氧化钼(纯度,99.9%)、五氧化二钽(纯度, 99.9%)、三氧化钨(纯度,99.9%)购买自安耐吉化学(energy chemical)。
85.2.材料制备
86.实施例1
‑‑
焦耳加热方法制备sic
87.1.1第一前驱体的制备
88.首先,将5g蔗糖溶于40ml去离子水中形成蔗糖水溶液。下一步, 将6g的mcm-41分子筛浸没入蔗糖水溶液中并超声处理30分钟。紧 接着,将超声处理后的混合物置于干燥器中并持续抽真空促进蔗糖分子 进入到分子筛的孔隙内(抽真空时间,2小时;抽真空压强,0.098mpa), 收集固体产物,然后置于真空电热干燥箱中分别在100℃和160℃依次各 干燥处理6小时,获得干燥所得的黑色粉末,即为第二前驱体(mcm
‑ꢀ
41@蔗糖)。
89.将第二前驱体盛放于坩埚中并置于管式炉恒温区域,在氮气气氛保 护下600℃热处理2小时,使蔗糖热解为碳质材料,所得的粉末产物即为 第一前驱体,将其命名为mcm-41@c(即被碳包覆的mcm-41)。mcm
‑ꢀ
41的全部表面被碳质材料包覆。
90.1.2sic的制备
91.将150mg的第一前驱体(mcm-41@c)压制成片径为9mm的薄 片,压机压强为30mpa。
将薄片放置在焦耳加热设备内。
92.图4的(a)示出了采用焦耳加热设备制备sic的示意图。焦耳加热 设备包括两个加热体202(两块碳布,长
×
宽=3cm
×
1.2cm),两个加热 体202的两端分别固定在两个铜电极上,铜电极通电后可在两个加热体 202之间产生焦耳热,从而对夹在两个加热体202之间的对象进行加热。 调整焦耳加热设备的电流(20a-60a)能够获得所需的目标温度。
93.将第一前驱体201置于两个加热体202之间,然后将它们至于低压 环境中,低压环境的压强参见表1。在低压环境下将第一前驱体201加热 至目标温度。实施例1中具体涉及实施例1.1至1.5共五个制备例,在实 施例1.1至实施例1.5中,加热的目标温度分别设定为1200℃到1600℃ (详见表1)。加热触发第一前驱体(mcm-41@c)中mcm-41与c的 碳热还原反应,保持加热30秒,收集加热后形成的粗产物。
94.在进一步表征之前,还对粗产物进行后处理。后处理包括:
95.a)煅烧除碳:对加热后制备的产物依次在650℃高温且处于空气氧 化性环境下煅烧2小时以去除未反应的碳。
96.b)酸刻蚀除sio2:上一步产物在20wt%质量比的氢氟酸水溶液中 搅拌12小时以完全去除残余的sio2。经后处理后,获得终产物sic。
97.图4的(b)示出了实施例1.2的低压碳热还原制备sic过程的温度 和压强变化。从温度曲线看,加热温度经2秒的快速升温后稳定在 ~1300℃,至第30秒停止加热。焦耳加热过程中利用真空泵持续工作以 提供低压环境,抽速为14.4l/分钟。从压强曲线看,压强的变化大致可 分为三个阶段:压强从约100pa以下上升到1000pa以上随后减少至约 100pa以下(阶段i,约12秒);随后,压强在100pa以下保持相对稳 定(阶段ii,约18秒),该低压环境有利于增强sio中间产物分子的扩 散,提高sic的生成速率;最后,当焦耳加热停止后,碳布表面的温度 和腔体内的压强均快速下降(阶段iii)。
98.对比例1
99.对比例1与实施例1的区别在于:在步骤1.2sic的制备步骤中,在 低压环境下将第一前驱体201加热至目标温度1100℃,保持加热30秒, 收集加热后形成的粗产物。其它步骤和参数与实施例1相同。
100.实施例2
‑‑
真空烧结炉宏量化制备sic
101.通过真空烧结炉(设备型号为shanghai chenhua,7w-25-20-xmu) 进行sic的宏量化制备。具体方法如下:
102.2.1提供mcm-41@c(方法同实施例1)
103.2.2将mcm-41@c压制为薄片(方法同实施例1)。将压制的mcm
‑ꢀ
41@c薄片放置于空心的圆柱状容器(直径7.5cm;高度2cm)中并加 盖。将圆柱状容器置于真空烧结炉内,在低压环境下(低压环境的压强参 见表1)将mcm-41@c样品以10℃/分钟的升温速率加热到1300℃并 保温5分钟,随后自然冷却,收集粗产物。粗产物含有未反应mcm-41、 未反应的碳粉和生成的sic。
104.对粗产物进行后处理。后处理包括:依次在650℃高温且处于空气气 氛中煅烧2小时以去除未反应的碳,并在20wt%质量比的氢氟酸水溶液 中搅拌12小时以完全去除残余的mcm-41。最终获得终产物sic。
105.对比例2
106.对比例2与实施例2的区别在于,采用高温烧结炉进行烧结,烧结 过程终采用氩气气氛,环境压力为常压(1个大气压)。其它步骤和参数 与实施例2相同。
107.实施例3
‑‑
难熔金属碳化物的制备
108.实施例3采用低压碳热还原的策略制备了金属碳化物,反应物分别 为对应的金属氧化物和碳粉。反应方程式如下:
[0109][0110]
mo
x
为金属氧化物,mcy为金属碳化物,x>0,y>0。
[0111]
下面实施例3.1至3.4分别采用低压碳热还原的策略制备制备金属碳 化物。
[0112]
实施例3.1将1.00g的碳粉与1.05g的nb2o5粉末持续研磨混合2 小时,获得混合粉末。将100mg的混合粉末分别在30mpa压强下被压 制成直径为9mm的薄片。最后,碳化物通过在持续抽真空的条件下以 焦耳加热触发的难熔金属氧化物与碳粉之间的碳热还原反应制备得到, 反应温度是约1300℃,反应加热时间为20秒,获得nbc。
[0113]
实施例3.2将1.00g的碳粉与1.14g的moo3粉末持续研磨混合2 小时,获得混合粉末。将100mg的混合粉末分别在30mpa压强下被压 制成直径为9mm的薄片。最后,碳化物通过在持续抽真空的条件下以 焦耳加热触发的难熔金属氧化物与碳粉之间的碳热还原反应制备得到, 反应温度是约1300℃,反应加热时间为20秒,获得mo2c。
[0114]
实施例3.3将1.00g的碳粉与1.75g的ta2o5粉末持续研磨混合2 小时,获得混合粉末。将100mg的混合粉末分别在30mpa压强下被压 制成直径为9mm的薄片。最后,碳化物通过在持续抽真空的条件下以 焦耳加热触发的难熔金属氧化物与碳粉之间的碳热还原反应制备得到, 反应温度是约1300℃,反应加热时间为20秒,获得tac。
[0115]
实施例3.4将1.00g的碳粉与1.61g的wo3粉末持续研磨混合2小 时,获得混合粉末。将100mg的混合粉末分别在30mpa压强下被压制 成直径为9mm的薄片。最后,碳化物通过在持续抽真空的条件下以焦 耳加热触发的难熔金属氧化物与碳粉之间的碳热还原反应制备得到,反 应温度是约1300℃,反应加热时间为20秒,获得wc。
[0116]
图4的(c)示出了实施例3.1的低压碳热还原制备金属碳化物过程 的温度和压强变化。从温度曲线看,加热温度经2秒的快速升温后稳定 在约1200℃,保持至第20秒停止加热,温度下降。从压强曲线看,在0 秒-12秒,压强从约100pa上升到1000pa以上随后减少至1000pa以 下;在12秒-20秒,压强从约1000pa下降至约100pa以下。
[0117]
实施例4
‑‑
vnbmotawc5和tivnbtawc5高熵碳化物的制备。
[0118]
难熔金属高熵碳化物的制备采用低压碳热还原的策略,反应物分别 为对应的多种难熔金属氧化物和碳粉混合粉末。
[0119]
实施例4.1制备vnbmotawc5高熵碳化物的反应物前驱体为0.432 g的碳粉,0.083g的vo2粉末,0.133g的nb2o5粉末,0.144g的moo3粉末,0.221g的ta2o5粉末以及0.232g的wo3粉末,称取上述混合粉 末后持续手动研磨混合4小时,获得混合粉末。随后,100mg的混合粉 末分别在30mpa压强下被压制成直径为9mm的薄片。最后,难熔金属 高熵碳化物通过在持续抽真空的条件下以焦耳加热触发的难熔金属氧化 物与碳粉之间的碳热还原反应制备得到,反应温度升高至~1400℃以促进 不同元素的扩散混合,反应加热时间为20秒。
[0120]
实施例4.2制备tivnbtawc5高熵碳化物的反应物前驱体为0.408 g的碳粉,0.078g
的tio2粉末,0.083g的vo2粉末,0.133g的nb2o5粉末,0.221g的ta2o5粉末和0.232g的wo3粉末,称取上述混合粉末 后持续手动研磨混合4小时,获得混合粉末。随后,100mg的混合粉末 分别在30mpa压强下被压制成直径为9mm的薄片。最后,难熔金属高 熵碳化物通过在持续抽真空的条件下以焦耳加热触发的难熔金属氧化物 与碳粉之间的碳热还原反应制备得到,反应温度升高至~1400℃以促进不 同元素的扩散混合,反应加热时间为20秒。
[0121]
图4的(d)示出了实施例4的低压碳热还原制备高熵碳化物过程的 温度和压强变化。从温度曲线看,加热温度经2秒的快速升温后稳定在 约1400℃,保持至第20秒停止加热,温度下降。从压强曲线看,在0秒
ꢀ‑
5秒,压强从100pa以下上升到1000pa以上然后减少至1000pa以下; 在5秒-20秒,压强从约1000pa下降至约100pa以下。
[0122]
测试与表征
[0123]
1.材料表征分析。
[0124]
焦耳加热所触发碳热还原反应过程的温度由配备高速数据采集工作 站的红外测温仪测量得到(型号,impac imga 740)。
[0125]
低压碳热还原反应过程中的压强变化由真空电阻规测量得到(型号, infitech vct160)。
[0126]
碳化物的相结构及结晶度分析由粉末x射线衍射仪测量分析得到 (xrd),型号为rigaku ultima-iv(cu靶,)。
[0127]
比表面积分析是通过bet法测试得到,型号为tristar ii 3020m。 样品的差热热重分析(tga-dsc)是在netzsch tg209f1仪器上测 试所得,测试气氛为氩气。
[0128]
样品的元素价态分析是通过配备单色铝靶的x射线光电子能谱 (xps)表征所得,仪器型号为escalab 250xi。
[0129]
样品的相貌、元素分布和微观结构表征是在fei tecnai tf20透射 电子显微镜(tem)上测试得到,加速电压为200kv。
[0130]
原料、前驱体、加热方式、加热温度、加热时间、压强、产物、产率 和比表面积参数如表1所示。
[0131]
[0132][0133]
结果分析:
[0134]
1、mcm-41的表征
[0135]
实施例1(即实施例1.1至1.5)选用mcm-41作为制备sic原料之 一。
[0136]
图1的(a)示出mcm-41的x射线衍射图谱。图1的(b)示出 mcm-41的实物图及结构示意图。如图1的(a)-(b)所示,mcm-41 的主要组分为具有多孔形貌结构的高比表面积sio2材料。
[0137]
图1的(c)示出mcm-41的n2分子吸脱附曲线,mcm-41的bet 比表面积为932.6m2/g-1
。
[0138]
图1的(d)示出mcm-41的孔径分布曲线,材料的孔径主要分布 在3.032nm附近。
[0139]
mcm-41作为制备sic的原料是有益的。这是因为mcm-41的微观 结构具有高比表面积(932.6m2/g)和多孔的结构特征,这样微观结构可 以作为外层碳材料的生长模板。
[0140]
mcm-41分子筛中sio2的壁厚可通过以下公式5.7进行计算:
[0141][0142]
图1的(a)示出的mcm-41的x射线衍射图谱,可以获得mcm
‑ꢀ
41的d(100)为3.831nm。图1的(d)示出的mcm-41的由孔尺寸为 3.032nm。因此,计算的mcm-41的壁厚为1.392nm。上述比较薄的壁 厚有利增强热量和物质传递过程,进而促进高温环境下sio2与c的充分 反应。
[0143]
图2的(a)和(b)分别示出mcm-41的扫描电子显微镜图像及 对应的edx元素分布图。图2的(c)(d)(e)分别示出不同倍数 mcm-41的透射电子显微镜图像。图2的(f)示出mcm-41的x射线 光电子能谱。图2的(g)示出mcm-41的si 2p的x射线光电子能谱。 图2的(h)示出mcm-41的o1s的x射线光电子能谱。
[0144]
如图2的(a)-(h)所示:sem及tem图像分析表明mcm-41 为近球形的尺寸约为~100nm的颗粒,组成元素为si和o。xps分析进 一步表明,si与o的原子比1:2.1,si元素的价态为 4价,氧的价态为
ꢀ‑
2价。以上说明mcm-41的主要成分为sio2。
[0145]
2、第一前驱体mcm-41@c的特征
[0146]
图3的(a)示出了mcm-41@c制备示意图,局域放大图为蔗糖分 子的结构示意图。如图所示,mcm-41在蔗糖溶液中充分浸泡并烘干后 获得mcm-41@蔗糖,其中mcm-41的表面被蔗糖分子充分覆盖。mcm
‑ꢀ
41@蔗糖再经碳化处理后获得mcm-41@c,得mcm-41的表面被碳覆 盖层充分地覆盖。
[0147]
mcm-41表面的碳覆盖层对于sic的形成是十分有利的。碳覆盖层 的一个作用是截留mcm-41表面上形成的sio中间反应物分子。具体而 言,在低压环境下,mcm-41表面上形成的sio中间反应物分子容易直 接被真空泵抽离腔体,进而无法被碳材料俘获并参与后续反应生成sic。 在上述实施例的mcm-41@c中,覆盖mcm-41表面的碳覆盖层能够有 效地俘获sio中间反应物分子,使其能够参与后续反应形成sic。
[0148]
图3的(b)示出了mcm-41@c的扫描电子显微镜图。如图所示, mcm-41@c仍具有明显的颗粒状形貌,保持了较高的比表面积。
[0149]
图3的(c)示出了mcm-41@c的n2分子吸脱附曲线,根据bet 理论计算,mcm-41@c的bet比表面积为840.2m2/g。
[0150]
3、sic产物的表征
[0151]
图5示出不同加热温度所得产物的x射线衍射图谱。图5右上角插 图为β-sic的原
子结构示意图(立方相;空间群:no.216);sic 是由sp3杂化的si-c键所构成的共价化合物。图5左侧的虚线点圈所标 注的衍射峰是由于sic晶粒中的堆垛位错引起的。如图5所示,1100℃ 所制备的样品(对比例1)未显现出任何sic晶面相对应的衍射峰,这主 要是因为该温度不足以驱动sio2和碳反应物前驱体跨越反应的能量势垒, 难以触发碳热还原反应。当制备温度由1100℃升高至1200℃时,在35.7
°ꢀ
位置出现了一个对应于β-sic相(111)面的衍射峰(pdf:03-065-0360)。 该xrd结果证实低压环境可以显著降低sic制备的热力学势垒,使得 以往需要1400℃以上高温环境来驱动反应发生的温度降低至1200℃,降 低了近200℃。而且,制备sic的反应速率大大提升,仅在30秒的时间 即可制备出sic。
[0152]
图6的(a)示出实施例1.2的产物sic的透射电子显微镜(tem) 图像。图6的(b)示出实施例1.2的产物sic的高分辨透射电子显微镜 (hr-tem)图像。图6的(c)示出实施例1.2的产物sic的选区电子 衍射图像。tem图像显示sic纳米颗粒呈现出不规则的形貌且表面粗糙 (见图6的a)。hr-tem(见图6的b)和电子衍射图(见图6的c) 进一步表明sic纳米颗粒由多晶的具有不同晶面取向的sic组成。此外, 在sic的晶粒中观测到锯齿状的堆垛层错(stacking fault,点圈标注, 见图6的b),这是由于sic相对较低的堆垛层错能所引起的典型结构 缺陷。
[0153]
4、金属碳化物的表征
[0154]
图8的(a)示出金属氧化物nb2o5和制备的金属碳化物nbc的x 射线衍射光谱。图8的(b)示出金属氧化物moo3和制备的金属碳化物 mo2c的x射线衍射光谱。图8的(c)示出金属氧化物nb2o5和制备的 金属碳化物nbc的x射线衍射光谱。图8的(d)示出金属氧化物nb2o5和制备的金属碳化物wc的x射线衍射光谱。
[0155]
如图8的(a)-(d)所示,实施例3.1-实施例3.4采用低压碳热还 原策略在较低的温度(1300℃)和较短的时间(20秒)内成功制备了金 属碳化物nbc、mo2c、nbc和wc。
[0156]
5.高熵碳化物
[0157]
图9的(a)示出vnbmotawc5高熵碳化物的x射线衍射图谱。 图9的(b)示出vnbmotawc5高熵碳化物的高倍透射电子显微镜图 像。图9的(c)示出vnbmotawc5的高角环形暗场扫描透射电子显微 镜图像及对应的edx元素分布图。
[0158]
x射线衍射图谱表明实施例4成功地制备出vnbmotawc5高熵碳 化物(图9的a)。高倍的tem照片(图9的b)显示高熵碳化物与未 反应的碳密切相接,晶粒的晶格条纹间距为0.256nm,对应于 vnbmotawc5高熵碳化物的(111)晶面,该结果表明着高熵碳化物的 的形成依次经历了碳热还原待碳化元素的氧化物,以及碳原子扩散进入 金属晶格的界面反应过程。高角环形暗场像-扫描透射电子显微镜 (haadf-stem)以及对应的元素分布(图9的c)表明v、nb、mo、 ta、w元素均匀地分布于同一碳化物颗粒中,再次验证了高熵碳化物的 成功制备。
[0159]
图10的(a)tivnbtawc5高熵碳化物的x射线衍射图谱,插图为 tivnbtawc5高熵碳化物的示意图。图10的(b)和(c)分别示出 tivnbtawc5高熵碳化物的透射电子显微镜图。图10的(d)示出10条 tivnbtawc5高熵碳化物的晶格条纹间距,经计算,平均晶格条纹间距 为0.257nm。
[0160]
图11(a)(b)(c)(d)分别示出tivnbtawc5高熵碳化物的 透射电子显微镜图像。
[0161]
由图9-图11所示,实施例4采用低压碳热还原策略在较低的温度 (1400℃)和较短
的时间(20秒)内成功制备了高熵碳化物 vnbmotawc5和tivnbtawc5。
[0162]
如图10-图11所示,tivnbtawc5高熵碳化物的形貌结构与 vnbmotawc5高熵碳化物类似。
[0163]
6.碳化硅水汽环境热稳定性测试。
[0164]
对实施例2采用真空烧结炉制备的碳化硅进行高比表面积碳化硅的 水汽环境热稳定性测试。
[0165]
图12的(a)展示了高温水蒸气和空气混合处理sic粉末在不同时 间(0h、25h、50h、100h、200h)后的实物图。如图所示,该测试在 650℃的水蒸气饱和环境下进行。首先,将碳化硅粉末置于刚玉坩埚中, 并将坩埚放入管式炉120的中心恒温区域。随后,按照图中箭头方向, 以空气为载气流过含有沸腾水的洗气瓶后通入管式炉120的石英管中, 加热至650℃进行保温并记录保温时间。长时间的稳定性测试采取间歇 性加热、冷却、取样、重复加热的方案进行。
[0166]
图12的(b)展示了高温水蒸气和空气混合处理sic粉末在不同时 间(0h、25h、50h、100h、200h)后的x射线衍射图谱;图12的(c) 示出实施例2的真空烧结炉实物图,及在1300℃、0.97pa条件下获得的 sic产物的实物称重图。
[0167]
图13的(a)示出sic的差热热重曲线。图13的(b)示出100小 时水汽高温热处理sic后n2分子的吸脱附曲线。图13的(c)示出650℃ 水汽饱和空气流高温处理不同时间(0h、100h、200h)sic后的x射 线光电子能谱,xps谱图显示了在漫长的加速氧化实验过程中sic逐渐 发生氧化;在200h后si-c峰消失,表明sic几乎被完全氧化。
[0168]
如图12-图13所示,低压碳热还原所制备高比表面积sic的水汽环 境热稳定性测试在650℃载有饱和水蒸气的持续空气流中进行。高温环 境下的水分子和氧分子对处于其中的材料具有强烈的腐蚀性,将加速sic 的快速氧化分解。差热热重分析(tga-dsc)首先被用于评估高比表面 积sic的热稳定性和抗氧化性,差热热重曲线表明sic在664℃之前保 持稳定,温度进一步升高后sic逐步发生氧化(见图13的a)。在100 小时的水汽环境热处理后,sic的xrd衍射峰和实物图几乎无变化(见 图12的b)。sic的bet比表面积测试显示其比表面积为536.4m2/g, 相较于初始569.9m2/g的比表面积保留了94.1%(图13的b)。这些结 果表明低压碳热还原策略所制备的高比表面积sic具有优异的抗氧化能 力和热稳定性,有望为工业异相催化领域提供理想的高比表面积、高化 学和热稳定性、机械性能优异的载体材料。当高温水汽热处理时间增长 至200h,sic的颜色有灰绿色变成浅灰色(图12的a),对应的xrd 衍射峰变弱,表明sic发生了氧化,对不同状态下sic样品的xps分析 (图13的c)进一步验证了该结果,如图13的c所示,sic样品由最 初的只有si-c特征峰(0h),转变为si-c特征峰与si-o特征峰共存 (100h),再转变为只有si-o特征峰(200h)。由此可见,在提高sic 载体比表面积的同时,改善sic的本征热稳定性对于实际工业化应用具 有同样重要的意义,例如可通过掺杂的策略来提高sic的抗氧化能力。
[0169]
综上所述,本技术为基于勒夏特列原理的快速合成难熔碳化物提供 了一种通用的低压碳热还原策略。本技术首次提出并通过实验验证了低 压碳热还原策略能够在更低的温度和更快的速率制备碳化物。
[0170]
本技术实施例1.2在约1300℃的加热温度,在30秒的时间,在约50 pa的低压环境下,合成高比表面积高达569.9m2/g的sic。
[0171]
本技术实施例2通过工业化的真空烧结炉,验证了低压碳热还原策 略在宏量化制备高比表面积sic的技术可行性,单次产量大于12g,这对 sic的工业应用具有重要意义。本技术制备的高比表面积sic具有良好 的热稳定性和抗氧化能力,能够在650℃载有饱和蒸汽的空气气流中保 持100小时的结构稳定。
[0172]
本技术实施例3将低压碳热还原策略应用在快速合成难熔金属碳化 物(nbc,mo2c,tac,wc)和实施例4制备了难熔金属高熵碳化物 (vnbmotawc5,tivnbtawc5)。这些难熔金属碳化物通常是非常难以 获得的。因此,对于低压碳热还原策略的深入理解和理性的控制,为多种 基于碳化物的先进功能材料的制备提供了一条理想的途径。
[0173]
尽管本发明的实施方式已经得到详细的描述,但本领域技术人员 将理解:根据已经公开的所有教导,可以对细节进行各种修改和变动, 并且这些改变均在本发明的保护范围之内。本发明的全部范围由所附 权利要求及其任何等同物给出。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。