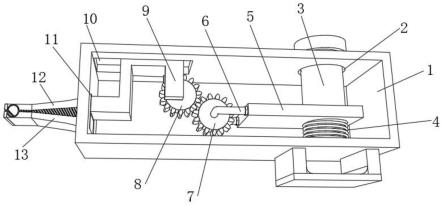
抑制酪蛋白激酶的方法
背景技术:
1.生物钟将我们每天的睡眠和活动周期与外部环境联系起来。生物钟失调与许多人类疾病有关,包括抑郁症、季节性情绪失调和代谢紊乱。例如,生物钟可调节多种下游节律,例如睡眠和觉醒、体温和激素分泌的节律(ko和takahashi,hum mol gen 15:r271-r277.)。此外,抑郁症、季节性情绪失调和代谢紊乱等疾病可能与昼夜节律有关(barnard和nolan,plos genet。2008年5月;4(5):e1000040。)。
2.生物钟蛋白的磷酸化是控制生物钟周期性节律的重要因素。酪蛋白激酶iδ(ck1δ)或ck1ε是密切相关的ser-thr蛋白激酶,它们作为关键的生物钟调节剂,如哺乳动物的突变所证实,各自均极大地改变着昼夜节律。因此,ck1δ/ε的抑制剂可用于治疗昼夜节律紊乱和其他相关疾病。
技术实现要素:
3.本技术提供抑制ck1δ和/或ck1ε的方法以及治疗相关疾病和/或病症的方法和组合物。
4.一方面,本技术提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式i化合物或其药学上可接受的盐:其中ar1为任选地经一个或多个(例如,1至2个或2个以上)r1取代基取代的芳基,且r1各自独立地选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、全氟烃氧基、羟基、巯基、羟基羰基、芳氧基、芳硫基、磺酰基或亚砜基,其中硫原子上的取代基为烃基、磺酰胺,其中磺酰胺基氮原子上的取代基为氢化物离子(hydrido)或烃基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烃氧基羰基烃氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、
杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基、n-单取代或n,n-二取代氨基烃基、其中氨基烃基氮原子上的取代基选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基形成5至8元杂环基或杂芳基环基,氨基和n-单取代或n,n-二取代氨基,其中氨基氮上的取代基选自以下组成的群组:氢化物离子、烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基、烃酰基、芳基磺酰基和烃基磺酰基,或其中所述氨基氮与同其连接的两个取代基形成5至8元杂环基或杂芳基环基;其中x1、x2和x3各自独立地为c或n;r7不存在或为-cn;a不存在、为或环a,其中r2为-nh2、-cn或z,其中z独立地选自以下组成的群组:氢化物离子、烃基、卤素、羧基、氰基、叠氮基、烃基磺酰基、羰基氧基烃基、羰基酰胺基和-x-y,其中-x为-o、-s或-nq,-y为氢化物离子、烃基或烃基芳基,q为氢化物离子、烃基、羟基烃基、2-、3-或4-吡啶基烃基或芳基烃基,其中r3为卤素或-cn,其中环a为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述环a任选地经r4取代基取代,且r4为=o;b不存在、为或环b,其中r5独立地选自以下组成的群组:-coo-c
1-c6烷基、-co-r
10
和r
52
,其中r
10
为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述r
10
任选地经r8取代基取代,所述r8取代基为c
1-c6烷基,其中r
52
独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、卤代烃基、全卤代烃基、烃氧基羰基、n-哌嗪基羰基、氨基羰基、哌嗪基和经一个或多个取代基取代的芳基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基-烃基、杂环氧基、羟基羰基-烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基
羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基-磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮上的取代基选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基形成5至8元杂环基或杂芳基环基,其中r6为c
1-c6烷基或r
61
,其中r
61
独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、烃基氨基、卤代烃基、全卤代烃基和任选地经一个或多个取代基取代的芳基取代基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮原子上的取代基选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基形成5至8元杂环基或杂芳基环基,其中环b为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述环b任选地经r9取代基取代,且所述r9为pmb。
5.在一些实施方式中,在式i化合物中,r1为f。
6.在一些实施方式中,式i化合物包含所述一个r1取代基。
7.在一些实施方式中,在式i化合物中,r3为f或-cn。
8.在一些实施方式中,在式i化合物中,a为以下之一:在一些实施方式中,在式i化合物中,a为以下之一:
9.在一些实施方式中,在式i化合物中,r5为-coo-ch2ch3或-co-r
10
。
10.在一些实施方式中,在式i化合物中,r
10
为
11.在一些实施方式中,在式i化合物中,r8为-ch3。
12.在一些实施方式中,在式i化合物中,r6为-ch3。
13.在一些实施方式中,在式i化合物中,b为以下之一:中,在式i化合物中,b为以下之一:
14.在一些实施方式中,式i化合物选自以下组成的群组:
15.在一些实施方式中,在式i化合物中,x1为c,x2为c,x3为n,r7不存在,a为且b为其中r2为z,z为-nr
23r24
,r
23
为氢化物离子或c
1-c6烃基,r
24
独立地选自以下组成的群组:氢化物离子、低级烃基、芳基低级烃基、羟基低级烃基和2-吡啶基低级烃基、3-吡啶基低级烃基和4-吡啶基低级烃基,ar1为经卤素或卤基、低级烃基或烃氧基取代的芳基,r6为r
61
,r
61
为氢化物离子或c
1-c6烃基,且r5为r
52
,r
52
为氢化物离子或低级烃基。
16.在一些实施方式中,在式i化合物中,x1为c,x2为c,x3为n,r7不存在,a为且b为其中r2为z,z为-or
25
,r
25
为氢化物离子、c
1-c6烃基或芳基低级烃基,ar1为独立地经卤素、低级烃基或烃氧基取代的芳基,r6为r
61
,r
61
为c
1-c6烃基,r5为r
52
,r
52
为氢化物离子。
17.在一些实施方式中,在式i化合物中,x1为c,x2为c,x3为n,r7不存在,a为且b为其中r2为-cn,ar1为经卤素、低级烃基或烃氧基取代的芳基,r6为r
61
,r
61
为低级烃基,且r5为r
52
,r
52
为氢化物离子或低级烃基。
18.另一方面,本技术提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式ii
化合物或其药学上可接受的盐:其中r1为卤素,n为0、1或2,x1、x2、x3、x4、x5和x6各自独立地为c或n,r2不存在或为=o,r3不存在或为-cn,a不存在、为或环a,其中r4为卤素,其中r5选自以下组成的群组:-nh2、c
1-c6烷基、c
1-c6烷基-nh-c
1-c6烷基、c
1-c6烷基-oh和c
1-c6烷基-o-co-c
1-c6烷基,其中环a为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述环a任选地经r6取代基取代,r6为=o,b为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述b任选地经一个或多个r7取代基取代,且其中r7各自独立地选自以下组成的群组:co-c
1-c6烷基、bn、=o、c
1-c6烷基、co-nh-c
1-c6烷基、co-c
1-c6烷基-cn、r8、r
8-c
1-c6烷基和co-r
8-c
1-c6烷基,其中r8为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换。
19.在一些实施方式中,在式ii化合物中,r1为f。
20.在一些实施方式中,在式ii化合物中,n为1。
21.在一些实施方式中,在式ii化合物中,n为2。
22.在一些实施方式中,在式ii化合物中,r4为f。
23.在一些实施方式中,在式ii化合物中,r5选自以下组成的群组:-nh2、-ch3、-ch
2-nh-ch3、-ch
2-oh和-ch
2-o-co-ch3。
24.在一些实施方式中,在式ii化合物中,a选自以下组成的群组:
25.在一些实施方式中,在式ii化合物中,r7选自以下组成的群组:-bn、=o、-ch3、-co-ch3、-co-ch
2-ch3、-co-ch-(ch3)2、-co-nh-ch3、-co-ch
2-cn、r8、r
8-ch3和-co-r
8-ch3,其中r8为
26.在一些实施方式中,在式ii化合物中,r7的数量为1或2。
27.在一些实施方式中,在式ii化合物中,b选自以下组成的群组:
28.在一些实施方式中,式ii化合物选自以下组成的群组:
29.另一方面,本技术还提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式iii化合物或其药学上可接受的盐:其中r1为卤素,n为0、1或2,x1和x2各自独立地为c或n,a不存在、为或环a,其中r2为c
1-c6烷基,环a为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述环c任选地经r3取代基取代,r3为=o,
c为或环c,其中r4为-cn、-coo-c
1-c6烷基或酰胺基,r5为c
1-c6烷基,且环c为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述环c任选地经r6取代基取代,r6为=o。
30.在一些实施方式中,在式iii化合物中,r1为f。
31.在一些实施方式中,在式iii化合物中,n为1。
32.在一些实施方式中,在式iii化合物中,r2为-ch3。
33.在一些实施方式中,在式iii化合物中,a为
34.在一些实施方式中,在式iii化合物中,r4为-cn、-co-nh2或-coo-ch3。
35.在一些实施方式中,在式iii化合物中,r5为-ch3。
36.在一些实施方式中,在式iii化合物中,c选自以下组成的群组:在一些实施方式中,在式iii化合物中,c选自以下组成的群组:
37.在一些实施方式中,式iii化合物选自以下组成的群组:
38.另一方面,本技术提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式iv化合物或其药学上可接受的盐:
其中r1为卤素,n为0、1或2,r2为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述r2为任选地经r3取代基取代,且r3为=o。
39.在一些实施方式中,在式iv化合物中,r1为f。
40.在一些实施方式中,在式iv化合物中,n为1。
41.在一些实施方式中,在式iv化合物中,r2为
42.在一些实施方式中,式iv化合物选自以下组成的群组:
43.在一些实施方式中,所述方法为活体外方法、离体方法或活体内方法。
44.另一方面,本技术还提供一种治疗哺乳动物神经和/或精神疾病或病症的方法,其包括向该哺乳动物施用治疗有效量的式i至iv中任一项的化合物或其药学上可接受的盐。
45.在一些实施方式中,所述疾病或病症为情绪障碍、睡眠障碍或昼夜节律紊乱。
46.在一些实施方式中,所述情绪障碍选自以下组成的群组:抑郁症和躁郁症。
47.在一些实施方式中,在本技术的方法中,施用选自以下组成的群组的化合物或其药学上可接受的盐:4-(2-(4-氟苯基)-4,5,6,7-四氢-2h-吲唑-3-基)嘧啶-2-胺、4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)呋喃并[3,4-b]吡啶-5(7h)-酮、4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)呋喃并[3,4-b]吡啶-7(5h)-酮、2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢-2h-吲唑、4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)吡啶、2-(4-氟苯基)-3-(吡啶-4-基)-2,4,5,6-四氢环戊[c]吡唑、2-(4-氟苯基)-5-(4-甲氧基苄基)-3-(吡啶-4-基)-4,5,6,7-四氢-2h-吡唑并[4,
3-c]吡啶、3-氟-4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)吡啶甲腈、4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)吡啶-2,3-二甲腈、4-(1-(4-氟苯基)-1h-1,2,3-三唑-5-基)吡啶、1-(4-氟苯基)-5-(吡啶-4-基)-1h-1,2,3-三唑-4-甲酸乙酯、(1-(4-氟苯基)-5-(吡啶-4-基)-1h-1,2,3-三唑-4-基)(4-甲基哌嗪-1-基)甲酮、1-(4-氟苯基)-5-(吡啶-4-基)-1h-吡咯-2-甲腈、4-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)嘧啶-2-胺、4-(2-(4-氟苯基)-5,6,7,8-四氢咪唑并[1,2-a]吡嗪-3-基)嘧啶-2-胺、4-(2-(4-氟苯基)-7-(1-甲基哌啶-4-基)-5,6,7,8-四氢咪唑并[1,2-a]吡嗪-3-基)嘧啶-2-胺、1-(3-(2-氨基嘧啶-4-基)-2-(4-氟苯基)-5,6,7,8-四氢咪唑并[1,2-a]吡嗪-7-基)乙-1-酮、2-(4-氟苯基)-3-(吡啶-4-基)咪唑并[1,2-a]吡嗪、2-(4-氟苯基)-3-(吡啶-4-基)-5,6,7,8-四氢咪唑并[1,2-a]吡嗪、1-(2-(4-氟苯基)-3-(吡啶-4-基)-5,6-二氢咪唑并[1,2-a]吡嗪-7(8h)-基)乙-1-酮、4-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)呋喃并[3,4-b]吡啶-5(7h)-酮、4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)嘧啶-2-胺、4-(2-(4-氟苯基)-5-(1-甲基哌啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)嘧啶-2-胺、1-(3-(2-氨基嘧啶-4-基)-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮、5-苄基-2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪、2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪、1-(2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮、4-(2-(4-氟苯基)-4,5,6,7-四氢-1h-苯并[d]咪唑-1-基)嘧啶-2-胺、2-(4-氟苯基)-1-(吡啶-4-基)-4,5,6,7-四氢-1h-苯并[d]咪唑、1-(2-(4-氟苯基)-3-(2-((甲氨基)甲基)吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-5-基)乙-1-酮、1-(2-(4-氟苯基)-3-(2-(羟基甲基)吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-5-基)乙-1-酮、(4-(2-(4-氟苯基)-5-(哌嗪-1-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)甲醇、1-(2-(4-氟苯基)-3-(嘧啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-5-基)乙-1-酮、1-(2-(4-氟苯基)-3-(哒嗪-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮、
酮、(r)-5-(3-(4-氟苯基)-4-(吡啶-4-基)异恶唑-5-基)二氢呋喃-2(3h)-酮,和(r)-4-(3-(4-氟苯基)-4-(吡啶-4-基)异恶唑-5-基)二氢呋喃-2(3h)-酮。
[0048]
本领域技术人员能够从下文的详细描述中容易地洞察到本技术的其它方面和优势。下文的详细描述中仅显示和描述了本技术的说明性实施方式。如将认识到的,本技术能够具有其他和不同的实施方式,并且其若干细节能够在各种明显方面进行修改,而所有这些均不背离本技术。相应地,附图和描述应视为示例性的,而非为限制性的。通过引用并入
[0049]
本说明书中提及的所有公布、专利和专利申请均以引用的方式并入本文,其程度与每个单独的公布、专利或专利申请被具体且单独地指示以引用的方式并入的程度相同。
附图说明
[0050]
本发明的新颖特征在随附的权利要求书中进行具体阐述。通过参考以下阐述说明性实施方式的详细描述,其中采用了本发明的原理,以及附图(本文中也称为“图”),将获得对本技术的特征和优势的更好理解,其中:
[0051]
图1说明化合物2-1的合成流程。
[0052]
图2说明化合物3-8的合成流程。
[0053]
图3说明化合物2-5、2-6、2-7和2-8的合成流程。
[0054]
图4说明化合物1-2的合成流程。
[0055]
图5说明化合物2-12、2-13和2-14的合成流程。
[0056]
图6说明化合物1-4的合成流程。
[0057]
图7说明化合物2-16的合成流程。
[0058]
图8说明化合物3-4、3-5、3-2和3-1的合成流程。
[0059]
图9说明化合物1-10的合成流程。
[0060]
图10说明化合物3-7的合成流程。
[0061]
图11说明化合物2-27的合成流程。
[0062]
图12说明化合物2-28的合成流程。
[0063]
图13说明化合物2-29和2-31的合成流程。
[0064]
图14说明化合物2-30的合成流程。
[0065]
图15说明化合物2-33的合成流程。
[0066]
图16说明化合物2-34的合成流程。
[0067]
图17说明化合物2-32的合成流程。
[0068]
图18说明化合物2-36的合成流程。
[0069]
图19说明化合物1-5的合成流程。
[0070]
图20说明化合物1-13的合成流程。
[0071]
图21-24说明化合物2-37至2-40的合成流程。
[0072]
图25说明化合物3-11的合成流程。
[0073]
图26说明化合物2-41的合成流程。
[0074]
图27说明化合物3-12的合成流程。
[0075]
图28-29说明化合物2-42至2-43的合成流程。
[0076]
图30说明化合物3-13的合成流程。
[0077]
图31-36说明化合物2-44至2-49的合成流程。
[0078]
图37说明化合物3-14的合成流程。
[0079]
图38-49说明化合物2-50至2-61的合成流程。
具体实施方式
[0080]
尽管本文已经示出和描述了本发明的各种实施方式,但对于本领域技术人员来说显而易见的是,这些实施方式仅作为示例提供。在不背离本发明的情况下,本领域技术人员可以想到多种变化、改变和替换。应了解,可以采用本文所述的本发明实施方式的各种替代方案。
[0081]
定义
[0082]
如本文所用,术语“烷基”通常是指含有1至20个碳原子的直链或分支链饱和烃基取代基(即,从烃中去除氢而得到的取代基);例如,含有1至12个碳原子;在另一示例中,含有1至10个碳原子;在另一种实施方式中,含有1至6个碳原子;以及在另一种实施方式中,含有1至4个碳原子(诸如,1个、2个、3个或3个以上碳原子)。这些取代基的实例包括,例如,甲基、乙基、丙基(包括正丙基和异丙基)、丁基(包括正丁基、异丁基、仲丁基和叔丁基)、戊基、异戊基、己基等。在一些情形下,烃基取代基(即,烷基、烯基、环烷基、芳基等)中碳原子的数量由前缀“c
a-c
b”表示,其中a为取代基中碳原子的最小数量且b为最大数量。因此,例如,“c
1-c6烷基”是指含有1至6个碳原子的烷基取代基。例如,烷基可任选地经进一步取代。
[0083]
如本文所用,术语“环烷基”通常是指从饱和碳环分子中去除氢得到的具有3至14个碳原子的碳环取代基。在一种实施方式中,环烷基取代基具有3至10个碳原子。环烷基可为单环,通常含有4至7个环原子。环烷基的实例包括环丙基、环丁基、环戊基和环己基。或者,环烷基可为稠合在一起的2个或3个环,诸如双环[4.2.0]辛烷和十氢萘基,而且也可以称为“双环烷基”。例如,环烷基可任选地经进一步取代。
[0084]
如本文所用,术语“环烷基”还包括与c
6-c
10
芳环或5至10元杂芳环稠合的取代基,其中具有这种稠合环烷基作为取代基的基团与环烷基的碳原子结合。当这种稠合环烷基经一个或多个取代基取代时,除非另有规定,否则该一个或多个取代基各自与环烷基的碳原子结合。稠合c
6-c
10
芳环或5至10元杂芳环可任选地经进一步取代。例如,环烷基可任选地经进一步取代。
[0085]
如本文所用,术语“卤素”通常是指氟(可表示为-f)、氯(可表示为-cl)、溴(可表示为-br)或碘(可表示为-i)。在一种实施方式中,卤素为氯。在另一种实施方式中,卤素为氟。在另一种实施方式中,卤素为溴。
[0086]
如本文所用,术语“杂环烷基”通常是指从总计含有4至14个环原子的饱和或部分饱和环结构中去除氢得到的取代基,其中至少一个环原子为杂原子(例如,非c原子),诸如氧、氮或硫。例如,如本文所用,术语“4至7元杂环烷基"是指取代基为总计具有4至7个成元的单环。或者,杂环烷基可包含稠合在一起的2个或3个环,其中至少一个这种环含有杂原子作为环原子(即,氮、氧或硫)。在具有杂环烷基取代基的基团中,与该基团结合的杂环烷基取代基的环原子可为该至少一个杂原子,或可为环碳原子,其中该环碳原子可与该至少一
个杂原子在同一环中或其中该环碳原子可与该至少一个杂原子位于不同的环中。类似地,如果杂环烷基取代基反过来经一个基团或取代基取代,则该基团或取代基可与该至少一个杂原子结合,或可与环碳原子结合,其中该环碳原子可与该至少一个杂原子在同一环中或其中该环碳原子可与该至少一个杂原子位于不同的环中。例如,杂环烷基可任选地经进一步取代。
[0087]
如本文所用,术语“杂环烷基”还包括与c
6-10
芳环或5至10元杂芳环稠合的取代基,其中具有这种稠合杂环烷基作为取代基的基团与杂环烷基的杂原子结合或与杂环烷基的碳原子结合。当这种稠合杂环烷基经一个或多个取代基取代时,除非另有规定,否则该一个或多个取代基各自与杂环烷基的杂原子结合或与杂环烷基的碳原子结合。稠合的c
6-c
10
芳环或5至10元杂芳环可任选地经进一步取代。例如,杂环烷基可任选地经进一步取代。
[0088]
如本文所用,术语“杂芳基”通常是指含有5至14个环原子的芳环结构,其中至少一个环原子为杂原子(例如,氧、氮或硫),剩余环原子独立地选自以下组成的群组:碳、氧、氮和硫。杂芳基可为单环或2或3个稠环。杂芳基取代基的实例包括(但不限于):6元环取代基,诸如吡啶基、吡唑基、嘧啶基和哒嗪基;5元环取代基,诸如三唑基、咪唑基、呋喃基、噻吩基、吡唑基、恶唑基、异恶唑基、噻唑基、1,2,3-、1,2,4-、1,2,5-或1,3,4-恶二唑基和异噻唑基;6/5元稠环取代基,诸如苯并硫代呋喃基、异苯并硫代呋喃基、苯并异恶唑基、苯并恶唑基、嘌呤基和邻氨基苯甲酰基;和6/6元稠环取代基,诸如喹啉基、异喹啉基、噌啉基、喹唑啉基和1,4-苯并恶嗪基。在具有杂芳基取代基的基团中,与该基团结合的杂芳基取代基的环原子可为该至少一个杂原子或可为环碳原子,其中该环碳原子可与该至少一个杂原子在同一环中或其中该环碳原子可与该至少一个杂原子位于不同的环中。类似地,如果杂芳基取代基反过来经一个基团或取代基取代,则该基团或取代基可与该至少一个杂原子结合,或可与环碳原子结合,其中该环碳原子可与该至少一个杂原子在同一环中或其中该环碳原子可与该至少一个杂原子位于不同的环中。例如,杂芳基可任选地经进一步取代。
[0089]
如本文所用,术语“杂芳基”还包括与如c5或c6碳环的c
4-10
碳环稠合的、或与4至10元杂环稠合的取代基,如吡啶基和喹啉基,其中具有该稠合杂芳基作为取代基的基团与杂芳基的芳碳或杂芳基的杂原子结合。当这种稠合杂芳基经一个或多个取代基取代时,除非另有规定,否则该一个或多个取代基各自与杂芳基的芳碳或杂芳基的杂原子结合。稠合的c
4-10
碳环或4至10元杂环可任选地经进一步取代。例如,杂芳基可任选地经进一步取代。
[0090]
如本文所用,术语“芳基”通常是指含有一个环或两个或三个稠环的芳香族取代基。芳基取代基可具有6至18个碳原子。举例来说,芳基取代基可具有6至14个碳原子。术语“芳基”可以是指诸如苯基、萘基和蒽基的取代基。术语“芳基”还可以包括与如c5或c6碳环的c
4-10
碳环稠合的、或与4至10元杂环稠合的取代基,如苯基、萘基和蒽基,其中具有该稠合芳基作为取代基的基团与芳基的芳碳结合。当这种稠合芳基经一个或多个取代基取代时,除非另有规定,否则该一个或多个取代基各自与稠合芳基的芳碳结合。稠合的c
4-10
碳环或4至10元杂环可任选地经进一步取代。芳基的实例相应地包括苯基、萘基、四氢萘基(也称为“1,2,3,4-四氢化萘基(tetralinyl)”)、茚基、异茚基、茚满基、蒽基、菲基、苯并萘亚甲基(benzonaphthenyl)(也称为“菔基(phenalenyl)”)和芴基。例如,芳基可任选地经进一步取代。
[0091]
在一些情形下,含有一个或多个杂原子的环状取代基(即,杂芳基或杂环烷基)中
的原子数量由前缀“x至y元”表示,其中x为形成该取代基环状部分的原子的最小数量且y为最大数量。因此,例如,5至8元杂环烷基是指杂环烷基的环状部分中含有5至8个原子(包括一个或多个杂原子)的杂环烷基。
[0092]
如本文所用,术语“氢”通常是指氢取代基,并且可表示为-h。
[0093]
如本文所用,术语“羟基”通常是指-oh。当与另一术语组合使用时,前缀“羟基”通常是指前缀连接的取代基经一个或多个羟基取代基取代。带有连接一个或多个羟基取代基的碳的化合物包括例如醇类、烯醇类和苯酚。例如,羟基可任选地经进一步取代。
[0094]
如本文所用,术语“氰基”(也称为“腈”)通常是指-cn。
[0095]
取代基如果包含至少一个与一个或多个氢原子键结的碳或氮原子,即为“可取代的”或可“经取代”。因此,例如氢、卤素和氰基均不在此定义内。
[0096]
如果取代基被描述为“经取代”,则为非氢取代基代替了该取代基的碳或氮上的氢取代基。因此,例如经取代的烷基取代基是其中至少一个非氢取代基代替了该烷基取代基上的氢取代基的烷基取代基。为进行说明,单氟烷基为经氟基取代基取代的烷基,且二氟烷基为经两个氟基取代基取代的烷基。应该认识到,如果取代基上有一个以上的取代,则每个非氢取代基可以是相同或不同的(除非另有规定)。
[0097]
如本文所用,术语“取代基”、“基”和“基团”可互换使用。
[0098]
如果取代基被描述为“任选地经取代”,则该取代基可(1)未经取代或(2)经取代。如果取代基的碳被描述为任选地经一系列取代基中的一个或多个取代,则该碳上的一个或多个氢(尽可能地)可单独地和/或一起经独立选择的任选取代基置换。如果取代基的氮被描述为任选地经一系列取代基中的一个或多个取代,则该氮上的一个或多个氢(尽可能地)可各自经独立选择的任选取代基置换。一种示例性的取代基可描述为-nr’r”,其中r’和r”连同其连接的氮原子一起可形成包含1或2个独立地选自氧、氮和硫的杂原子的杂环,其中所述杂环烷基部分可任选地经取代。由r’和r”连同其连接的氮原子一起形成的杂环可以是部分或完全饱和的,或是芳香族的。在一种实施方式中,杂环由4至10个原子组成。
[0099]
如果取代基被描述为“独立地选自”某一群组,则每个取代基的选择都与另一(其他)取代基无关。因此,每个取代基可以与另一(其他)取代基相同或不同。
[0100]
如本文所用,术语“式i”(或式ii、式iii或式iv)在下文中可称为“本发明的化合物”。这些术语也定义为包括所有形式的式i(或式ii、式iii或式iv)化合物,包括其水合物、溶剂合物、异构体、结晶和非结晶形式、同构体、多晶型和代谢物。例如,式i化合物或其药学上可接受的盐可存在非溶剂化形式和溶剂化形式。当溶剂或水紧密结合时,复合物将具有明确的化学计量,不受湿度影响。然而,当溶剂或水结合较弱时,如在通道溶剂化物和吸湿性化合物中,水/溶剂的含量将取决于湿度和干燥条件。在这些情形下,非化学计量将成为常态。
[0101]“式i”(或式ii、式iii或式iv)的化合物可具有不对称的碳原子。式i化合物的碳-碳键在本文中可用实线、实心楔形或虚线楔形来表示。使用实线表示与不对称碳原子的键意思是表示包括该碳原子的所有可能的立体异构体(例如,具体为对映异构体、外消旋混合物等)。使用实心楔形或虚线楔形表示与不对称碳原子的键意思是表示只打算包括所示的立体异构体。本技术的化合物可能可含有一个以上的不对称碳原子。在那些化合物中,使用实线表示与不对称碳原子的键意思是表示打算包括所有可能的立体异构体。例如,除非另
有规定,否则意思是式i(或式ii、式iii或式iv)的化合物可存在其对映异构体和非对映异构体或外消旋体及其混合物的形式。使用实线表示与式i(或式ii、式iii或式iv)的化合物中的一个或多个不对称碳原子的键以及使用实心楔形或虚线楔形表示与同一化合物中的其他不对称碳原子的键意思是表示存在非对映异构体的混合物。
[0102]
本技术的化合物(例如,式i、式ii、式iii、式iv、式v或式vi的化合物)可以包合物或其他复合物的形式存在。本发明的范畴中包括复合物,诸如包合物、药物-宿主包涵复合物,其中与前述溶剂合物相反,药物和宿主以化学计量或非化学计量的量存在。还包括含有两种或两种以上可为化学计量或非化学计量的量的有机和/或无机组分的式i(或式ii、式iii或式iv)的复合物。得到的复合物可以是离子化的、部分离子化的或非离子化的。关于这些复合物的综述,参考haleblian的j.pharm.sci.,64(8),第1269-1288页(1975年8月)。
[0103]
式i(或式ii、式iii或式iv)的立体异构体包括式i(或式ii、式iii或式iv)的化合物的顺式和反式异构体、光学异构体(如r和s对映异构体)、非对映异构体、几何异构体、旋转异构体、构象异构体和互变异构体,包括展示一种以上异构类型的化合物;以及其混合物(诸如外消旋体和非对映异构对)。还包括酸加成盐和碱加成盐,其中抗衡离子是光学活性的,例如d-乳酸盐或l-赖氨酸,或是外消旋性的,例如,dl-酒石酸盐或dl-精氨酸。
[0104]
当任何外消旋体结晶时,可能形成两种不同类型的晶体。第一种类型是上文提到的外消旋化合物(真外消旋体),其中产生含有等摩尔量的两种对映异构体的一种均质形式的晶体。第二种类型是外消旋混合物或聚集体,其中产生等摩尔量的两种形式的晶体,各自包含单一的对映异构体。
[0105]
式i(或式ii、式iii或式iv)的化合物可展示互变异构和结构异构现象。例如,式i化合物可存在几种互变异构形式,包括烯醇和亚胺形式、酮和烯胺形式以及几何异构体及其混合物。所有这些互变异构形式都包括在式i(或式ii、式iii或式iv)化合物的范畴内。互变异构体以互变异构集合在溶液中的混合物形式存在。固体形式时,通常一种互变异构体占优势。尽管可能描述一种互变异构体,但本技术包括式i(或式ii、式iii或式iv)化合物的所有互变异构体。
[0106]
本发明还包括经同位素标记的化合物,其与上文式i(或式ii、式iii或式iv)中列举的化合物相同,但实际上一个或多个原子被具有与自然界中通常发现的原子质量或质量数不同的原子质量或质量数的原子置换。可并入式i(或式ii、式iii或式iv)化合物中的同位素的实例包括氢、碳、氮、氧、磷、氟和氯的同位素,诸如(但不限于)2h、3h、
13
c、
14
c、
15
n、
18
o、
17
o、
31
p、
32
p、
35
s、
18
f和
36
cl。某些经同位素标记的式i(或式ii、式iii或式iv)化合物,例如并入诸如3h和
14
c等放射性同位素的化合物,适用于药物和/或底物组织分布测定。氚化(即3h)和碳-14(即
14
c)同位素因其易于制备和可检测性而被使用。此外,经较重同位素(如氘,即2h)取代可提供因代谢稳定性更大而产生的某些治疗优势,例如体内半衰期增加或剂量需求减少,且因此可用于某些情形中。经同位素标记的式i(或式ii、式iii或式iv)化合物通常可通过进行下文流程和/或实施例中公开的程序,通过用经同位素标记的试剂代替未经同位素标记的试剂来制备。
[0107]
本技术的化合物可以源自无机酸或有机酸的盐的形式来使用。根据特定的化合物,化合物的盐因该盐的一种或多种物理性质而可能是有利的,诸如在不同温度和湿度下的药物稳定性增强、或在水或油中的溶解性有利。在一些情形下,化合物的盐也在化合物的
分离、纯化和/或解析中用作助剂。
[0108]
除非另有说明,否则如本文所用的术语“治疗(treating)”通常意思是逆转、缓解、抑制该术语所适用的病症或病况的进程、或预防该病症或病况或该病症或病况的一种或多种症状。除非另有说明,否则如本文所用的术语“治疗(treatment)”通常是指如前文定义的“治疗(treating)”的治疗行为。术语“治疗(treating)”也可包括对受试者的辅助性和新辅助性治疗。
[0109]
在意欲将盐施用于患者的情况下(与例如在体外环境中使用相反),该盐可以是药学上可接受的。术语“药学上可接受的盐”通常是指通过将本技术的化合物(例如,式i至iv的任一种化合物)与酸或碱组合制备的盐,所述酸的阴离子或碱的阳离子通常认为适合人类食用。药学上可接受的盐因其相对于母化合物具有更大的水溶解度而尤其适合作为本技术方法的产物。对于医学使用而言,本技术的化合物的盐是无毒的“药学上可接受的盐”。术语“药学上可接受的盐”中所涵盖的盐通常是指通常通过使游离碱与合适的有机或无机酸反应制备的本技术的化合物的无毒盐。
[0110]
本技术的化合物通常以对治疗本文所述病况有效的量来施用。本发明的化合物通过任何合适途径、以适合该途径的药物组合物形式,并以对预期治疗有效的剂量来施用。本领域一般技术人员使用在医学领域中熟知的临床前和临床方法,可以容易地确定治疗医学病况进程所需的化合物的治疗有效剂量。如本文中所用的术语“治疗有效量”通常是指在某种程度上能够缓解正在治疗的病症的一种或多种症状的所施用的化合物的量。
[0111]
用途和方法
[0112]
本技术提供一种抑制ck1δ或ck1ε活性的方法。所述方法可包括施用有效量的式i化合物或其药学上可接受的盐:其中ar1可为可任选地经一个或多个(例如,1至2个或2个以上)r1取代基取代的芳基,且r1可各自独立地选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、全氟烃氧基、羟基、巯基、羟基羰基、芳氧基、芳硫基、磺酰基和亚砜基,其中硫原子上的取代基可为烃基、磺酰胺,其中磺酰胺基氮原子上的取代基可为氢化物离子或烃基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烃氧基羰基烃氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨
基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基、n-单取代或n,n-二取代氨基烃基、其中氨基烃基氮原子上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,氨基和n-单取代或n,n-二取代氨基,其中氨基氮上的取代基可选自以下组成的群组:氢化物离子、烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基、烃酰基、芳基磺酰基和烃基磺酰基,或其中所述氨基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基;其中x1、x2和x3可各自独立地为c或n;r7可不存在或为-cn;a可不存在,为或环a,其中r2可为-nh2、-cn或z,其中z可独立地选自以下组成的群组:氢化物离子、烃基、卤素、羧基、氰基、叠氮基、烃基磺酰基、羰基氧基烃基、羰基酰胺基和-x-y,其中-x可为-o、-s或-nq,-y可为氢化物离子、烃基或烃基芳基、q可为氢化物离子、烃基、羟基烃基、2-、3-或4-吡啶基烃基或芳基烃基,其中r3可为卤素或-cn,其中环a可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环a可任选地经r4取代基取代,且r4可为=o;b可不存在,为或环b,其中r5可独立地选自以下组成的群组:-coo-c
1-c6烷基、-co-r
10
和r
52
,其中r
10
可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述r
10
可任选地经r8取代基取代,所述r8取代基可为c
1-c6烷基,其中r
52
可独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、卤代烃基、全卤代烃基、烃氧基羰基、n-哌嗪基羰基、氨基羰基、哌嗪基和可经一个或多个取代基取代的芳基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基-烃基、杂环氧基、羟基羰基-烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、
杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基-磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,其中r6可为c
1-c6烷基或r
61
,其中r
61
可独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、烃基氨基、卤代烃基、全卤代烃基和可任选地经一个或多个取代基取代的芳基取代基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮原子上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,其中环b可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环b可任选地经r9取代基取代,且所述r9可为pmb。
[0113]
在一些情形下,式i化合物可包含式ia化合物:其中r1可各自独立地选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟
烃基、三氟甲基烃基、全氟烃氧基、羟基、巯基、羟基羰基、芳氧基、芳硫基、磺酰基和亚砜基,其中硫原子上的取代基可为烃基、磺酰胺,其中磺酰胺基氮原子上的取代基可为氢化物离子或烃基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烃氧基羰基烃氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基、n-单取代或n,n-二取代氨基烃基、其中氨基烃基氮原子上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,氨基和n-单取代或n,n-二取代氨基,其中氨基氮上的取代基可选自以下组成的群组:氢化物离子、烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基、烃酰基、芳基磺酰基和烃基磺酰基,或其中所述氨基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基;其中x1、x2和x3可各自独立地为c或n;r7可不存在或为-cn;a可不存在,为或环a,其中r2可为-nh2、-cn或z,其中z可独立地选自以下组成的群组:氢化物离子、烃基、卤素、羧基、氰基、叠氮基、烃基磺酰基、羰基氧基烃基、羰基酰胺基和-x-y,其中-x可为-o、-s或-nq,-y可为氢化物离子、烃基或烃基芳基,q可为氢化物离子、烃基、羟基烃基、2-、3-或4-吡啶基烃基或芳基烃基,其中r3可为卤素或-cn,其中环a可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环a可任选地经r4取代基取代,且r4可为=o;b可不存在,为或环b,其中r5可独立地选自以下组成的群组:-coo-c
1-c6烷基、-co-r
10
和r
52
,
其中r
10
可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述r
10
可任选地经r8取代基取代,所述r8取代基可为c
1-c6烷基,其中r
52
可独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、卤代烃基、全卤代烃基、烃氧基羰基、n-哌嗪基羰基、氨基羰基、哌嗪基和可经一个或多个取代基取代的芳基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基-烃基、杂环氧基、羟基羰基-烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基-磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,其中r6可为c
1-c6烷基或r
61
,其中r
61
可独立地选自以下组成的群组:叠氮基、氢化物离子、烃基、酰胺基、烃基氨基、卤代烃基、全卤代烃基和可任选地经一个或多个取代基取代的芳基取代基,所述一个或多个取代基选自以下组成的群组:卤素、烃基、烃氧基、硝基、氰基、全氟烃基、三氟甲基烃基、羟基、巯基、羟基羰基、芳氧基、芳硫基、芳基氨基、芳基烃基、芳基、杂芳氧基、杂芳硫基、杂芳基氨基、杂芳基烃基、烃氧基羰基烃基、杂环氧基、羟基羰基烃基、杂环硫基、杂环氨基、环烃氧基、环烃硫基、环烃基氨基、杂芳基烃氧基、杂芳基烃硫基、杂芳基烃基氨基、芳基烃氧基、芳基烃硫基、芳基烃基氨基、杂环基、杂芳基、羟基羰基烃氧基、烷氧基羰基烷氧基、烃酰基、芳基羰基、芳基烃酰基、烃酰氧基、芳基烃酰氧基、羟基烃基、羟基烃氧基、烃硫基、烃氧基烃硫基、烃氧基羰基、羟基羰基烃氧基、烃氧基羰基烃基、烃基羟基羰基烃硫基、烃氧基羰基烃氧基、烃氧基羰基烃硫基、氨基、烃基羰基氨基、芳基羰基氨基、环烃基羰基氨基、杂环烃基羰基氨基、芳基烃基羰基氨基、杂芳基羰基氨基、杂芳基烃基羰基氨基、杂环烃氧基、烃基磺酰基氨基、芳基磺酰基氨基、芳基烃基磺酰基氨基、杂芳基磺酰基氨基、杂芳基烃基磺酰基氨基、环烃基磺酰基氨基、杂环烃基磺酰基氨基和n-单取代或n,n-二取代氨基烃基,其中氨基烃基氮原子上的取代基可选自以下组成的群组:烃基、芳基、芳基烃基、环烃基、芳基烃氧基羰基、烃氧基羰基和烃酰基,或其中所述氨基烃基氮与同其连接的两个取代基可形成5至8元杂环基或杂芳基环基,其中环b可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环b可任选地经r9取代基取代,且所述r9可为pmb。
[0114]
在一些情形下,式i化合物可包含式ia化合物:其中r1可各自独立地为卤素,n可为0、1或2,x1、x2和x3可各自独立地为c或n;r7可不存在或为-cn,a可不存在,为或环a,r2可为-nh2或-cn,r3可为卤素或-cn,环a可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环a可任选地经r4取代基取代,且r4为=o;b可不存在,为或环b,r5可独立地选自以下组成的群组:-coo-c
1-c6烷基和-co-r
10
,其中r
10
可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述r
10
可任选地经r8取代基取代,所述r8取代基可为c
1-c6烷基,其中r6可为c
1-c6烷基,其中环b可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环b可任选地经r9取代基取代,且所述r9可为pmb。
[0115]
在一些情形下,在式i或式ia化合物中,r1可为f。
[0116]
在一些情形下,在式ia化合物中,n可为1。
[0117]
在一些情形下,在式i或式ia化合物中,r3可为f或-cn。
[0118]
在一些情形下,在式i或式ia化合物中,a可为以下之一:在一些情形下,在式i或式ia化合物中,a可为以下之一:
[0119]
在一些情形下,在式i或式ia化合物中,r5可为-coo-ch2ch3或-co-r
10
。
[0120]
在一些情形下,在式i或式ia化合物中,r
10
可为
[0121]
在一些情形下,在式i或式ia化合物中,r8可为-ch3。
[0122]
在一些情形下,在式i或式ia化合物中,r6可为-ch3。
[0123]
在一些情形下,在式i或式ia化合物中,b可为以下之一:在一些情形下,在式i或式ia化合物中,b可为以下之一:
[0124]
在一些情形下,式i化合物或式ia可为表1中的化合物。
[0125]
表1.式i或式ia化合物
[0126]
在一些情形下,在式i化合物中,x1可为c,x2可为c,x3可为n,r7可不存在,a可为且b可为其中r2可为z,z可为-nr
23r24
,r
23
可为氢化物离子或c
1-c6烃基,r
24
可独立地选自以下组成的群组:氢化物离子、低级烃基、芳基低级烃基、羟基低级烃基和2-吡啶基低级烃基、3-吡啶基低级烃基和4-吡啶基低级烃基;ar1可为可经卤素或卤基、低级烃基或烃氧基取代的芳基;r6可为r
61
,r
61
可为氢化物离子或c
1-c6烃基;且r5可为r
52
,r
52
可为氢化物离子或低级烃基。
[0127]
在一些情形下,在式i化合物中,x1可为c,x2可为c,x3可为n,r7可不存在,a可为且b可为其中r2可为z,z可为-or
25
,r
25
可为氢化物离子、c
1-c6烃基或芳基低级烃基;ar1可为可独立地经卤素、低级烃基或烃氧基取代的芳基;r6可为r
61
,r
61
可为c
1-c6烃基;且r5可为r
52
,r
52
可为氢化物离子。
[0128]
在一些情形下,在式i化合物中,x1可为c,x2可为c,x3可为n,r7可不存在,a可为且b可为其中r2可为-cn;ar1可为可经卤素、低级烃基或烃氧基取代的芳基;r6可为r
61
,r
61
可为低级烃基;且r5可为r
52
,r
52
可为氢化物离子或低级烃基。
[0129]
式i化合物还可以包括wo 99/58523中所述的任何化合物,wo 99/58523以全文引
用的方式并入本文中。
[0130]
本技术还提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式ii化合物或其药学上可接受的盐:其中r1可为卤素;n可为0、1或2;x1、x2、x3、x4、x5和x6可各自独立地为c或n;r2可不存在或为=o;r3可不存在或为-cn;a可不存在,为或环a,其中r4可为卤素,其中r5可选自以下组成的群组:-nh2、c
1-c6烷基、c
1-c6烷基-nh-c
1-c6烷基、c
1-c6烷基-oh和c
1-c6烷基-o-co-c
1-c6烷基,其中环a可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环a可任选地经r6取代基取代,r6可为=o;b可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述b可任选地经一个或多个r7取代基取代,其中r7取代基可各自独立地选自以下组成的群组:co-c
1-c6烷基、bn、=o、c
1-c6烷基、co-nh-c
1-c6烷基、co-c
1-c6烷基-cn、r8、r
8-c
1-c6烷基和co-r
8-c
1-c6烷基,其中r8可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换。
[0131]
在一些情形下,在式ii化合物中,r1可为f。
[0132]
在一些情形下,在式ii化合物中,n可为1。
[0133]
在一些情形下,在式ii化合物中,n可为2。
[0134]
在一些情形下,在式ii化合物中,r4可为f。
[0135]
在一些情形下,在式ii化合物中,r5可选自以下组成的群组:-nh2、-ch3、-ch
2-nh-ch3、-ch
2-oh和-ch
2-o-co-ch3。
[0136]
在一些情形下,在式ii化合物中,a可选自以下组成的群组:
[0137]
在一些情形下,在式ii化合物中,r7可各自独立地选自以下组成的群组:-bn、=o、-ch3、-co-ch3、-co-ch
2-ch3、-co-ch-(ch3)2、-co-nh-ch3、-co-ch
2-cn、r8、r
8-ch3和-co-r
8-ch3,其中r8可为
[0138]
在一些情形下,在式ii化合物中,r7的数量可为1或2。
[0139]
在一些情形下,在式ii化合物中,b可选自以下组成的群组:
[0140]
在一些情形下,其中所述a可为
[0141]
在一些情形下,其中所述a可为
[0142]
在一些情形下,其中x1可为c。
[0143]
在一些情形下,其中x6可为n。
[0144]
在一些情形下,其中所述化合物可具有如下结构:
[0145]
在一些情形下,其中r1可为f,n可为1,x1和x3可为c,x2和x6可为n,r2和r3可不存在,r
n1
和r
n2
可独立地任选地经取代,b可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述b可任选地经取代。
[0146]
在一些情形下,其中r1可为f,n可为1,x1和x3可为c,x2和x6可为n,r2和r3可不存在,b可为4至7元环烷基或杂环烷基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述b可任选地经取代。
[0147]
在一些情形下,其中r1可为f,n可为1,x1和x3可为c,x2和x6可为n,r2和r3可不存在,b可选自以下组成的群组:且所述b可任选地经取代。
[0148]
在一些情形下,其中r1可为f,n可为1,x1和x3可为c,x2和x6可为n,r2和r3可不存在,b可为且所述b可任选地经c
1-c6烷基或co-c
1-c6烷基取代。
[0149]
在一些情形下,式ii化合物可为表2-1至2-2中的化合物之一。
[0150]
表2-1.式ii化合物
[0151]
表2-2.式ii化合物
[0152]
本技术还提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式iii化合物或其药学上可接受的盐:
其中r1可为卤素;n可为0、1或2;x1和x2可各自独立地为c或n;a可不存在,为或环a;其中r2可为c
1-c6烷基,环a可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环c可任选地经r3取代基取代,r3可为=o;c可为或环c;其中r4可为-cn、-coo-c
1-c6烷基或酰胺基,r5可为c
1-c6烷基,且环c可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子可经选自=n-和-o-的杂原子置换,且所述环c可任选地经r6取代基取代,r6可为=o。
[0153]
在一些情形下,在式iii化合物中,r1可为f。
[0154]
在一些情形下,在式iii化合物中,n可为1。
[0155]
在一些情形下,在式iii化合物中,r2可为-ch3。
[0156]
在一些情形下,在式iii化合物中,其中所述环a可为5元杂芳基。
[0157]
在一些情形下,在式iii化合物中,其中所述a的2个碳原子可经选自=n-和-o-的杂原子置换。
[0158]
在一些情形下,在式iii化合物中,其中所述环a可为在一些情形下,在式iii化合物中,其中所述环a可为
[0159]
在一些情形下,其中化合物可具有如下结构:其中r1可为卤素,n可为0、1或2,r
a1
、r
a2
和r
a3
可独立地选自c和n。
[0160]
在一些情形下,其中r
a1
可为n。
[0161]
在一些情形下,在式iii化合物中,a可为
[0162]
在一些情形下,在式iii化合物中,r4可为-cn、-co-nh2或-coo-ch3。
[0163]
在一些情形下,在式iii化合物中,r5可为-ch3。
[0164]
在一些情形下,在式iii化合物中,c可选自以下组成的群组:在一些情形下,在式iii化合物中,c可选自以下组成的群组:
[0165]
在一些情形下,式iii化合物可为表3中的化合物之一。
[0166]
表3.式iii化合物
[0167]
本技术还提供一种抑制ck1δ或ck1ε活性的方法,包括施用有效量的式iv化合物或其药学上可接受的盐:其中r1可为卤素;n可为0、1或2;r2可为4至7元环烷基或杂环烷基或5至6元杂芳基,其中至多2个碳原子经选自=n-和-o-的杂原子置换,且所述r2可任选地经r3取代基取代,r3可为=o。
[0168]
在一些情形下,在式iv化合物中,r1可为f。
[0169]
在一些情形下,在式iv化合物中,n可为1。
[0170]
在一些情形下,在式iv化合物中,r2可为
[0171]
在一些情形下,式iv化合物可为表4中的化合物之一。
[0172]
表4.式iv化合物
[0173]
在一些情形下,所述方法可为活体外方法、离体方法或活体内方法。例如,本技术的化合物(例如,式i至式iv化合物)可活体外施用于一个或多个细胞(或细胞系)以控制这些细胞的生长或活性。在另一示例中,本技术的化合物(例如,式i至式iv化合物)可施用于有需要的患者(例如,哺乳动物,诸如人类患者)。
[0174]
本技术还提供一种治疗哺乳动物神经和/或精神疾病或病症的方法,其包括向该哺乳动物施用治疗有效量的式i至iv中任一项的化合物或其药学上可接受的盐。
[0175]
在一些情形下,所述疾病或病症为情绪障碍、睡眠障碍或昼夜节律紊乱。
[0176]
在一些情形下,所述情绪障碍选自以下组成的群组:抑郁症和躁郁症。
[0177]
本技术的方法还可以包括向有需要的受试者施用包含式i、ii、iii或iv的一种或多种化合物或其药学上可接受的盐以及任选地药学上可接受的载剂的药物组合物。
[0178]
本技术的化合物可口服。口服可涉及到吞咽,以便化合物进入胃肠道,或者可采用口颊或舌下施药,通过这种方式,化合物从口中直接进入血流。
[0179]
在另一种实施方式中,本技术的化合物也可以直接施用至血流、肌肉或内部器官中。适合肠胃外施药的方式包括静脉内、动脉内、腹腔内、鞘内、脑室内、尿道内、胸骨内、颅内、肌肉内和皮下。适合肠胃外施药的装置包括针式(包括微针)注射器、无针注射器和输液技术。
[0180]
在另一种实施方式中,本技术的化合物也可以局部施用于皮肤或粘膜,也就是经真皮或透皮施用。在另一种实施方式中,本技术的化合物也可以鼻内或通过吸入来施用。在另一种实施方式中,本技术的化合物可以经直肠或阴道施用。在另一种实施方式中,本技术的化合物也可以直接施用于眼睛或耳朵。
[0181]
化合物和/或含有化合物的组合物的剂量方案基于多种因素,包括患者的类型、年龄、体重、性别和医学病况;病况的严重性;施药途径;以及所采用特定化合物的活性。因此,剂量方案可广泛变化。每天每公斤体重约0.01mg至约100mg级别的剂量水平适用于治疗上述病况。在一种实施方式中,本发明的化合物的总的每日剂量(单次施药或分开施药)通常为约0.01至约100mg/kg,诸如约0.1至约50mg/kg、约0.5至约30mg/kg(即,每公斤体重的本技术化合物的mg数)。在一种实施方式中,剂量为每天0.01至10mg/kg。在另一种实施方式中,剂量为每天0.1至1.0mg/kg。剂量单位组合物可含有所述量或其因数来组成每日剂量。在许多情形下,一天内可重复多次化合物施药(通常不超过4次)。如果需要,通常可使用每天多次给药来增加总的每日剂量。
[0182]
对于口服来说,组合物可以含有0.01、0.05、0.1、0.5、1.0、2.5、5.0、10.0、15.0、25.0、50.0、75.0、100、125、150、175、200、250或500mg有效成分的锭剂形式提供以根据症状调整对患者的剂量。药物通常含有约0.01mg至约500mg的有效成分,或在另一种实施方式中,含有约1mg至约100mg的有效成分。静脉注射的剂量在恒速输注期间可以在每分钟约0.1至约10mg/kg的范围内。
[0183]
根据本技术的合适受试者包括哺乳动物受试者。根据本发明的哺乳动物包括(但不限于)犬科、猫科、牛科、山羊类、马类、绵羊类、猪类、啮齿动物类、兔类、灵长动物类等,并且涵盖在子宫内的哺乳动物。在一种实施方式中,人类为合适的受试者。人类受试者可以是任一性别并处于任何发展阶段。
[0184]
在另一种实施方式中,本技术包括本技术的一种或多种化合物用于制备治疗本文列举的病况的药物的用途。
[0185]
为治疗上文提到的病况,本技术的化合物可以化合物本身的形式施用。或者,药学上可接受的盐因其相对于母化合物具有更大的水溶解度而适用于医学应用。
[0186]
在另一种实施方式中,本技术包括药物组合物。所述药物组合物包含与药学上可接受的载剂一起存在的本技术的化合物。载剂可为固体、液体或两者均可,并且可以与化合物配制成可含有0.05重量%至95重量%的活性化合物的单位剂量组合物,例如锭剂。本技术的化合物可与合适的聚合物偶联作为靶向药物载剂。也可以存在其他药理活性物质。
[0187]
本技术的化合物可通过任何合适途径、以适合该途径的药物组合物形式,并以对预期治疗有效的剂量来施用。活性化合物和组合物例如可口服、经直肠、肠胃外或局部施用。
[0188]
本技术的化合物可单独或与其他治疗剂组合用于治疗各种病况或疾病状态。本技术的化合物与其他治疗剂可同时施用(在同一剂型或单独的剂型中)或依次施用。
[0189]“组合”施用两种或两种以上化合物是指两种化合物的施药时间足够紧密,以至于一种化合物的存在会改变另一种化合物的生物学效应。两种或两种以上化合物可同时、共同或依次施用。另外,可以通过在施药之前将化合物混合或在同一时间点但在不同解剖部位或使用不同的施药途径施用化合物来进行同时施药。
[0190]
短语“共同施药”、“共施药”、“同时施药”和“进行同时施药”意思是组合施用化合物。实施例
[0191]
阐述以下实施例是为了向本领域一般技术人员提供关于如何制造和使用本技术的完整公开和描述,并不旨在限制本发明人视为其发明的范围,也并非旨在表示以下实验是全部或唯一进行的实验。已努力确保所使用的数字(例如,数量、温度等)的准确性,但应考虑到一些实验误差和偏差。除非另有说明,否则部分是指重量份,分子量是指重量平均分子量,温度为摄氏度,且压力为大气压或接近大气压。可以使用标准缩写,例如bp,碱基对;kb,千碱基;pl,皮升;s或sec,秒;min,分钟;h或hr,小时;aa,氨基酸;nt,核苷酸;i.m.,肌肉内;i.p.,腹膜内;s.c.,皮下;等。实施例1.制备化合物2-1
[0192]
图1说明化合物2-1的合成流程。如图1中所示,具体合成步骤如下:
[0193]
步骤1:2-溴-3-(4-氟苯基)-3-氧代丙酸甲酯
[0194]
将nbs(9.98g,56.1mmol)和aibn(0.73g,5.1mmol)加入3-(4-氟苯基)-3-氧代丙酸甲酯(10g,51mmol)于130ml chcl3中的溶液中。将混合物在62℃下搅拌15小时。将反应混合物在压力下浓缩。随后将粗物质加入硅胶柱并用pe/ea(20:1)洗脱。化学式:计算值(m h
)c
10
h8brfo3:275.07,实验值:274.7。
[0195]
步骤2:2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-甲酸甲酯
[0196]
将2-溴-3-(4-氟苯基)-3-氧代丙酸甲酯(14g,50.9mmol)和吡嗪-2-胺(9.68g,101.8mmol)溶解于100ml etoh中。将混合物加热至回流持续18h。在真空下去除溶剂,将残余物通过硅胶柱色谱法纯化并用dcm/meoh(50:1)洗脱。化学式:计算值(m h
)c
14h10
fn3o2:271.25,实验值:271.8。
[0197]
步骤3:2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-甲酸
[0198]
将2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-甲酸甲酯(0.7g,2.6mmol)溶解于meoh(40ml)中。添加溶解于10ml水中的lioh(0.31g,13mmol)。将反应混合物在室温下搅拌15h。用hcl(1mol/l)将反应混合物调整至ph=5-6且随后在减压下浓缩,并将粗产物溶解于乙酸乙酯中并用水洗涤。浓缩溶剂,将粗物质加入硅胶柱并用dcm/meoh(20:1)洗脱。化学式:计算值(m h
)c
13
h8fn3o2:257.22,实验值:257.8。
[0199]
步骤4:2-(4-氟苯基)-n-甲氧基-n-甲基咪唑并[1,2-a]吡嗪-3-甲酰胺
[0200]
将2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-甲酸(0.7g,2.7mmol)和socl2(0.026ml,3.5mmol)在无水dce(30ml)中加热至回流持续1h,之后将混合物蒸发并蒸馏。将所得酰氯溶解于甲苯中并在0℃下经20min时间逐滴添加至n,o-二甲基羟基胺(0.17g,2.7mmol)和k2co3(0.82g,5.8mmol)于h2o
–
甲苯混合物(1:1,100ml)中的搅拌溶液中。搅拌2h后,将有机相用稀的naoh水溶液和h2o洗涤。在真空下蒸发溶剂。将粗物质加入硅胶柱并用pe/ea(10:1)洗脱。化学式:计算值(m h
)c
15h13
fn4o2:300.29,实验值:300.8。
[0201]
步骤5:1-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)乙-1-酮
[0202]
在-78℃下将ch3mgbr于四氢呋喃(thf)(0.26g,2.2mmol)中的3m溶液加入2-(4-氟苯基)-n-甲氧基-n-甲基咪唑并[1,2-a]吡嗪-3-甲酰胺(0.5g,1.7mmol)于thf(20ml)中的搅拌溶液中。使混合物在室温下静置且随后搅拌16h。添加氯化铵饱和溶液,并将混合物用
etoac萃取。在真空下蒸发溶剂,将粗物质加入硅胶柱并用pe/ea(1:2)洗脱。化学式:计算值(m h
)c
14h10
fn3o:255.25,实验值:255.8。
[0203]
步骤6:4-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)嘧啶-2-胺
[0204]
将1,1-二甲氧基-n,n-二甲基甲胺(0.31g,2.5mmol)连续加入1-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)乙-1-酮(100mg,0.4mmol)于正丙醇(20ml)中的溶液中。将反应混合物在92℃下加热3h。然后添加胍(0.12g,1.9mmol)和k2co3(0.25g,1.8mmol)。将反应混合物在92℃下搅拌16h。添加氢氧化钠溶液(5n,50mg)。将反应混合物在92℃下搅拌8h。然后在真空下去除溶剂,并将残余物溶解于etoac中、用水洗涤。通过ptlc纯化残余物以获得产物。化学式:计算值(m h
)c
16h11
fn6:306.3,实验值:306.9。1h nmr(400mhz,cdcl3)δ9.35(dd,j=4.7hz,1.4hz,1h),9.22(s,1h),8.22(d,j=5.2hz,1h),8.05(d,j=4.7hz,1h),7.71-7.67(m,2h),7.18(t,j=8.7hz,2h),6.60(d,j=5.3hz,1h)。
[0205]
实施例2.制备化合物3-8
[0206]
图2说明化合物3-8的合成流程。如图2中所示,具体合成步骤如下:
[0207]
步骤1:(z)-3-((烯丙氧基)亚氨基)-3-(4-氟苯基)丙腈
[0208]
将o-烯丙基羟基氯化铵(1.35g,12.3mmol)和koac(1.2g,12.3mmol)在meoh(5ml)中去中心化。将混合物在25℃下搅拌30min。同时,经5分钟时间逐滴添加溶解于meoh(15ml)中的3-(4-氟苯基)-3-氧代丙腈(2g,12.3mmol)。然后将反应混合物在60℃下搅拌16h。然后在真空下浓缩溶剂。将粗物质加入硅胶柱并用pe/ea(20:2)洗脱。化学式:计算值(m h
)c
12h11
fn2o:218.23,实验值:218.9。
[0209]
步骤2:2-(4-氟苯基)-4-甲基-1h-吡咯-3-甲腈
[0210]
将[(cod)ircl]2(0.31g,0.4mmol)、agotf(0.24g,0.9mmol)和nabh4(35mg,0.9mmol)在thf(10ml)中去中心化。然后将此混合物在氮气氛下在25℃下搅拌20min。添加溶解于thf(20ml)中的(z)-3-((烯丙氧基)亚氨基)-3-(4-氟苯基)丙腈(2g,9.2mmol)。然后将反应混合物在25℃下搅拌18h,且随后加热至50℃持续24h。然后在真空下浓缩溶剂。将粗物质加入硅胶柱并用pe/ea(10:2)洗脱。化学式:计算值(m h
)c
12
h9fn2:200.22,实验值:200.9。
[0211]
步骤3:2-(4-氟苯基)-4-甲基-1-(吡啶-4-基)-1h-吡咯-3-甲腈
[0212]
将2-(4-氟苯基)-4-甲基-1h-吡咯-3-甲腈(100mg,0.49mmol)、4-碘吡啶(204mg,1.00mmol)、k2co3(207mg,1.50mmol)和cui(9.5mg,0.05mmol)的混合物与nmp(4ml)混合。将系统抽空并用氩气氛置换。然后将混合物在微波上在200℃下搅拌2.5h。将混合物用etoac萃取,在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
17h12
fn3:277.30,实验值:277.8。1h nmr(400mhz,cdcl3)δ8.60(s,2h),7.28
–
7.20(m,2h),7.12
–
7.05(m,2h),7.02(d,j=5.5hz,2h),6.79(d,j=1.0hz,1h),2.30(d,j=0.9hz,3h)。
[0213]
实施例3.制备化合物2-5、2-6、2-7和2-8
[0214]
图3说明化合物2-5、2-6、2-7和2-8的合成流程。如图3中所示,具体合成步骤如下:
[0215]
步骤1:2-(4-氟苯基)咪唑并[1,2-a]吡嗪
[0216]
将2-溴-1-(4-氟苯基)乙酮(10g,46.1mmol)和nahco3(11.6g,138.3mmol)添加至吡嗪-2-胺(4.38g,46.1mmol)于300ml etoh中的搅拌溶液中。将混合物加热至回流持续4h。然后在真空下浓缩溶剂。将粗物质加入硅胶柱并用dcm/meoh(10:1)洗脱。化学式:计算值(m
h
)c
12
h8fn3:213.22,实验值:213.9。
[0217]
步骤2:3-溴-2-(4-氟苯基)咪唑并[1,2-a]吡嗪
[0218]
将nbs(1g,4.7mmol)溶解于ch3cn(30ml)中,在0℃下搅拌溶液。然后添加2-(4-氟苯基)咪唑并[1,2-a]吡嗪(1g,5.6mmol)。将混合物在0℃下搅拌2h。将溶剂浓缩并溶解于ea中,用水洗涤且在真空下浓缩溶剂。将粗物质加入硅胶柱并用pe/ea(1:2)洗脱。化学式:计算值(m h
)c
12
h7brfn3:292.11,实验值:291.7。
[0219]
步骤3:2-(4-氟苯基)-3-(吡啶-4-基)咪唑并[1,2-a]吡嗪
[0220]
将吡啶-4-基硼酸(63mg,0.51mmol)和pd(pph3)4(79mg,0.068mmol)加入3-溴-2-(4-氟苯基)咪唑并[1,2-a]吡嗪(100mg,0.34mmol)于etoh(3ml)中的溶液中。添加甲苯(1ml)和碳酸钠溶液(2n,0.5ml)。然后将混合物用氮气吹扫脱气。将混合物在微波上在100℃下搅拌3h。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
17h11
fn4:290.30,实验值:290.8。1h nmr(400mhz,cdcl3)δ9.22(d,j=1.2hz,1h),8.85(d,j=5.9hz,2h),8.05(dd,j=4.7,1.4hz,1h),7.97(d,j=4.7hz,1h),7.70
–
7.58(m,2h),7.44(dd,j=4.5,1.5hz,2h),7.09(t,j=8.7hz,2h)。
[0221]
步骤4:2-(4-氟苯基)-3-(吡啶-4-基)-5,6,7,8-四氢咪唑并[1,2-a]吡嗪
[0222]
将nabh4(26mg,0.69mmol)加入4-[2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基]吡啶(100mg,0.35mmol)于etoh(5ml)中的溶液中。将混合物在80℃下搅拌16h。然后浓缩溶剂,并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
17h15
fn4:294.33,实验值:294.9。1h nmr(400mhz,cdcl3)δ8.68(dd,j=4.5,1.5hz,2h),7.50
–
7.34(m,2h),7.26(dd,j=4.5,1.6hz,2h),6.97(dd,j=9.7,7.8hz,2h),4.28(s,2h),3.86(t,j=5.4hz,2h),3.29(t,j=5.4hz,2h)。
[0223]
步骤5:1-(2-(4-氟苯基)-3-(吡啶-4-基)-5,6-二氢咪唑并[1,2-a]吡嗪-7(8h)-基)乙-1-酮
[0224]
向4-[2-(4-氟苯基)-5h,6h,7h,8h-咪唑并[1,2-a]吡嗪-3-基]吡啶(20mg)于thf(5ml)中的溶液中添加乙酸酐(14mg)。将反应混合物在25℃下搅拌6h。然后浓缩溶剂,并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
19h17
fn4o:336.37,实验值:336.9。1h nmr(400mhz,cdcl3)δ8.70(d,j=5.7hz,2h),7.49
–
7.33(m,2h),7.28
–
7.18(m,2h),6.98(dd,j=12.0,5.5hz,2h),4.91(s,2h),4.06(t,j=5.3hz,2h),3.90(t,j=5.3hz,2h),2.26(s,3h)。
[0225]
步骤6:2-(4-氟苯基)-3-(三丁基甲锡烷基)咪唑并[1,2-a]吡嗪
[0226]
在氮气氛下在-78℃下搅拌3-溴-2-(4-氟苯基)咪唑并[1,2-a]吡嗪(100mg,0.34mmol)和n-buli(39mg,0.34mmol)于thf(5ml)中的溶液,然后添加2.4n tmeda(0.43ml)。将反应混合物在-78℃下搅拌1h。然后添加三丁基氯锡烷(166mg,0.51mmol)。将反应混合物在室温下搅拌1h。将反应混合物用饱和氯化铵溶液淬灭并用乙醚洗涤数次,然后通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
24h34
fn3sn:502.27,实验值:503.7。
[0227]
步骤7:4-(2-(4-氟苯基)咪唑并[1,2-a]吡嗪-3-基)呋喃并[3,4-b]吡啶-5(7h)-酮
[0228]
将4-溴呋喃并[3,4-b]吡啶-5(7h)-酮(8.5mg,0.04mmol)和pd(pph3)4(9mg,
0.008mmol)加入2-(4-氟苯基)-3-(三丁基甲锡烷基)咪唑并[1,2-a]吡嗪(20mg,0.04mmol)于1,4-二恶烷(5ml)中的溶液中。将反应混合物在氮气氛下在80℃下搅拌16h。用etoac萃取混合物,在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
19h11
fn4o2:346.32,实验值:346.8。1h nmr(400mhz,cdcl3)δ9.15(d,j=5.0hz,1h),7.93(dd,j=8.8,5.4hz,2h),7.81
–
7.69(m,2h),7.66(s,1h),7.55(dd,j=5.7,3.3hz,1h),7.15(t,j=8.7hz,2h),5.52(s,2h)。
[0229]
实施例4.制备化合物1-2
[0230]
图4说明化合物1-2的合成流程。如图4中所示,具体合成步骤如下:
[0231]
步骤1:4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)呋喃并[3,4-b]吡啶-5(7h)-酮
[0232]
将4-溴-7h-呋喃并[3,4-b]吡啶-5-酮(169.5007mg,0.792mmol)、na2co3(139.9200mg,1.32mmol)和pd(dppf)cl2(48.2460mg,0.066mmol)加入1-(4-氟苯基)-3-甲基-5-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)吡唑(200mg,0.66mmol)于meph/h2o(11ml)中的溶液中。将反应混合物在100℃下搅拌16h。过滤并浓缩反应混合物以提供粗产物。通过combi-flash(pe/ea=1:1)纯化残余物以提供白色固体产物状的4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)呋喃并[3,4-b]吡啶-5(7h)-酮(170mg,79.1%产率),通过lcms对其进行测定,ms(esi):质量计算值c
17h12
fn3o
2 309.1,m/z实验值310.0[m h]
。1h nmr(400mhz,dmso)δ8.87(d,j=5.1hz,1h),7.35(d,j=5.1hz,1h),7.25-7.14(m,4h),6.68(s,1h),5.38(s,2h),2.33(s,3h)。
[0233]
实施例5.制备化合物2-12、2-13和2-14
[0234]
图5说明化合物2-12、2-13和2-14的合成流程。如图5中所示,具体合成步骤如下:
[0235]
步骤1:3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯
[0236]
在室温下将2-乙氧基-2-氧代乙烷重氮盐(19.2g,0.167mmol)加入中间物1(20.0g,0.167mol)于甲苯(200ml)中的溶液中,并所得混合物在130℃下搅拌3h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(3:1)洗脱以提供15.6g(40%)白色固体产物。化学式:计算值(m h
)c
12h11
fn2o2:235.08,实验值:234.9。
[0237]
步骤2:1-(2-溴乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯
[0238]
在室温下将1,2-二溴乙烷(15g)加入5-(4-氟苯基)-2h-吡唑-3-甲酸乙酯(15.6g)和碳酸钾(18.4g)于乙腈(200ml)中的溶液中。将所得混合物在90℃下搅拌4h。将反应混合物冷却至室温并在压力下浓缩、用水(200ml)稀释。将反应混合物用dcm(50ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(10:1)洗脱以提供13.5g(59.5%)白色固体产物。化学式:计算值(m h
)c
14h14
brfn2o2:341.02,实验值:340.7。
[0239]
步骤3:5-苄基-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮
[0240]
在室温下将苯基甲胺(4.7g)和nahco3(3.7g)加入2-(2-溴乙基)-5-(4-氟苯基)吡唑-3-甲酸乙酯(13.5g)和碘化钾(13g)于乙腈(200ml)中的溶液中。将所得混合物在90℃下搅拌16h。将反应混合物冷却至室温并在压力下浓缩、用水(100ml)稀释。将反应混合物用dcm(30ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(3:1)洗脱以提供8.9g(70.0%)白色固体产物。化学式:计算值(m h
)c
19h16
fn3o:322.13,实验值:321.8。
[0241]
步骤4:5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0242]
在0℃下将lah(2.1g)加入2-(4-氟苯基)-5-(1-甲基苯基)-6h,7h-吡唑并[1,5-a]
吡嗪-4-酮(8.9g)于thf(100ml)中的溶液中。将所得混合物在25℃下搅拌过夜。将反应混合物用冰冷水淬灭、用水(100ml)稀释。将反应混合物用dcm(30ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(4:1)洗脱以提供4.3g(50.3%)白色固体产物。化学式:计算值(m h
)c
19h18
fn3:308.15,实验值:307.8。
[0243]
步骤5:5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0244]
在冰冷却下将nbs(1270mg,7.14mmol)加入2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪(2000mg,6.49mmol)于乙腈(50ml)中的搅拌溶液中。将混合物搅拌1h、用水(100ml)稀释。将反应混合物用ea(50ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(4:1)洗脱以提供1.3g(51.7%)白色固体产物。化学式:计算值(m h
)c
19h17
brfn3:386.06,实验值:385.7。
[0245]
步骤6:5-苄基-2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0246]
将pd(dppf)cl2(377mg)、吡啶-4-基硼烷二醇(476mg)、k3po4(1.6g)加入3-溴-2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪(1g)于dmf(30ml)中的溶液中,使混合物在n2下在80℃下反应16h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(20:1)洗脱以提供0.55g(55.3%)白色固体产物。化学式:计算值(m h
)化学式:c
24h21
fn4:385.18,实验值:384.8。
[0247]
步骤7:2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0248]
将甲酸铵(454mg)、pd/c(140mg)加入4-[2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶(140mg)于meoh(20ml)中的溶液中,将混合物加热至50℃持续3h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(20:1)洗脱以提供60mg(56.6%)白色固体产物。化学式:计算值(m h
)化学式:c
17h15
fn4:295.13,实验值:294.9。
[0249]
步骤8:1-(2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0250]
将乙酸乙酰酯加入4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶于thf中的溶液中,将混合物在室温下搅拌1h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(20:1)洗脱以提供30mg(33%)白色固体产物。化学式:计算值(m h
)化学式:c
19h17
fn4o:337.14,实验值:336.8。1h nmr(400mhz,cdcl3)δ8.60(dd,j=15.4,5.2hz,2h),7.42
–
7.36(m,2h),7.14(dd,j=12.9,5.4hz,2h),7.04(t,j=8.7hz,1h),4.91(s,1h),4.77(s,1h),4.35(dt,j=28.1,5.5hz,2h),4.19(t,j=5.5hz,1h),4.04(t,j=5.5hz,1h),2.28(s,3h)。
[0251]
实施例6.制备化合物1-4
[0252]
图6说明化合物1-4的合成流程。如图6中所示,具体合成步骤如下:
[0253]
步骤1:2-(4-氟苯基)-2,3a,4,5,6,7-六氢-3h-吲唑-3-酮
[0254]
将2-氧代环己烷-1-甲酸乙酯(10.3911g,0.0610mol)和diea(14.319g,0.111mol)加入(4-氟苯基)肼(7g,0.0555mol)于etoh(100ml)中的溶液中。将反应混合物在100℃下搅拌16h。反应完成并通过lcms进行检查。将混合物浓缩以提供粗产物。将混合物用h2o(50ml)淬灭、过滤并浓缩以提供黄色固体产物2-(4-氟苯基)-2,3a,4,5,6,7-六氢-3h-吲唑-3-酮(9.5g,62.7%产率),通过lcms对其进行测定,ms(esi):质量计算值c
13h13
fn2o 232.1,m/z实
验值233.1[m h]
。
[0255]
步骤2:3-溴-2-(4-氟苯基)-4,5,6,7-四氢-2h-吲唑
[0256]
将pobr3(11.0682g,0.0387mol)加入2-(4-氟苯基)-4,5,6,7-四氢-3ah-吲唑-3-酮(3g,0.0129mol)于acn(30ml)中的溶液中。将反应混合物在90℃下搅拌16h。反应完成并通过lcms进行检查。使用na2co3将混合物(60%sma和30%产物)的ph调整至7-8。用ea(30ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。通过combi-flash(pe/ea=5:1)纯化残余物以提供黄色油状产物3-溴-2-(4-氟苯基)-4,5,6,7-四氢-2h-吲唑(0.6g,13.2%产率),通过lcms对其进行测定,ms(esi):质量计算值c
13h12
brfn
2 294.0,m/z实验值294.9[m h]
。
[0257]
步骤3:2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢-2h-吲唑
[0258]
将吡啶-4-基硼烷二醇(62.6882mg,0.51mmol)、k3po4(216.2400mg,1.02mmol)和pd(dppf)cl2(24.8540mg,0.034mmol)加入3-溴-2-(4-氟苯基)-4,5,6,7-四氢吲唑(100mg,0.34mmol)于dmf(11ml)中的溶液中。将反应混合物在100℃下搅拌5h。反应完成并通过lcms进行检查。用h2o(30ml)淬灭反应。浓缩混合物。然后用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。通过combi-flash(pe/ea=1:1)纯化残余物以提供白色固体产物2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢-2h-吲唑(24mg,24.1%产率),通过lcms对其进行测定,ms(esi):质量计算值c
18h16
fn
3 293.1,m/z实验值293.9[m h]
。1h nmr(400mhz,dmso)δ8.56(dd,j=4.4、1.6hz,2h),7.29-7.21(m,2h),7.15(dd,j=4.4、1.6hz,1h),2.68(t,j=6.0hz,1h),2.59(t,j=6.0hz,1h),1.82(d,j=5.6hz,1h),1.74(d,j=5.6hz,1h)。
[0259]
实施例7.制备化合物2-16
[0260]
图7说明化合物2-16的合成流程。如图7中所示,具体合成步骤如下:
[0261]
步骤1:2-(4-氟苯基)-4,5,6,7-四氢-1h-苯并[d]咪唑
[0262]
将环己烷-1,2-二酮(2.7079g,0.02415mol)和nh4oac(6.1985g,0.0805mol)加入4-氟苯甲醛(2g,0.0161mol)于etoh(30ml)中的溶液中。将反应混合物在80℃下搅拌2h。反应完成并通过lcms进行检查。将混合物浓缩以提供粗产物。通过combi-flash(pe/ea=0:1)纯化残余物以提供棕色固体产物2-(4-氟苯基)-4,5,6,7-四氢-1h-苯并[d]咪唑(3.4g,92.6%产率),通过lcms对其进行测定,ms(esi):质量计算值c
13h13
fn
2 216.1,m/z实验值217.1[m h]
。
[0263]
步骤2:2-(4-氟苯基)-1-(吡啶-4-基)-4,5,6,7-四氢-1h-苯并[d]咪唑
[0264]
在微波下将4-碘吡啶(568.783mg,2.7746mmol)、k2co3(574.342mg,4.1619mmol)和cui(26.3587mg,0.13873mmol)加入2-(4-氟苯基)-4,5,6,7-四氢-1h-1,3-苯并二唑(300mg,1.3873mmol)于nmp(4ml)中的溶液中。将反应混合物在220℃下搅拌2.5小时。反应完成并通过lcms进行检查。用水(20ml)缓慢淬灭反应,用ea(30ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩。通过combi-flash(pe/ea=1:1)纯化残余物以提供白色固体产物2-(4-氟苯基)-1-(吡啶-4-基)-4,5,6,7-四氢-1h-苯并[d]咪唑(5mg,1.20%产率),通过lcms对其进行测定,ms(esi):质量计算值c
18h16
fn
3 293.1,m/z实验值293.9[m h]
。1h nmr(400mhz,cdcl3)δ8.71(dd,j=4.5,1.6hz,2h),7.37-7.32(m,2h),7.11(dd,j=4.5,1.6hz,2h),7.01-6.95(m,2h),2.76(t,j=5.8hz,2h),2.48(t,j=
5.8hz,2h),1.93-1.85(m,4h)。
[0265]
实施例8.制备化合物3-1、3-2、3-4和3-5
[0266]
图8说明化合物3-1、3-2、3-4和3-5的合成流程。如图8中所示,具体合成步骤如下:
[0267]
步骤1:(e)-3-(4-氟苯基)-2-(吡啶-4-基)丙烯腈
[0268]
将4-氟苯甲醛(3.1462g,0.0253mol)和k2co3(1.3993g,0.0101mol)加入2-(吡啶-4-基)乙腈(2g,0.0169mol)于meoh(30ml)中的溶液中。将反应混合物在80℃下搅拌4h。反应完成并通过lcms进行检查。将混合物浓缩以提供粗产物。然后添加水,过滤反应混合物并浓缩滤饼以提供棕色固体产物(e)-3-(4-氟苯基)-2-(吡啶-4-基)丙烯腈(1.8g,40.2%产率),通过lcms对其进行测定,ms(esi):质量计算值c
14
h9fn
2 224.1,m/z实验值225.0[m h]
。
[0269]
步骤2:3-(4-氟苯基)-4-(吡啶-4-基)-1h-吡咯-2-甲酸甲酯
[0270]
在室温下将dbu(1322.4000mg,8.7mmol)加入(2e)-3-(4-氟苯基)-2-(吡啶-4-基)丙-2-烯腈(1300mg,5.80mmol)和2-异氰基乙酸甲酯(574.7140mg,5.8mmol)于thf(210ml)中的混合物中。将混合物在75℃下搅拌16h。完成反应混合物并通过lcms进行检查,然后浓缩混合物。用水(20ml)淬灭反应。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩。通过combi-flash(pe/ea=0:1)纯化残余物以提供棕色固体产物3-(4-氟苯基)-4-(吡啶-4-基)-1h-吡咯-2-甲酸甲酯(0.35g,0.02%产率),通过lcms对其进行测定,ms(esi):质量计算值c
17h13
fn2o
2 296.1,m/z实验值296.9[m h]
。
[0271]
步骤3:3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酸甲酯
[0272]
将dabco(11.3120mg,0.101mmol)和dmf(0.5ml)加入3-(4-氟苯基)-4-(吡啶-4-基)-1h-吡咯-2-甲酸甲酯(300mg,1.01mol)于dmc(10ml)中的溶液中。将所得混合物加热至90℃并在此温度下搅拌16h。lcms(enb190609-146-r1)显示反应完成。用水(20ml)缓慢淬灭反应,用ea(50ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供棕色固体产物3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酸甲酯(120mg,36.8%产率),通过lcms对其进行测定,ms(esi):质量计算值c
18h15
fn2o
2 310.1,m/z实验值311.0[m h]
。
[0273]
步骤4:3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酸
[0274]
在25℃下将koh(53.7600mg,0.96mmol)加入3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)吡咯-2-甲酸甲酯(100mg,0.32mmol)于meoh/h2o=1:1(6ml)中的溶液中。将混合物在80℃下搅拌15小时。lcms显示反应完成。使用4n hcl将混合物的ph调整至7-8。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供棕色固体产物3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酸(60mg,60.1%产率),通过lcms对其进行测定,ms(esi):质量计算值c
17h13
fn2o
2 296.1,m/z实验值297.0[m h]
。
[0275]
步骤5:3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酰胺
[0276]
在25℃下将nh4cl(31.8mg,0.6mmol)、diea(77.4mg,0.6mmol)和hatu(91.2mg,0.24mmol)加入3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)吡咯-2-甲酸(60mg,0.2mmol)于dmf(10ml)中的溶液中。将混合物在25℃下搅拌2小时。lcms显示反应完成。用水(20ml)淬灭反应,用ea(50ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供棕色固体产物3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲酰胺(70mg,118.5%产率),通过lcms对其进行测定,ms(esi):质量计算值c
17h14
fn3o 295.1,m/z
实验值296.0[m h]
。1h nmr(400mhz,cdcl3)δ8.44(s,2h),7.32(dd,j=8.7,5.4hz,2h),7.17(t,j=8.6hz,2h),7.12(s,1h),7.00(d,j=11.8hz,2h),5.17(s,2h),4.06(s,3h)。
[0277]
步骤6:3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲腈
[0278]
在25℃下将pocl3(93.2688mg,0.6096mmol)和tea(61.5696mg,0.6096mmol)加入3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)吡咯-2-甲酰胺(60mg,0.02mmol)于thf(10ml)中的溶液中。将混合物在25℃下搅拌2小时。lcms显示反应完成。用水(20ml)淬灭反应,用ea(50ml*3)萃取混合物。将合并的有机层用盐水(30ml)洗涤、经硫酸钠干燥、过滤并浓缩。通过combi-flash(pe/ea=0:1)纯化残余物以提供棕色固体产物3-(4-氟苯基)-1-甲基-4-(吡啶-4-基)-1h-吡咯-2-甲腈(7mg,11.9%产率),通过lcms对其进行测定,ms(esi):质量计算值c
17h12
fn
3 277.1,m/z实验值278.0[m h]
。1h nmr(400mhz,cdcl3)δ8.49(d,j=5.3hz,2h),7.32-7.29(m,2h),7.13-7.08(m,3h),7.07-7.05(m,2h),3.91(s,3h)。
[0279]
实施例9.制备化合物1-10
[0280]
图9说明化合物1-10的合成流程。如图9中所示,具体合成步骤如下:
[0281]
步骤1:1-叠氮基-4-氟苯
[0282]
在0℃下将4-氟苯胺(5g,45mmol)溶解于3m hcl水溶液(50ml)中。缓慢添加nano2(3.72g,54mmol)并将所得溶液在同一温度下搅拌0.5h。然后逐份添加溶解于水(20ml)中的nan3(4.38g,67.5mmol)并将混合物在室温下搅拌0.5h。用mtbe(50ml*2)萃取反应混合物,并将合并的有机层经na2so4干燥、滤除并在真空下蒸发溶剂以提供粗产物。
[0283]
步骤2:4-((三甲基甲硅烷基)乙炔基)吡啶
[0284]
将tea(9.9mg,98mmol)、pph3(77mg,0.2mmol)和pdcl2(pph3)2(344mg,0.4mmol)加入4-碘吡啶(2g,9.8mmol)于thf(30ml)中的溶液中。将此混合物抽空并用n2回填数次以除去溶液中的氧。将反应混合物在25℃下搅拌1h。添加cui(56mg,0.2mmol)和乙炔基三甲基硅烷(1.44g,14.7mmol)。将反应混合物在25℃下搅拌15h。添加水(50ml)。用ea(30ml*3)萃取残余物。将反应混合物浓缩以提供粗产物。化学式:计算值(m h
)c
10h14
nsi,分子量:176.31,实验值:176.0。1h nmr(400mhz,cdcl3)δ8.57(d,j=5.6hz,2h),7.31(dd,j=4.5,1.6hz,2h),0.27(s,9h)。
[0285]
步骤3:4-(1-(4-氟苯基)-1h-1,2,3-三唑-5-基)吡啶
[0286]
将4-((三甲基甲硅烷基)乙炔基)吡啶(639mg,3.65mmol)和t-buok(409mg,3.65mmol)加入1-叠氮基-4-氟苯(500mg,3.65mmol)于dmf(20ml)中的溶液中。将反应混合物在25℃下搅拌15h。添加水(20ml)。用ea(2*20ml)萃取残余物。将粗物质经硫酸钠干燥,并在radleys排出装置中在氮气流下干燥以提供粗产物。将反应混合物在压力下浓缩。将粗产物加入硅胶柱并用ch2cl2/meoh(15:1)洗脱。通过通用制备型hplc纯化残余物以获得产物。化学式:计算值(m h
)c
13h10
fn4,分子量:240.24,实验值:240.9。1h nmr(400mhz,meod)δ8.58(dd,j=4.7,1.4hz,2h),8.23(s,1h),7.57-7.45(m,2h),7.42-7.26(m,4h)。
[0287]
实施例10.制备化合物3-7
[0288]
图10说明化合物3-7的合成流程。如图10中所示,具体合成步骤如下:
[0289]
步骤1:3-(4-氟苯基)-1h-吡咯-2-甲酸甲酯
[0290]
将2-异氰基乙酸甲酯(6.18g,62.4mmol)和ag2co3(1.15g,4.2mmol)加入1-乙炔基-4-氟苯(5g,42mmol)于nmp(20ml)中的溶液中。将混合物在氮气氛下在80℃下搅拌1h。将混
合物用etoac萃取。在真空下蒸发溶剂,并将粗物质加入硅胶柱,用pe/ea(20:1)洗脱。化学式:计算值(m h )c
12h10
fno2:219.22,实验值:219.9。
[0291]
步骤2:3-(4-氟苯基)-1-甲基-1h-吡咯-2-甲酸甲酯
[0292]
将nah(0.66g,27.4mmol)于dmf(5ml)中的溶液在干冰水浴中冷却至0℃。添加3-(4-氟苯基)-1h-吡咯-2-甲酸甲酯(3g,13.7mmol)的dmf溶液(10ml)。将混合物在0℃下搅拌1h,然后添加ich3(2.9g,20.5mmol)。将反应混合物在25℃下搅拌1h。添加5ml水,并将混合物用etoac萃取。在真空下蒸发溶剂,将粗物质加入硅胶柱并用pe/ea(20:1)洗脱。化学式:计算值(m h )c
13h12
fno2:233.24,实验值:233.9。
[0293]
步骤3:3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酸甲酯
[0294]
将icl(8.4g,5.1mmol)的ccl4溶液加入3-(4-氟苯基)-1-甲基-1h-吡咯-2-甲酸甲酯(1g,4.3mmol)于ccl4(5ml)中的溶液中。将混合物在0℃下搅拌15min。在真空下蒸发溶剂,将粗物质加入硅胶柱并用pe/ea(20:1)洗脱。化学式:计算值(m h )c
13h11
fino2:359.14,实验值:359.6。
[0295]
步骤4:3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酸
[0296]
将naoh(0.22g,5.5mmol)加入3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酸甲酯(0.4g,1.1mmol)于meoh/h2o(6ml)中的溶液。将混合物在60℃下搅拌5h。用hcl(6mol/l)将反应调整至ph 5-6并用etoac萃取。在真空下蒸发溶剂以提供粗产物。化学式:计算值(m h )c
12
h9fino2:345.11,实验值:345.7。
[0297]
步骤5:3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酰胺
[0298]
将hatu(502mg,1.32mmol)、diea(506mg,3.3mmol)和nh4cl(177mg,3.3mmol)加入3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酸(380mg,1.1mmol)于dmf(10ml)中的溶液中。将混合物在室温下搅拌1h。添加5ml水,并将混合物用etoac萃取。在真空下蒸发溶剂以提供粗产物。化学式:计算值(m h )c
12h10
fin2o:344.13,实验值:344.7。
[0299]
步骤6:3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲腈
[0300]
将pocl3(468mg,3.05mmol)和et3n(309mg,3.05mmol)加入3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲酰胺(350mg,1.02mmol)于thf(5ml)中的溶液中。将混合物在室温下搅拌1h。真空下蒸发溶剂,将粗物质加入硅胶柱并用pe/ea(20:1)洗脱。化学式:计算值(m h )c
12
h8fin2:326.11,实验值:326.6。
[0301]
步骤7:3-(4-氟苯基)-1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1h-吡咯-2-甲腈
[0302]
在氮气氛下在-78℃下搅拌3-(4-氟苯基)-4-碘-1-甲基-1h-吡咯-2-甲腈(50mg,0.15mmol)于thf(3ml)中的溶液,然后添加2.4m n-buli(0.19ml)。将反应混合物在-78℃下搅拌1h。然后添加2-异丙氧基-4,4,5,5-四甲基-1,3,2-二恶硼烷(86mg,0.46mmol)。将反应混合物在室温下搅拌1h。将反应用饱和氯化铵溶液淬灭并用乙醚洗涤数次。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h )c
18h20
bfn2o2:326.18,实验值:326.8。
[0303]
步骤8:3-(4-氟苯基)-1-甲基-4-(5-氧代-5,7-二氢呋喃并[3,4-b]吡啶-4-基)-1h-吡咯-2-甲腈
[0304]
将4-溴呋喃并[3,4-b]吡啶-5(7h)-酮(38mg,0.18mmol)、na2co3(38mg,0.36mmol)
和pd(dppf)cl2(13mg,0.018mmol)加入3-(4-氟苯基)-1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1h-吡咯-2-甲腈(60mg,0.018mmol)于甲苯/h2o(5.5ml)中的溶液中。将混合物在氮气氛下在100℃下搅拌16h。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h )c
19h12
fn3o2:333.32,实验值:333.7。1h nmr(400mhz,cdcl3)δ8.50(d,j=5.3hz,1h),7.69(s,1h),7.35
–
7.22(m,2h),7.10(t,j=8.6hz,2h),6.88(d,j=5.3hz,1h),5.33(s,2h),3.93(s,3h)。
[0305]
实施例11.制备化合物2-27
[0306]
图11说明化合物2-27的合成流程。如图11中所示,具体合成步骤如下:
[0307]
步骤1:1-(2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)丙-1-酮
[0308]
将et3n加入4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]-2-甲基吡啶的dcm溶液中。然后添加丙酰氯,将混合物在室温下搅拌2h。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
20h19
fn4o:350.40,实验值:350.9。1h nmr(400mhz,cdcl3)δ8.59(d,j=10.7hz,2h),7.46-7.35(m,2h),7.11(s,2h),7.07-6.96(m,2h),4.90(s,1h),4.76(s,1h),4.37(s,1h),4.26(d,j=43.4hz,2h),4.04(s,1h),2.65-2.20(m,2h),1.36-1.06(m,3h)。
[0309]
实施例12.制备化合物2-28
[0310]
图12说明化合物2-28的合成流程。如图12中所示,具体合成步骤如下:
[0311]
步骤1:1-(2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)-2-甲基丙-1-酮
[0312]
将et3n加入4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]-2-甲基吡啶的dcm溶液中。然后添加2-甲基丙酰氯,将混合物在室温下搅拌2h。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
21h22
fn4o:364.42,实验值:364.8。1h nmr(400mhz,cdcl3)δ8.58(s,2h),7.42-7.38(m,2h),7.12(d,j=3.7hz,2h),7.08-7.00(m,2h),4.85(d,j=37.0hz,2h),4.24(dd,j=80.9,32.3hz,2h),1.96(s,1h),1.39-1.04(m,2h)。
[0313]
实施例13.制备化合物2-29和2-31
[0314]
图13说明化合物2-29和2-31的合成流程。如图13中所示,具体合成步骤如下:
[0315]
步骤1:3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯
[0316]
在室温下将2-乙氧基-2-氧代乙烷重氮盐加入1-乙炔基-4-氟苯于甲苯(300ml)中的溶液中,并将所得混合物在130℃下搅拌3h。将混合物冷却至室温。将反应混合物在压力下浓缩。将粗物质加入快速色谱并用pe/ea(3:1)洗脱。化学式:计算值(m h
)c
12h11
fn2o2:234.23,实验值:234.9。
[0317]
步骤2:1-(2-溴乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯
[0318]
在室温下将1,2-二溴乙烷加入3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯和碳酸钾于乙腈(200ml)中的溶液中,将所得混合物在90℃下搅拌4h。将混合物冷却至室温。将反应混合物在压力下浓缩。将粗物质加入快速色谱并用pe/ea(10:1)洗脱。化学式:计算值(m h
)c
14h14
brfn2o2:341.18,实验值:342.02。
[0319]
步骤3:5-苄基-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮
[0320]
在室温下将苯基甲胺和nahco3加入1-(2-溴乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯和碘化钾于乙腈(200ml)中的溶液中,将所得混合物在90℃下搅拌16h。将混合物冷却至室温。将反应混合物在压力下浓缩。将粗物质加入快速色谱并用pe/ea(3:1)洗脱。化学式:计算值(m h
)c
19h16
fn3o:324.36,实验值:321.9。
[0321]
步骤4:5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0322]
在0℃下将lah加入5-苄基-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮于thf(100ml)中的溶液中,并将所得混合物在25℃下搅拌过夜。将反应混合物用冰冷水淬灭。过滤混合物并用dcm萃取滤液。在真空下蒸发溶剂,将粗物质加入快速色谱并用pe/ea(4:1)洗脱。化学式:计算值(m h
)c
19h18
fn3:307.37,实验值:307.9。
[0323]
步骤5:5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0324]
在冰冷却下将nbs加入5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪于乙腈中的搅拌溶液中。将混合物搅拌2h。在真空下蒸发溶剂,将粗物质加入快速色谱并用pe/ea(4:1)洗脱。化学式:计算值(m h
)c
19h17
brfn3:386.27,实验值:387.7。
[0325]
步骤6:5-苄基-2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0326]
将pd(dppf)cl2、吡啶-4-基硼烷二醇、k3po4加入5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪的dmf溶液中,使混合物在n2下在80℃下反应3h。用dcm萃取混合物。在真空下蒸发溶剂,将粗物质加入快速色谱并用pe/ea(1:1)洗脱。化学式:计算值(m h
)c
25h23
fn4:398.49,实验值:398.9。
[0327]
步骤7:2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0328]
将甲酸铵(454mg)、pd/c(140mg)加入5-苄基-2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(140mg)于meoh(20ml)中的溶液中,将混合物加热至50℃持续3h。在真空下蒸发溶剂,将粗物质加入快速色谱并用dcm/meoh(20:1)洗脱。化学式:计算值(m h
)c
18h17
fn4:308.36,实验值:308.9。
[0329]
步骤8:1-(2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0330]
将et3n加入2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪的dcm溶液中。然后添加乙酰氯,将混合物在室温下搅拌1h。在真空下蒸发溶剂,将粗物质加入快速色谱并用dcm/meoh(20:1)洗脱。化学式:计算值(m h
)c
20h19
fn4o:350.40,实验值:350.9。1h nmr(400mhz,cdcl3)δ8.47(dd,j=20.0,5.2hz,1h),7.47-7.36(m,2h),7.02(t,j=8.7hz,2h),6.93(dd,j=26.4,6.0hz,2h),4.87(s,1h),4.74(s,1h),4.37(t,j=5.4hz,1h),4.30(t,j=5.4hz,1h),4.18(t,j=5.3hz,1h),4.02(t,j=5.4hz,1h),2.55(s,3h),2.22(d,j=37.2hz,3h)。
[0331]
步骤9:4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基吡啶-1-氧化物
[0332]
将1-[2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4h,6h,7h-吡唑并溶解于dcm中并冷却至0℃,添加间氯过氧苯甲酸并将反应混合物在室温下搅拌1h。将混合物用饱和na2co3水溶液洗涤、经na2so4干燥、过滤并浓缩以提供所需产物。化学式:计算值(m h
)c
20h19
fn4o2:366.40,实验值:366.8。1h nmr(400mhz,cdcl3)δ8.28(dd,j=13.4,6.6hz,1h),7.47-7.33
(m,2h),7.08-6.94(m,4h),4.88(s,1h),4.74(s,1h),4.37(t,j=5.4hz,1h),4.30(d,j=5.8hz,1h),4.19(d,j=5.4hz,1h),4.03(t,j=5.4hz,1h),2.54(s,3h),2.24(d,j=32.6hz,3h)。
[0333]
实施例14.制备化合物2-30
[0334]
图14说明化合物2-30的合成流程。如图14中所示,具体合成步骤如下:
[0335]
步骤1:1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸甲酯
[0336]
将1-溴-2-氯乙烷(5.5213g,0.0385mol)和k2co3(3.1878g,0.0231mol)加入5-(4-氟苯基)-2h-吡唑-3-甲酸乙酯(1.8g,0.0077mol)于丙酮(20ml)中的溶液中。将反应混合物在70℃下搅拌4h。反应完成并通过lcms进行检查。浓缩混合物,然后添加h2o、过滤并浓缩滤饼。化学式:计算值(m h
)c
13h13
clfn2o2,分子量:283.70,实验值:283.0。
[0337]
步骤2:(1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-基)甲醇
[0338]
在0℃下将lah(0.2160g,0.0054mol)加入1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸甲酯(1.6g,0.0054mol)于四氢呋喃(30ml)中的溶液中。将反应混合物在0-25℃下搅拌2h。反应完成并通过lcms进行检查。用h2o(50ml)淬灭反应。将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。化学式:计算值(m h
)c
12h13
clfn2o,分子量:255.69,实验值:255.0。
[0339]
步骤3:2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0340]
在0℃下将nah(84.72mg,3.53mmol)加入(1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-基)甲醇(900mg,3.53mmol)于dmf(10ml)中的溶液中并将反应混合物在0-25℃下搅拌2h。反应完成并通过lcms进行检查。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。化学式:计算值(m h
)c
12h11
fn2o,分子量:219.23,实验值:219.0。
[0341]
步骤4:3-溴-2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0342]
将nbs(284.8mg,1.6mmol)加入2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪(350mg,1.6mmol)于dcm(10ml)中的溶液中。将反应混合物在25℃下搅拌16h。反应完成并通过lcms进行检查。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。化学式:计算值(m h
)c
12h11
brfn2o,分子量:298.13,实验值:298.9。
[0343]
步骤5:2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0344]
将吡啶-4-基硼烷二醇(184.3770mg,1.5mmol)、k3po4(106mg,0.5mmol)、x-phos(47.7000mg,0.1mmol)和pd2(dba)3(45.8mg,0.05mmol)加入3-溴-2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪(150mg,0.50mmol)于1,4-二恶烷(10ml)中的溶液中。将反应混合物在90℃下搅拌16h。反应完成并通过lcms进行检查,然后过滤并浓缩。1h nmr(400mhz,dmso)δ8.50(dd,j=4.5,1.6hz,2h),7.43-7.36(m,2h),7.25-7.17(m,2h),7.10(dd,j=4.5,1.6hz,2h),4.92(s,2h),4.22(t,j=5.0hz,2h),4.14(t,j=5.1hz,2h)化学式:计算值(m h
)c
17h15
fn3o,分子量:296.32,实验值:296.0。
[0345]
实施例15.制备化合物2-33
[0346]
图15说明化合物2-33的合成流程。如图15中所示,具体合成步骤如下:
[0347]
步骤1:(2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)
(1-甲基哌啶-4-基)甲酮
[0348]
将1-甲基哌啶-4-甲酸的草酰氯溶液加热至70℃持续1h,然后在真空下去除溶剂。添加et3n,4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶,将混合物在室温下搅拌1h。在真空下蒸发溶剂并通过ptlc纯化粗物质以获得产物。化学式:计算值(m h
)c
24h26
fn5o:419.21,实验值:419.8。1h nmr(400mhz,cdcl3)δ8.57(s,2h),7.47-7.36(m,2h),7.18-6.94(m,4h),4.83(d,j=43.8hz,2h),4.37(s,2h),4.13(d,j=45.8hz,2h),3.10-2.91(m,2h),2.37(s,2h),1.96(s,2h),1.66(s,4h)。
[0349]
实施例16.制备化合物2-34
[0350]
图16说明化合物2-34的合成流程。如图16中所示,具体合成步骤如下:
[0351]
步骤1:n-甲基-1h-咪唑-1-甲酰胺
[0352]
将cdi(500mg,3.08mmol)和六亚甲基四胺(95.6mg,3.08mmol)溶解于dmf(1ml)和乙腈(3ml)中。将溶液在室温下搅拌2h,之后在空气流中浓缩成稠状油。快速色谱(4%meoh/ch2cl2)提供产物。
[0353]
步骤2:2-(4-氟苯基)-n-甲基-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-甲酰胺
[0354]
将n-甲基-1h-咪唑-1-甲酰胺(8.7mg,0.07mol)和tea(7.78mg,0.077mol)加入2-(4-氟苯基)-3-(吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(20mg,0.07mol)于dcm(2ml)中的溶液中。将反应混合物在25℃下搅拌2h。将混合物浓缩,然后通过制备型tlc和制备型hplc纯化以提供产物。化学式:计算值(m h
)c
19h19
fn5o,分子量:351.39,实验值:351.8。1h nmr(400mhz,cdcl3)δ8.57(s,2h),7.40(dd,j=8.6,5.4hz,2h),7.22(s,2h),7.05(t,j=8.7hz,2h),4.85(s,1h),4.71(s,2h),4.34(t,j=5.3hz,2h),4.01(t,j=5.3hz,2h),2.88(s,3h)。
[0355]
实施例17.制备化合物2-32
[0356]
图17说明化合物2-32的合成流程。如图17中所示,具体合成步骤如下:
[0357]
步骤1:(4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)乙酸甲酯
[0358]
将4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基吡啶-1-氧化物(30mg)于ac2o(2ml)中的溶液在100℃下搅拌1h。将混合物浓缩并通过制备型hplc纯化以提供产物。化学式:计算值(m h
)c
22h22
fn4o3,分子量:408.43,实验值:408.8。1h nmr(400mhz,cdcl3)δ8.59(dd,j=9.9、5.1hz,1h),7.40(dd,j=7.9,5.8hz,2h),7.15(s,1h),7.06(dd,j=12.0,5.4hz,3h),5.22(d,j=3.5hz,2h),4.91(s,1h),4.78(s,1h),4.38(t,j=5.3hz,1h),4.31(d,j=5.6hz,1h),4.20(d,j=5.5hz,1h),4.07-4.00(m,1h),2.24(d,j=32.1hz,3h),2.10(d,j=3.5hz,3h)。
[0359]
实施例18.制备化合物2-36
[0360]
图18说明化合物2-36的合成流程。如图18中所示,具体合成步骤如下:
[0361]
步骤1:5-苄基-2-(4-氟苯基)-3-(3-氟吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0362]
将sm2(110mg,0.78mmol)、cs2co3(508mg,1.56mmol)和pd(dppf)cl2(38mg,0.052mmol)加入5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(200mg,
0.52mmol)于二恶烷(5ml)和水(0.5ml)中的溶液中。将反应混合物在100℃下搅拌3h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)化学式:c
24h21
f2n4,分子量:402.45,实验值:402.8。
[0363]
步骤2:2-(4-氟苯基)-3-(3-氟吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0364]
将hcoonh4(156mg,2.48mmol)和pd/c(10mg)加入5-苄基-2-(4-氟苯基)-3-(3-氟吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(100mg,0.248mmol)于(5ml)中的溶液中。将反应混合物在50℃下搅拌5h。过滤溶液并收集滤液。将反应混合物在压力下浓缩。通过lcms(与其一致)证明产物。化学式:计算值(m h
)化学式:c
17h15
f2n4,分子量:312.32,实验值:312.8。
[0365]
步骤3:1-(2-(4-氟苯基)-3-(3-氟吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0366]
将ac2o(45.8mg,0.448mmol)加入2-(4-氟苯基)-3-(3-氟吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(70mg,0.224mmol)于thf(5ml)中的溶液中。将反应混合物在25℃下搅拌15h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用ch2cl2/meoh(10:1)洗脱。通过制备型tlc(ch2cl2/meoh=6:1)纯化残余物以提供产物。通过手性制备型hplc纯化残余物以提供产物。通过通用制备型hplc纯化残余物以提供产物。1h nmr(400mhz,meod)δ8.54(d,j=68.1hz,2h),7.49-7.26(m,3h),7.08(t,j=8.8hz,2h),4.81(d,j=14.3hz,2h),4.34(dt,j=36.5,5.5hz,2h),4.15(dt,j=11.0,5.5hz,2h),2.22(d,j=35.9hz,3h)。化学式:计算值(m h
)化学式:c
19h17
f2n4o,分子量:354.36,实验值:354.9。
[0367]
实施例19.制备化合物1-5
[0368]
图19说明化合物1-5的合成流程。如图19中所示,具体合成步骤如下:
[0369]
步骤1:4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)吡啶
[0370]
将吡啶-4-基硼烷二醇(71.907mg,0.585mmol)、k3po4(165.36mg,0.78mmol)和pd(dppf)cl2(28.509mg,0.039mmol)加入5-溴-1-(4-氟苯基)-3-甲基吡唑(100mg,0.39mmol)于dmf(11ml)中的溶液中。将反应混合物在100℃下搅拌5h。反应完成并通过lcms进行检查。用h2o(30ml)淬灭反应,然后将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以提供粗产物。通过combi-flash(pe/ea=1:1)纯化残余物以提供白色固体产物4-(1-(4-氟苯基)-3-甲基-1h-吡唑-5-基)吡啶(60mg,509.5%产率),通过lcms对其进行测定,ms(esi):质量计算值c
15h12
fn3253.1,m/z实验值254.1[m h]
。1h nmr(400mhz,dmso)δ8.54(dd,j=4.6,1.4hz,2h),7.31(t,j=7.1hz,4h),7.18(dd,j=4.6,1.4hz,2h),6.69(s,1h),2.29(s,3h)。
[0371]
实施例20.制备化合物1-13
[0372]
图20说明化合物1-13的合成流程。如图20中所示,具体合成步骤如下:
[0373]
步骤1:1-(4-氟苯基)吡咯-2-甲腈
[0374]
在0℃下将[(氯磺酰基)亚氨基]甲酮(2897mg,20.47mmol)缓慢加入1-(4-氟苯基)吡咯(3000mg,18.6mmol)于can(30ml)中的溶液中。将反应混合物在0℃下搅拌1h。向反应混合物中逐滴添加dmf(3ml)。将混合物在同一温度下搅拌1h。添加水(20ml)。用mtbe(30ml*3)萃取残余物。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(10:1)洗脱。化学式:计算值(m h
)化学式:c
11
h8fn2,分子量:186.19,实验值:187.0。
[0375]
步骤2:5-溴-1-(4-氟苯基)吡咯-2-甲腈
[0376]
在0℃下将nbs(48mg,0.27mmol)加入1-(4-氟苯基)吡咯-2-甲腈(50mg,0.27mmol)于thf(3ml)中的溶液中。将反应混合物在25℃下搅拌3h。将反应混合物在压力下浓缩。通过制备型tlc(pe/etoac=5:1)纯化残余物以提供产物。
[0377]
步骤3:1-(4-氟苯基)-5-(吡啶-4-基)-1h-吡咯-2-甲腈
[0378]
将吡啶-4-基硼烷二醇(138mg,1.125mmol)、pd(dppf)cl2(55mg,0.075mmol)和k3po4(318mg,1.5mmol)加入5-溴-1-(4-氟苯基)吡咯-2-甲腈(200mg,0.75mmol)于dmf(10ml)和水(1ml)中的溶液中。将反应混合物在100℃下搅拌15h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(3:2)洗脱以提供产物。化学式:计算值(m h
)化学式:c
16h11
fn3,分子量:263.28,实验值:263.9。1h nmr(400mhz,meod)δ8.51(dd,j=4.7,1.6hz,2h),8.01(d,j=1.5hz,1h),7.71(dd,j=4.9,1.4hz,2h),7.68
–
7.62(m,3h),7.37(t,j=8.6hz,2h)。
[0379]
实施例21.制备化合物2-38
[0380]
如图21中所示,具体合成步骤如下:
[0381]
步骤1:3-(4-氟苯基)-1-(环氧乙烷-2-基甲基)-1h-吡唑-5-甲酸乙酯
[0382]
向5-(4-氟苯基)-2h-吡唑-3-甲酸乙酯(2.34g,10mmol)于dmf(30ml)中的混合物中添加2-(氯甲基)环氧乙烷(9.26g,100mmol)、cs2co3(4.89g,15mmol)。将反应在25℃下搅拌16h。lcms显示检测到所需的ms为主峰。用ea(50ml*2)和h2o(50ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,然后通过柱色谱(pe/ea=2/1)纯化以产生白色固体(1.96g,产率:67.5%)。化学式:计算值(m h
)c
15h15
fn2o3:290.11,实验值:291.1。1h nmr(400mhz,dmso)δ7.94
–
7.90(m,2h),7.41(s,1h),7.26(t,j=8.9hz,2h),4.87(dd,j=14.5,3.6hz,1h),4.61(dd,j=14.5,5.5hz,1h),4.34(q,j=7.1hz,2h),3.42
–
3.38(m,1h),2.80
–
2.77(m,1h),2.52-2.51(m,1h),1.34(t,j=7.1hz,3h)。
[0383]
步骤2:1-(3-(4-氟苯基)-5-(羟基甲基)-1h-吡唑-1-基)丙-2-醇
[0384]
在0℃下向5-(4-氟苯基)-2-(环氧乙烷-2-基甲基)吡唑-3-甲酸乙酯(1.45g,5.0mmol)于thf(20ml)中的混合物中添加氢化铝锂(380mg,10.0mmol)。将反应在0℃下搅拌4h。lcms显示检测到所需的ms为主峰。用冰淬灭反应,用ea(50ml*2)和h2o(50ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩以产生白色固体(1.29g,纯度:90%,产率:92%)。化学式:计算值(m h
)c
13h15
fn2o2:250.11,实验值:251.0。1h nmr(400mhz,dmso)δ7.82-7.77(m,2h),7.21(t,j=8.9hz,2h),6.59(s,1h),5.29(t,j=5.6hz,1h),5.00(d,j=4.7hz,1h),4.56(dd,j=11.1,6.3hz,2h),4.07
–
4.01(m,3h),1.10(d,j=5.9hz,3h)。
[0385]
步骤3:2-(4-氟苯基)-6-甲基-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0386]
向1-[3-(4-氟苯基)-5-(羟基甲基)吡唑-1-基]丙-2-醇(1.0g,4.0mmol)于甲苯(20ml)中的混合物中添加硫酸(1.96g,20mmol)。将反应在110℃下搅拌4h。将混合物浓缩以去除有机物、用h2o(20ml)稀释、用1m naoh处理至ph约为7,然后用ea(50ml*2)和h2o(50ml)萃取。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,通过柱色谱(dcm/meoh=30/1)纯化以产生白色固体(305mg,产率:32%)。化学式:计算值(m h
)c
13h13
fn2o:232.10,实验值:233.1。1h nmr(400mhz,dmso)δ7.82
–
7.78(m,2h),7.22(t,j=8.9hz,2h),
6.50(s,1h),4.94(d,j=15.0hz,1h),4.75(d,j=15.1hz,1h),4.22(dd,j=12.4,3.1hz,1h),4.08-4.01(m,1h),3.76(dd,j=12.1,10.7hz,1h),1.31(d,j=6.2hz,3h)。
[0387]
步骤4:3-溴-2-(4-氟苯基)-6-甲基-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0388]
向2-(4-氟苯基)-6-甲基-4h,6h,7h-吡唑并[3,2-c][1,4]恶嗪(296mg,1.27mmol)于dcm(5ml)中的混合物中添加nbs(340mg,1.905mmol)并在室温下搅拌16h。lcms显示检测到所需的ms为主峰。用dcm(50ml*2)和h2o(50ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,然后通过柱色谱(pe/ea=2/1)纯化以产生白色固体(340mg,产率:86%)。化学式:计算值(m h
)c
13h12
brfn2o:310.01,实验值:310.9。1h nmr(400mhz,dmso)δ7.87
–
7.81(m,2h),7.34
–
7.28(m,2h),4.85(d,j=15.2hz,1h),4.72(d,j=15.2hz,1h),4.23(dd,j=12.4,3.0hz,1h),4.12-4.04(m,1h),3.80(dd,j=12.2,10.6hz,1h),1.32(d,j=6.2hz,3h)。
[0389]
步骤5:2-(4-氟苯基)-6-甲基-3-(吡啶-4-基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0390]
向3-溴-2-(4-氟苯基)-6-甲基-4h,6h,7h-吡唑并[3,2-c][1,4]恶嗪(155mg,0.55mmol)于二恶烷/h2o(20ml/4ml)中的溶液中添加吡啶-4-基硼烷二醇(185mg,1.5mmol)、cs2co3(245mg,0.75mmol)和pd(dppd)cl2(82mg,0.1mmol),并将混合物在干燥n2下在100℃下搅拌4h。将混合物浓缩以去除有机物,用h2o(20ml)稀释,然后用ea(50ml*2)和h2o(50ml)萃取。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,通过柱色谱(dcm/meoh=30/1)纯化以产生粗产物,然后通过sfc(chiralpak-oj,co
2-meoh(dea))纯化以产生化合物2-38(粗物质)和化合物2-39(27.2mg,hplc:99.47%)。将化合物2-38通过hplc(gemini-c18 150
×
21.2mm,5um,acn
–
h2o 0.1%fa,梯度10%至40%)纯化以产生化合物2-38(44.8mg,hplc:100%)。化学式:计算值(m h
)c
18h16
fn3o:309.13,实验值:310.1。
[0391]
化合物2-38:1h nmr(400mhz,dmso)δ8.50(d,j=5.3hz,2h),7.41
–
7.36(m,2h),7.23
–
7.18(m,2h),7.10(d,j=6.0hz,2h),4.93(t,j=19.5hz,2h),4.30(dd,j=12.6,3.1hz,1h),4.16-4.08(m,1h),3.88
–
3.81(m,1h),1.34(d,j=6.2hz,3h)。
[0392]
实施例22.制备化合物2-37
[0393]
如图22中所示,具体合成步骤如下:
[0394]
步骤1:(r)-1-(1-((叔丁氧羰基)氨基)丙-2-基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(2)
[0395]
将于thf(50ml)中的pph3(3360mg,12.81mmol)和diad(5180mg,25.62mmol)以及(r)-1-(boc-氨基)-2-丙醇(1795mg,10.25mmol)加入1-(3-(4-氟苯基)-1h-吡唑-5-基)乙-1-酮溶液(2000mg,8.54mmol)中。将反应混合物在25℃下搅拌15h。添加饱和nh4cl水溶液(50ml)。用ea(30ml*2)萃取残余物。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(4:1)洗脱以产生产物。
[0396]
化学式:计算值(m h
):c
20h26
fn3o4,分子量:391.44,实验值:391.8。
[0397]
步骤2:(r)-1-(1-((叔丁氧羰基)氨基)丙-2-基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(3)
[0398]
将tfa(1747mg,15.32mmol)加入(r)-1-(1-((叔丁氧羰基)氨基)丙-2-基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(3000mg,7.66mmol)于dcm(30ml)中的溶液中。将反应混合物
在25℃下搅拌15h。将反应混合物在压力下浓缩以产生产物。
[0399]
化学式:计算值(m h
):c
15h18
fn3o2,分子量:291.33,实验值:291.8。
[0400]
步骤3:(r)-2-(4-氟苯基)-7-甲基-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮(4)
[0401]
将tea(1390mg,13.74mmol)加入(r)-1-(1-((叔丁氧羰基)氨基)丙-2-基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(2000mg,6.87mmol)于meoh(5ml)中的溶液中。将反应混合物在50℃下搅拌5h。将反应混合物在压力下浓缩以产生产物。
[0402]
化学式:计算值(m h
):c
13h12
fn3o,分子量:245.26,实验值:245.7。
[0403]
步骤4:(r)-3-溴-2-(4-氟苯基)-7-甲基-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮(5)
[0404]
将nbs(878mg,4.935mmol)加入(r)-2-(4-氟苯基)-7-甲基-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮(1000mg,3.29mmol)于thf(20ml)中的溶液中。将反应混合物在50℃下搅拌20h。将反应混合物在压力下浓缩以产生产物。
[0405]
化学式:计算值(m h
):c
13h11
brfn3o,分子量:324.15,实验值:324.7。
[0406]
步骤5:(r)-2-(4-氟苯基)-7-甲基-3-(吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮
[0407]
向(r)-3-溴-2-(4-氟苯基)-7-甲基-6,7-二氢吡唑并[1,5-a]吡嗪-4(5h)-酮(1000mg,3.09mmol)于二甲氧基乙烷(20ml)和水(3ml)中的溶液中添加吡啶-4-基硼酸(570mg,4.63mmol)、pd(pph3)4(357mg,0.309mmol)和na2co3(667mg,6.18mmol)。将反应混合物在100℃下搅拌5h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用ch2cl2/meoh(10:1)洗脱以产生产物。
[0408]
化学式:计算值(m h
):c
18h15
fn4o,分子量:322.34,实验值:322.8。
[0409]
实施例23.制备化合物2-39
[0410]
如图23中所示,具体合成步骤与实施例21类似。
[0411]
化合物2-39:1h nmr(400mhz,dmso)δ8.50(dd,j=4.5,1.6hz,2h),7.41
–
7.36(m,2h),7.21(t,j=8.9hz,2h),7.10(dd,j=4.5,1.6hz,2h),4.95(d,j=23.8hz,2h),4.30(dd,j=12.5,3.1hz,1h),4.14
–
4.09(m,1h),3.87
–
3.82(m,1h),1.35(d,j=6.2hz,3h)。
[0412]
实施例24.制备化合物2-40
[0413]
如图24中所示,具体合成步骤如下:
[0414]
步骤1:1-(3-氯丙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯
[0415]
向5-(4-氟苯基)-2h-吡唑-3-甲酸乙酯(2.34g,10mmol)于dmf(20ml)中的混合物中添加1-溴-3-氯丙烷(2.37g,15mmol)、cs2co3(4.89g,15mmol)。将反应在25℃下搅拌16h。lcms显示检测到所需的ms为主峰。用ea(50ml*2)和h2o(50ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,然后通过柱色谱(pe/ea=2/1)纯化以产生白色固体(1.9g,产率:58%)。化学式:计算值(m h
)c
15h16
clfn2o2:310.09,实验值:311.1。1h nmr(400mhz,dmso)δ7.94
–
7.89(m,2h),7.38(s,1h),7.29-7.23(m,2h),4.67(t,j=6.9hz,2h),4.34(q,j=7.1hz,2h),3.67(t,j=6.3hz,2h),2.32-2.24(m,2h),1.34(t,j=7.1hz,3h)。
[0416]
步骤2:(1-(3-氯丙基)-3-(4-氟苯基)-1h-吡唑-5-基)甲醇和3-(3-(4-氟苯基)-5-(羟基甲基)-1h-吡唑-1-基)丙-1-醇
[0417]
在0℃下向1-(3-氯丙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(1.24g,4.0mmol)于thf(20ml)中的混合物中添加氢化铝锂(304mg,8.0mmol)。将反应在0℃下搅拌4h。lcms显示检测到所需的ms为主峰。用冰淬灭反应,用ea(50ml*2)和h2o(50ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩以产生两种白色固体(化合物3,730mg)和(化合物3a,80mg)。化合物3:化学式:计算值(m h
)c
13h14
clfn2o:268.08,实验值:269.1。1h nmr(400mhz,dmso)δ7.82
–
7.78(m,2h),7.24
–
7.19(m,2h),6.62(s,1h),5.50-5.15(m,1h),4.54(s,2h),4.25(t,j=6.9hz,2h),3.69(t,j=6.5hz,2h),2.27(p,j=6.6hz,2h)。化合物3a:化学式:计算值(m h
)c
13h15
fn2o2:250.11,实验值:251.1。1h nmr(400mhz,dmso)δ7.79(dd,j=8.9,5.6hz,2h),7.21(t,j=8.9hz,2h),6.58(s,1h),5.35(t,j=5.5hz,1h),4.61(t,j=5.1hz,1h),4.53(d,j=5.5hz,2h),4.16(t,j=7.1hz,2h),3.45-3.39(m,2h),1.98-1.91(m,2h)。
[0418]
步骤3:2-(4-氟苯基)-7,8-二氢-4h,6h-吡唑并[5,1-c][1,4]氧氮杂卓
[0419]
向3-[3-(4-氟苯基)-5-(羟基甲基)吡唑-1-基]丙-1-醇(80mg,0.31mmol)于甲苯(10ml)中的混合物中添加硫酸(157mg,1.59mmol)。将反应在110℃下搅拌4h。将混合物浓缩以去除有机物,用h2o(20ml)稀释,用1m naoh处理至ph约为7,然后用ea(30ml*2)和h2o(30ml)萃取。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,通过柱色谱(dcm/meoh=30/1)纯化以产生白色固体(20mg,产率:27%)。化学式:计算值(m h
)c
13h13
fn2o:232.10,实验值:233.1。1h nmr(400mhz,dmso)δ7.79
–
7.75(m,2h),7.21(t,j=8.9hz,2h),6.63(s,1h),4.63(s,2h),4.46
–
4.42(m,2h),3.98
–
3.95(m,2h),1.89
–
1.84(m,2h)。
[0420]
步骤4:3-溴-2-(4-氟苯基)-7,8-二氢-4h,6h-吡唑并[5,1-c][1,4]氧氮杂卓
[0421]
向2-(4-氟苯基)-4h,6h,7h,8h-吡唑并[3,2-c][1,4]氧杂氮杂环庚烷(20mg,0.09mmol)于dcm(5ml)中的混合物中添加nbs(25mg,0.135mmol)并在室温下搅拌16h。lcms显示检测到所需的ms为主峰。用dcm(30ml*2)和h2o(30ml)萃取混合物。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,然后通过柱色谱(pe/ea=2/1)纯化以产生白色固体(16mg,产率:56%)。化学式:计算值(m h
)c
13h12
brfn2o:310.01,实验值:311.0。1h nmr(400mhz,dmso)δ7.84
–
7.79(m,2h),7.33
–
7.27(m,2h),4.68(s,2h),4.53
–
4.49(m,2h),4.02
–
3.98(m,2h),1.94-1.87(m,2h)。
[0422]
步骤5:2-(4-氟苯基)-3-(吡啶-4-基)-7,8-二氢-4h,6h-吡唑并[5,1-c][1,4]氧氮杂卓
[0423]
向3-溴-2-(4-氟苯基)-7,8-二氢-4h,6h-吡唑并[5,1-c][1,4]氧氮杂卓(16mg,0.05mmol)于二恶烷/h2o(5ml/1ml)中的溶液中添加吡啶-4-基硼烷二醇(19mg,0.15mmol)、cs2co3(25mg,0.075mmol)和pd(dppd)cl2(9mg,0.01mmol),并将混合物在干燥n2下在100℃下搅拌4h。将混合物浓缩以去除有机溶剂、用h2o(20ml)稀释、然后用ea(30ml
×
2)和h2o(30ml)萃取。将有机层用盐水(50ml)洗涤、经无水na2so4干燥、过滤并浓缩,通过柱色谱(dcm/meoh=30/1)纯化以产生粗产物,然后通过hplc(gemini-c18 150
×
21.2mm,5um,acn
–
h2o 0.1%fa,梯度10%至40%)纯化以产生白色固体(3.2mg,产率:20%,hplc:99.72%)。化学式:计算值(m h
)c
18h16
fn3o:309.13,实验值:310.1。1h nmr(400mhz,dmso)δ8.52(s,2h),7.39
–
7.34(m,2h),7.22(d,j=4.7hz,2h),7.08(t,j=8.8hz,2h),4.71(s,2h),4.61
–
4.58(m,2h),4.13
–
4.10(m,2h),2.08-2.02(m,2h)。
[0424]
实施例25.制备化合物3-11
[0425]
如图25中所示,具体合成步骤如下:
[0426]
步骤1:3-(4-氟苯基)-1-甲基-4-(1h-吡唑并[3,4-b]吡啶-4-基)-1h-吡咯-2-甲腈
[0427]
将4-溴-1h-吡唑并[3,4-b]吡啶(46mg,0.23mmol)、2m na2co3水溶液(0.5ml)和pd(dppf)cl2(22mg,0.03mmol)加入3-(4-氟苯基)-1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1h-吡咯-2-甲腈(50mg,0.15mmol)于etoh/甲苯=8:2(5ml)中的溶液中。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。通过制备型tlc(pe/ea=1:10)纯化残余物以产生产物。化学式:计算值(m h
)c
18h12
fn5:317.33,实验值:317.8。1h nmr(400mhz,cdcl3)δ12.43(s,1h),8.49(d,j=4.9hz,1h),7.76(s,1h),7.31(dd,j=6.0,2.8hz,2h),7.25(s,1h),7.04(t,j=8.7hz,2h),6.87(d,j=4.9hz,1h),3.97(s,3h)。
[0428]
实施例26.制备化合物2-41
[0429]
如图26中所示,具体合成步骤如下:
[0430]
步骤1:1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-甲酸乙酯(2)
[0431]
向5-(4-氟苯基)-2h-吡唑-3-甲酸乙酯(1.8g,0.0077mol)于丙酮(20ml)中的溶液中添加1-溴-2-氯乙烷(5.5213g,0.0385mol)和k2co3(3.1878g,0.0231mol)。将反应混合物在70℃下搅拌4h。通过lcms检测到混合物完成。将混合物浓缩,然后添加h2o,过滤并浓缩滤饼。化学式:计算值(m h
)c
13h12
clfn2o2:282.1,实验值:283.0。
[0432]
步骤2:(1-(2-氯乙基)-3-(4-氟苯基)-1h-吡唑-5-基)甲醇(3)
[0433]
在0℃下向2-(2-氯乙基)-5-(4-氟苯基)吡唑-3-甲酸乙酯(1.6g,0.0054mol)于四氢呋喃(30ml)中的溶液中添加lah(0.2160g,0.0054mol)。将反应混合物在0-25℃下搅拌2h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生粗产物。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
12h12
clfn2o:254.1,实验值:255.0。
[0434]
步骤3:2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪(4)
[0435]
在0℃下向[2-(2-氯乙基)-5-(4-氟苯基)吡唑-3-基]甲醇(900mg,3.53mmol)于dmf(10ml)中的溶液中添加nah(84.72mg,3.53mmol),并将反应混合物在0-25℃下搅拌2h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生粗产物。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
12h11
fn2o:218.1,实验值:219.0。
[0436]
步骤4:3-溴-2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪(5)
[0437]
向2-(4-氟苯基)-4h,6h,7h-吡唑并[3,2-c][1,4]恶嗪(350mg,1.6mmol)于dcm(10ml)中的溶液中添加nbs(284.8mg,1.6mmol)。将反应混合物在25℃下搅拌16h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应。将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生粗产物。将粗物质加入硅胶
柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
12h10
brfn2o:290.0,实验值:296.9。
[0438]
步骤5:2-(4-氟苯基)-3-(吡啶-4-基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪
[0439]
向2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[3,2-c][1,4]恶嗪(50mg,0.15mmol)于1,2-二甲氧基乙烷(5ml)中的溶液中添加4-溴哒嗪(47.70mg,0.3mmol)、na2co3(47.70mg,0.4499mmol)和pd(dtbpf)cl2(9.76mg,0.015mmol)。将反应混合物在90℃下搅拌16h。通过lcms检测到反应完成。将混合物过滤并浓缩以产生产物。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。通过通用制备型hplc纯化残余物以产生产物。
[0440]1h nmr(400mhz,dmso)δ9.24(dd,j=5.3,1.2hz,1h),9.05(dd,j=2.3,1.2hz,1h),7.56(dd,j=5.3,2.4hz,1h),7.46(dd,j=15.2,8.7hz,1h),7.39(dd,j=8.8,5.5hz,2h),7.23(t,j=8.9hz,2h),5.82(d,j=15.2hz,1h),5.70(s,1h),5.07(d,j=8.7hz,1h),4.53(d,j=5.5hz,2h)。
[0441]
实施例27.制备化合物3-12
[0442]
如图27中所示,具体合成步骤如下:
[0443]
步骤1:3-(4-氟苯基)-1-甲基-4-(1h-吡咯并[2,3-b]吡啶-4-基)-1h-吡咯-2-甲腈
[0444]
将4-溴-1h-吡咯并[2,3-b]吡啶(46mg,0.23mmol)、2m na2co3水溶液(0.5ml)和pd(dppf)cl2(22mg,0.03mmol)加入3-(4-氟苯基)-1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1h-吡咯-2-甲腈(50mg,0.15mmol)于etoh/甲苯=8:2(5ml)中的溶液中。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。通过制备型tlc(pe/ea=1:10)纯化残余物以产生产物。化学式:计算值(m h
)c
19h13
fn4:316.34,实验值:316.8。1h nmr(400mhz,cdcl3)δ9.66(s,1h),7.32
–
7.29(m,2h),7.27(d,j=2.0hz,2h),7.21(s,1h),7.01(t,j=8.7hz,2h),6.81(s,1h),6.31(s,1h),3.95(s,3h)。
[0445]
实施例28.制备化合物2-42
[0446]
如图28中所示,具体合成步骤如下:
[0447]
步骤1:4-(2-(4-氟苯基)-6,7-二氢-4h-吡唑并[5,1-c][1,4]恶嗪-3-基)吡啶-2-胺
[0448]
向3-溴-2-(4-氟苯基)-4h,6h,7h-吡唑并[3,2-c][1,4]恶嗪(500mg,1.68mmol)于etoh/phme/h2o=8:4:1(13ml)中的溶液中添加4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)吡啶-2-胺(739.46mg,3.36mmol)、na2co3(534.24mg,5.04mmol)和pd(dppf)cl2(245.62mg,0.336mmol)。将反应混合物在100℃下搅拌1h。通过lcms检测到反应完成。将混合物过滤并浓缩以产生产物。将粗物质加入硅胶柱并用etoac洗脱。1h nmr(301mhz,dmso)δ7.81(dd,j=4.8,1.2hz,1h),7.45
–
7.38(m,2h),7.22
–
7.15(m,2h),6.21
–
6.17(m,2h),5.87(s,2h),4.81(s,2h),4.17(d,j=4.8hz,2h),4.13(d,j=4.8hz,2h)。
[0449]
实施例29.制备化合物2-43
[0450]
如图29中所示,具体合成步骤如下:
[0451]
步骤1:4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺
[0452]
将4-(3,3,4,4-四甲基环戊基)吡啶-2-胺(102mg,0.46mmol)、2m na2co3水溶液
(0.5ml)和pd(dppf)cl2(45mg,0.06mmol)加入5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(120mg,0.31mmol)于etoh/甲苯=8:2(5ml)中的溶液中。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。通过制备型tlc(pe/ea=1:4)纯化残余物以产生产物。化学式:计算值(m h
)c
24h22
fn5:399.47,实验值:399.8。
[0453]
步骤2:4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺
[0454]
将hcoonh4(157mg,2.5mmol)和pd/c(200mg)加入4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺(100mg,0.25mmol)于meoh(10ml)中的溶液中。将混合物在60℃下搅拌2h。过滤溶液、收集滤液并将溶液在压力下浓缩。化学式:计算值(m h
)c
17h16
fn5:309.35,实验值:309.8。
[0455]
步骤3:1-(3-(2-氨基吡啶-4-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0456]
将acoac(29mg,0.29mmol)和et3n(88mg,0.87mmol)加入4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺(90mg,0.29mmol)于dcm(10ml)中的溶液中。将混合物在室温下搅拌2h。将反应混合物在压力下浓缩。通过制备型tlc(dcm/meoh=20:1)纯化残余物以产生产物。化学式:计算值(m h
)c
19h18
fn5o:351.39,实验值:351.8。1h nmr(400mhz,cdcl3)δ7.71(d,j=16.3hz,1h),7.39(dd,j=8.4,5.0hz,2h),7.04(dd,j=20.6,12.0hz,2h),6.59(d,j=10.3hz,1h),6.39(d,j=5.6hz,1h),4.85(d,j=39.9hz,2h),4.34(dd,j=20.7,15.4hz,2h),4.23
–
3.95(m,2h),2.37
–
2.14(m,3h)。
[0457]
实施例30.制备化合物3-13
[0458]
如图30中所示,具体合成步骤如下:
[0459]
步骤1:3-(4-氟苯基)-4-(3h-咪唑并[4,5-b]吡啶-7-基)-1-甲基-1h-吡咯-2-甲腈
[0460]
将7-溴-3h-咪唑并[4,5-b]吡啶(137mg,0.69mmol)、2m na2co3水溶液(0.5ml)和pd(dppf)cl2(67mg,0.092mmol)加入3-(4-氟苯基)-1-甲基-4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1h-吡咯-2-甲腈(150mg,0.46mmol)于etoh/甲苯=8:2(5ml)中的溶液中。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。通过制备型tlc(pe/ea=1:1)纯化残余物以产生产物。化学式:计算值(m h
)c
18h12
fn5:317.33,实验值:317.8。1h nmr(400mhz,cdcl3)δ8.19(s,1h),7.93(d,j=8.4hz,1h),7.32(td,j=5.6,2.1hz,3h),7.04(dd,j=12.0,5.3hz,2h),6.98(d,j=8.4hz,1h),3.90(s,3h)。
[0461]
实施例31.制备化合物2-44
[0462]
如图31中所示,具体合成步骤如下:
[0463]
步骤1:1-(2-(4-氟苯基)-3-(2-甲基嘧啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0464]
将pd(dppf)cl2(28.5mg,0.039mmol)、4-溴-2-甲基嘧啶(101.2mg,0.585mmol)、cs2co3(381mg,1.17mmol)加入1-(2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮(150mg,0.39mmol)于二恶烷/h2o(20:4ml)中的溶液中。使混合物在n2下在90℃下反应16h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(25:1)洗脱以产生黄色固体产物(36mg,产率:25.7%)。化学式:计算值(m h
)化学式:c
19h18
fn5o:351.15,实验值:352。1h nmr
(400mhz,cdcl3)δ8.37(d,j=5.4hz,1h),7.52
–
7.40(m,2h),7.12(q,j=8.7hz,2h),6.77(dd,j=14.4,5.3hz,1h),5.17(d,j=8.5hz,2h),4.42
–
4.23(m,2h),4.16(t,j=5.4hz,1h),4.01(t,j=5.4hz,1h),2.74(d,j=4.6hz,3h),2.28(d,j=11.0hz,3h)。
[0465]
实施例32.制备化合物2-45
[0466]
如图32中所示,具体合成步骤如下:
[0467]
步骤1:1-(3-(2-乙基吡啶-4-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0468]
将pd(dppf)cl2(41.7mg,0.057mmol)、4-溴-2-乙基吡啶(159mg,0.855mmol)、cs2co3(557mg,1.71mmol)加入1-[2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-5-基]乙酮(220mg,0.57mmol)于二恶烷/h2o(20:4ml)中的溶液中。使混合物在n2下在90℃下反应16h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(25:1)洗脱以产生黄色固体产物(15.5mg,产率:7.4%)。化学式:计算值(m h
)化学式:c
21h21
fn4o:364.17,实验值:365。1h nmr(400mhz,cdcl3)δ8.50(dd,j=17.6,4.7hz,1h),7.39(dd,j=8.6,5.5hz,2h),7.01(t,j=8.7hz,2h),6.93(s,2h),4.81(d,j=51.7hz,2h),4.32(dt,j=27.2,5.5hz,2h),4.09(dt,j=61.0,5.5hz,2h),2.79(q,j=7.5hz,2h),2.21(d,j=36.5hz,3h),1.25(t,j=7.6hz,3h)。
[0469]
实施例33.制备化合物2-46
[0470]
如图33中所示,具体合成步骤如下:
[0471]
步骤1:1-(3-(6-氨基嘧啶-4-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0472]
将pd(dppf)cl2(28.5mg,0.039mmol)、4-溴-2-异丙基吡啶(117mg,0.585mmol)、cs2co3(381mg,1.17mmol)加入1-[2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-5-基]乙酮(150mg,0.39mmol)于二恶烷/h2o(20:4ml)中的溶液中。使混合物在n2下在90℃下反应16h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(25:1)洗脱以产生黄色固体产物(52mg,产率:35%)。化学式:计算值(m h
)化学式:c
22h23
fn4o:378.19,实验值:379。1h nmr(400mhz,cdcl3)δ8.53(dd,j=15.1,5.0hz,1h),7.39(dd,j=8.2,5.7hz,2h),7.06
–
6.91(m,4h),4.82(d,j=51.0hz,2h),4.32(dt,j=27.0,5.5hz,2h),4.09(dt,j=60.3,5.5hz,2h),3.03(dt,j=13.8,6.9hz,1h),2.21(d,j=35.0hz,3h),1.23(d,j=6.9hz,6h)。
[0473]
实施例34.制备化合物2-47
[0474]
如图34中所示,具体合成步骤如下:
[0475]
步骤1:3-(4-氟苯基)-1h-吡唑-5-醇
[0476]
在25℃下向3-(4-氟苯基)-3-氧代丙酸甲酯(20g,101.9mmol)于etoh(250ml)中的混合物中添加氢重氮烯(6.3g,203.8mmol)。将反应在60℃下搅拌3h。lcms显示检测到所需的ms为主峰。将混合物浓缩并通过柱色谱(pe/ea=6/1)纯化以产生紫色固体(17.2g,产率:98%)。化学式:计算值(m h
)c9h7fn2o:178.05,实验值:179。
[0477]
步骤2:6-(4-氟苯基)-2,3-二氢吡唑并[5,1-b]恶唑
[0478]
在25℃下向3-(4-氟苯基)-1h-吡唑-5-醇(1200mg,6.74mmol)于acn(50ml)中的混合物中添加1,2-二溴乙烷(2500mg,13.48mmol)和k2co3(3728mg,27mmol)。将反应在80℃下
搅拌3h。lcms显示检测到所需的ms为主峰。将反应混合物冷却至室温并在压力下浓缩、用水(50ml)稀释、用dcm(50ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(10:1)洗脱以产生粉色固体产物(3700mg,产率:80.6%)。化学式:计算值(m h
)c
11
h9fn2o:204.07,实验值:205。
[0479]
步骤3:7-溴-6-(4-氟苯基)-2,3-二氢吡唑并[5,1-b]恶唑
[0480]
在冰冷却下向6-(4-氟苯基)-2,3-二氢吡唑并[5,1-b]恶唑(3600mg,17.63mmol)于乙腈(100ml)中的搅拌溶液中添加nbs(1883mg,10.57mmol)。将混合物在25℃下搅拌1h。lcms显示检测到所需的ms为主峰。将反应混合物在压力下浓缩并用水(50ml)稀释、用ea(50ml*3)萃取、过滤并在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(4:1)洗脱以产生黄色固体产物(1080mg,产率:19.48%)。化学式:计算值(m h
)c
11
h8brfn2o:283.18,实验值:284。
[0481]
步骤4:4-(6-(4-氟苯基)-2,3-二氢吡唑并[5,1-b]恶唑-7-基)吡啶-2-胺
[0482]
将pd(dppf)cl2(38.78mg,0.053mmol)、4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)吡啶-2-胺(134.2mg,0.61mmol)、cs2co3(518mg,1.59mmol)加入7-溴-6-(4-氟苯基)-2,3-二氢吡唑并[5,1-b]恶唑(150mg,0.53mmol)于二恶烷/h2o(15:3ml)中的溶液中。使混合物在n2下在90℃下反应16h。将反应混合物冷却至室温并在压力下浓缩。将粗物质加入硅胶柱并用meoh/dcm(25:1)洗脱以产生黄色固体产物(14.9mg,产率:9.3%)。化学式:计算值(m h
)化学式:c
16h13
fn4o:296.11,实验值:297。1h nmr(400mhz,cdcl3)δ7.70(d,j=6.0hz,1h),7.51
–
7.43(m,2h),7.11(t,j=8.7hz,2h),6.58(d,j=6.0hz,1h),6.46(s,1h),5.71(s,2h),5.26
–
5.19(m,2h),4.46
–
4.40(m,2h)。
[0483]
实施例35.制备化合物2-48
[0484]
如图35中所示,具体合成步骤如下:
[0485]
步骤1:(2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)(1-甲基哌啶-4-基)甲酮
[0486]
将甲基哌啶-4-甲酸(41.8mg,0.29mmol)、t3p(155mg,0.49mmol)和dipea(150.9mg,1.17mmol)加入2-(4-氟苯基)-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(60mg,0.19mmol)于dcm(5ml)中的溶液中。将混合物在25℃下搅拌3h。将反应混合物在压力下浓缩。通过制备型tlc(dcm/meoh=5:1)纯化残余物以产生产物。化学式:计算值(m h
)c
25h28
fn5o:433.53,实验值:433.8。1h nmr(400mhz,cdcl3)δ8.48(d,j=25.6hz,1h),8.34(s,1h),7.37(s,2h),7.12
–
6.84(m,3h),4.79(d,j=43.8hz,2h),4.33(d,j=28.8hz,2h),4.11(d,j=48.7hz,2h),3.30(s,2h),3.03(s,2h),2.66(d,j=20.2hz,4h),2.54(s,3h),2.40
–
1.92(m,4h)。
[0487]
实施例36.制备化合物2-49
[0488]
如图36中所示,具体合成步骤如下:
[0489]
步骤1:2-(4-氟苯基)-5-甲基-3-(2-甲基吡啶-4-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪
[0490]
向4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]-2-甲基吡啶(50mg,0.16mmol)于hcooh(3ml)中的溶液中添加hcho(24mg,0.8mmol)。将反应混合物在70℃下搅拌2h。通过lcms检测到反应未完成。将反应混合物在压力下浓缩。用na2co3将混合物
调整至ph=8。将有机相用水(5ml)洗涤。用ea(5ml*3)萃取残余物。将溶剂经硫酸钠干燥,并在radleys排出装置中在氮气流下干燥以产生粗产物。过滤溶液、收集滤液。将反应混合物在压力下浓缩。通过通用制备型hplc纯化残余物以产生产物。
[0491]1h nmr(400mhz,cdcl3)δ8.42(d,j=5.2hz,1h),7.42
–
7.36(m,2h),7.05
–
6.97(m,2h),6.94(s,1h),6.90(d,j=4.8hz,1h),4.30(t,j=5.6hz,2h),3.67(s,2h),3.01
–
2.94(m,2h),2.54(d,j=4.0hz,6h)。
[0492]
实施例37.制备化合物3-14
[0493]
如图37中所示,具体合成步骤如下:
[0494]
步骤1:3-(4-氟苯基)-1h-吡咯-2-甲酸甲酯
[0495]
向1-乙炔基-4-氟苯(3000mg,24.97mmol)于nmp(50ml)中的溶液中添加2-异氰基乙酸甲酯(3711.3410mg,37.455mmol)、ag2co3(2756.6880mg,9.988mmol)。将反应混合物在80℃下搅拌1h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生粗产物。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
12h10
fno2:219.1,实验值:220.0。
[0496]
步骤2:3-(4-氟苯基)-4-碘-1h-吡咯-2-甲酸甲酯
[0497]
然后在0℃下向3-(4-氟苯基)-1h-吡咯-2-甲酸甲酯[800mg,3.65mmol]于ccl4[10ml]中的溶液中添加乙酰氯[592.6030mg,3.65mmol]。将反应混合物在0℃下搅拌15min。通过lcms检测到反应完成。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
12
h9fino2:345.0,实验值:345.9。
[0498]
步骤3:1-(2-((叔丁氧羰基)氨基)乙基)-3-(4-氟苯基)-4-碘-1h-吡咯-2-甲酸甲酯
[0499]
然后向3-(4-氟苯基)-4-碘-1h-吡咯-2-甲酸甲酯[400mg,1.16mmol]于dmf[10ml]中的溶液中添加n-(2-氯乙基)氨基甲酸叔丁酯[416.776mg,2.32mmol]、k2co3[480.24mg,3.48mmol]。将反应混合物在100℃下搅拌16h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。化学式:计算值(m na
)c
19h22
fin2o4:488.1,实验值:510.9。
[0500]
步骤4:1-(2-氨基乙基)-3-(4-氟苯基)-4-碘-1h-吡咯-2-甲酸甲酯
[0501]
然后向1-(2-{[(叔丁氧基)羰基]氨基}乙基)-3-(4-氟苯基)-4-碘吡咯-2-甲酸甲酯[500mg,1.0240mmol]于dcm[3ml]中的溶液中添加hcl-二恶烷[3ml]。将反应混合物在25℃下搅拌2h。通过lcms检测到反应完成。将反应混合物在压力下浓缩。用na2co3将混合物调整至ph=8。用ea(
×
5ml)萃取残余物。将溶剂经硫酸钠干燥,并在radleys排出装置中在氮气流下干燥以产生粗产物。过滤溶液、收集滤液。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
14h14
fin2o2:388.0,实验值:388.9。
[0502]
步骤5:8-(4-氟苯基)-7-碘-3,4-二氢吡咯并[1,2-a]吡嗪-1(2h)-酮
[0503]
然后向1-(2-氨基乙基)-3-(4-氟苯基)-4-碘吡咯-2-甲酸甲酯[300mg,0.77mmol]于meoh[5ml]中的溶液中添加k2co3[159.39mg]。将反应混合物在50℃下搅拌2h。通过lcms检测到反应完成。用h2o(50ml)淬灭反应,将混合物浓缩。用ea(50ml*3)萃取混合物。将合并的
有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。计算值(m h
)c
13h10
fin2o:356.0,实验值:356.8。
[0504]
步骤6:8-(4-氟苯基)-7-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-3,4-二氢吡咯并[1,2-a]吡嗪-1(2h)-酮
[0505]
然后向8-(4-氟苯基)-7-碘-2h,3h,4h-吡咯并[1,2-a]吡嗪-1-酮[120mg,0.34mmol]于1,4-二恶烷[5ml]中的溶液中添加4,4,5,5-四甲基-2-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-1,3,2-二恶硼烷[129.5089mg,0.51mmol]、kaco[99.9600mg,1.02mmol]、pd(dppf)cl2[49.7080mg]。将反应混合物在100℃下搅拌16h。通过lcms检测到反应完成。用ea(50ml*3)萃取混合物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。计算值(m h
)c
19h22
bfn2o3:356.2,实验值:357.0。
[0506]
步骤7:4-(8-(4-氟苯基)-1-氧代-1,2,3,4-四氢吡咯并[1,2-a]吡嗪-7-基)呋喃并[3,4-b]吡啶-5(7h)-酮
[0507]
然后向8-(4-氟苯基)-7-(4,4,5-三甲基-1,3,2-二恶硼烷-2-基)-2h,3h,4h-吡咯并[1,2-a]吡嗪-1-酮[50mg,0.14mmol]于1,4-二恶烷[5ml]中的溶液中添加4-溴-7h-呋喃并[3,4-b]吡啶-5-酮[44.94mg,0.2100mmol]、na2co3[44.52mg,0.42mmol]、pd(dppf)cl2[20.47mg,0.0280mmol]。将反应混合物在100℃下搅拌16h。通过lcms检测到反应完成。过滤溶液、收集滤液。将反应混合物在压力下浓缩。通过通用制备型hplc纯化残余物以产生产物。
[0508]1h nmr(400mhz,cdcl3)δ8.39(d,j=5.2hz,1h),8.00(s,1h),7.69(s,1h),7.52(s,1h),7.01(dd,j=10.0,6.8hz,2h),6.72(d,j=5.2hz,1h),5.82(s,1h),5.28(s,1h),4.37
–
4.23(m,2h),3.76(s,2h)。
[0509]
实施例38.制备化合物2-50
[0510]
如图38中所示,具体合成步骤如下:
[0511]
步骤1:1-(4-氟苯基)-6-羟基己烷-1,3-二酮
[0512]
在0℃下在氩气下向nah(60%,0.56g,14.4mmol)于无水乙醚(15ml)中的混合物中添加乙醇(2ml),接着添加二氢呋喃-2(3h)-酮(0.62g,7.2mmol)。然后在0℃下缓慢添加1-(4-氟苯基)乙-1-酮(1g,7.2mmol)于乙醚(5ml)中的溶液。并且使所得悬浮液缓慢温至室温。将反应混合物在25℃下搅拌72h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
12h13
fo3:224.23,实验值:224.8。
[0513]
步骤2:3-(3-(4-氟苯基)-1h-吡唑-5-基)丙-1-醇
[0514]
向1-(4-氟苯基)-6-羟基己烷-1,3-二酮(500mg,2.23mmol)于etoh(20ml)中的溶液中添加水合肼(wt 80%,178mg,4.46mmol)。将混合物在25℃下搅拌1h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
12h13
fn2o:220.25,实验值:220.8。
[0515]
步骤3:3-(3-(4-氟苯基)-1h-吡唑-5-基)甲磺酸丙酯
[0516]
在0℃下向3-(3-(4-氟苯基)-1h-吡唑-5-基)丙-1-醇(500mg,2.27mmol)于dcm(20ml)中的溶液中添加甲磺酰氯(0.264ml,3.4mmol)和dipea(880mg,6.8mmol)。将混合物在25℃下搅拌1h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
13h15
fn2o3s:298.33,实验值:298.8。
[0517]
步骤4:2-(4-氟苯基)-5,6-二氢-4h-吡咯并[1,2-b]吡唑
[0518]
在0℃下向3-(3-(4-氟苯基)-1h-吡唑-5-基)甲磺酸丙酯(300mg,1.0mmol)于dmf(15ml)中的溶液中添加nai(15.08mg,0.1mmol)和nah(29mg,1.2mmol)。在0℃下添加混合物持续0.5h,然后在25℃下搅拌1h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
12h11
fn2:202.23,实验值:202.8。
[0519]
步骤5:3-溴-2-(4-氟苯基)-5,6-二氢-4h-吡咯并[1,2-b]吡唑
[0520]
向2-(4-氟苯基)-5,6-二氢-4h-吡咯并[1,2-b]吡唑(200mg,0.99mmol)于ch3cn(15ml)中的溶液中添加nbs(264mg,1.48mmol)。将混合物在25℃下搅拌2h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
12h10
brfn2:281.13,实验值:280.8。
[0521]
步骤6:4-(2-(4-氟苯基)-5,6-二氢-4h-吡咯并[1,2-b]吡唑-3-基)吡啶-2-胺
[0522]
向3-溴-2-(4-氟苯基)-5,6-二氢-4h-吡咯并[1,2-b]吡唑(250mg,0.89mmol)于etoh/甲苯=8:2(15ml)中的溶液中添加4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)吡啶-2-胺(294mg,1.3mmol)、2m na2co3水溶液(1.5ml)和pd(dppf)cl2(130mg,0.18mmol)。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。化学式:计算值(m h
)c
17h15
fn4:294.33,实验值:294.8。1h nmr(400mhz,cdcl3)δ8.45(s,2h),7.65
–
7.50(m,1h),7.48
–
7.35(m,2h),7.07(ddd,j=8.7,5.3,1.9hz,2h),6.61
–
6.36(m,2h),4.25(t,j=7.1hz,2h),3.09(t,j=7.1hz,2h),2.80
–
2.64(m,2h)。
[0523]
实施例39.制备化合物2-51
[0524]
如图39中所示,具体合成步骤如下:
[0525]
步骤1:4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺
[0526]
将4-(3,3,4,4-四甲基环戊基)吡啶-2-胺(102mg,0.46mmol)、2m na2co3水溶液(0.5ml)和pd(dppf)cl2(45mg,0.06mmol)加入5-苄基-3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(120mg,0.31mmol)于etoh/甲苯=8:2(5ml)中的溶液中。将混合物在氮气氛下在微波上在100℃下搅拌1h。将反应混合物在压力下浓缩。通过制备型tlc(pe/ea=1:4)纯化残余物以产生产物。化学式:计算值(m h
)c
24h22
fn5:399.47,实验值:399.8。
[0527]
步骤2:4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺
[0528]
将hcoonh4(157mg,2.5mmol)和pd/c(200mg)加入4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺(100mg,0.25mmol)于meoh(10ml)中的溶液中。将混合物在60℃下搅拌2h。过滤溶液、收集滤液并将溶液在压力下浓缩。化学式:计算值(m h
)c
17h16
fn5:309.35,实验值:309.8。
[0529]
实施例40.制备化合物2-52
[0530]
如图40中所示,具体合成步骤如下:
[0531]
步骤1:(4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯
[0532]
向4-[2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶-2-胺(2000mg,4.99mmol)于dcm(20ml)中的溶液中添加(boc)2o(1631.7300mg,7.485mmol)、dipea(1287.4200mg,9.98mmol)、dmap(121.7560mg,0.9980mmol)。将反应混合物在25℃下搅拌2h。通过lcms检测到反应完成。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
29h30
fn5o2:499.2,实验值:500.1。
[0533]
步骤2:(4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯
[0534]
向(4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯(1200mg,2.39mmol)于ea(10ml)中的溶液中添加pd/c(800mg)和acoh(0.5ml)。将反应混合物在h2下在25℃下搅拌2h。通过lcms检测到反应完成。过滤溶液、收集滤液。将反应混合物在压力下浓缩。将有机相用2m碳酸钠溶液(5ml)洗涤。用ea(10ml*3)萃取残余物。将合并的有机层用盐水(10ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。化学式:计算值(m h
)c
22h24
fn5o2:409.2,实验值:410.1。
[0535]
步骤3:(4-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯
[0536]
向(4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯(230mg,0.5603mmol)于thf(5ml)中的溶液中添加甲醛(84.05mg,2.8015mmol)。将反应混合物在70℃下搅拌16h。添加三乙酰氧基硼氢化钠(142.54mg,0.6723mmol)。将反应混合物在25℃下搅拌2h。通过lcms检测到反应完成。将反应混合物在压力下浓缩。将有机相用水(5ml)洗涤。用ea(5ml*3)萃取残余物。将合并的有机层用盐水(10ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱。化学式:计算值(m h
)c
23h26
fn5o2:423.2,实验值:424.0。
[0537]
步骤4:4-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-胺
[0538]
然后向(4-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)氨基甲酸叔丁酯(200mg,0.47mmol)于dcm(4ml)中的溶液中添加hcl-二恶烷(4ml)。将反应混合物在25℃下搅拌2h。通过lcms检测到反应完成。将反应混合物在压力下浓缩。将有机相用2m碳酸钠溶液(5ml)洗涤。用ea(5ml*3)萃取残余物。将合并的有机层用盐水(10ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。通过通用制备型hplc纯化残余物以产生产物。
[0539]1h nmr(400mhz,dmso)δ7.83(d,j=5.2hz,1h),7.46
–
7.37(m,2h),7.21
–
7.14(m,2h),6.21(dd,j=7.4,2.0hz,2h),5.90(s,2h),4.16(t,j=5.4hz,2h),3.57(s,2h),2.90(t,j=5.5hz,2h),2.41(s,3h)。
[0540]
实施例41.制备化合物2-53
[0541]
如图41中所示,具体合成步骤如下:
[0542]
步骤1:1-(2-(4-氟苯基)-3-(1h-吡咯并[2,3-b]吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0543]
向1-[2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-5-基]乙酮(1000mg,2.5958mmol)于1,4-二恶烷/h2o=10:1(30ml)中的溶液中添加4-溴-1h-吡咯并[2,3-b]吡啶(767.18mg,3.8937mmol)、na2co3(825.46mg,7.7874mmol)、pd(dppf)cl2(379.51mg,0.5191mmol)。将反应混合物在100℃下搅拌16h。通过lcms检测到反应完成。过滤溶液、收集滤液。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用dcm/meoh(10:1)洗脱。化学式:计算值(m h
)c
21h18
fn5o:375.1,实验值:376.0。
[0544]
步骤2:4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-3,
3-二溴-1,3-二氢-2h-吡咯并[2,3-b]吡啶-2-酮
[0545]
在室温下经6h向1-(2-(4-氟苯基)-3-(1h-吡咯并[2,3-b]吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮(500mg,1.3319mmol)于t-buoh(10ml)中的溶液中分小份添加py-hbr3(1704.83mg,5.3276mmol)。将反应混合物在压力下浓缩。将有机相用水(5ml)洗涤。用ea(5ml*3)萃取残余物。将合并的有机层用盐水(50ml)洗涤、经硫酸钠干燥、过滤并浓缩以产生产物。化学式:计算值(m h
)c
21h16
br2fn5o2:547.0,实验值:547.5。
[0546]
步骤3:4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-1,3-二氢-2h-吡咯并[2,3-b]吡啶-2-酮
[0547]
向4-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-3,3-二溴-1,3-二氢-2h-吡咯并[2,3-b]吡啶-2-酮(220mg,0.4006mmol)于ea(5ml)中的溶液中添加pd/c(100mg)。将反应混合物在h2下在25℃下搅拌5h。通过lcms检测到反应完成。过滤溶液、收集滤液。将反应混合物在压力下浓缩。通过通用制备型hplc纯化残余物以产生产物。
[0548]1h nmr(400mhz,meod)δ8.09(dd,j=50.2,26.5hz,2h),7.45
–
7.34(m,2h),7.09(t,j=8.8hz,2h),7.02(dd,j=14.8,5.2hz,1h),4.78(d,j=12.4hz,2h),4.31(dt,j=36.0,5.6hz,2h),4.12(dt,j=11.0,5.6hz,2h),2.86(s,2h),2.19(d,j=39.6hz,3h)。
[0549]
实施例42.制备化合物2-54
[0550]
如图42中所示,具体合成步骤如下:
[0551]
步骤1:(4-溴吡啶-2-基)乙酸
[0552]
在-78℃下向4-溴-2-甲基吡啶(4.0g,0.0233mol)于thf(30ml)中的溶液中添加碳酸二甲酯(2.52g,0.0279mol)和lda(13.8ml)。将混合物在-78℃下搅拌1h,并在室温下搅拌5h。lcms显示反应完成。将反应用饱和nh4cl水溶液淬灭并用水洗涤、用ea萃取、经na2so4干燥。通过硅胶柱(pe:etoac=4:1)纯化粗产物以产生产物。化学式:c8h8brno2。计算值(m h
)
:
230.06,实验值:231.1。
[0553]
步骤2:2-{4-[2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶-2-基}乙酸甲酯
[0554]
在室温下向2-(4-溴吡啶-2-基)乙酸甲酯(150mg,0.652mmol)于dmf(5ml)中的溶液中添加2-(4-氟苯基)-5-(1-甲基苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪(566.37mg,1.304mmol)、pd(dppf)cl2(53.2mg,0.0652mmol)和k2co3(180.23mg,1.304mmol)。将混合物在n2下在90℃下搅拌3h。lcms显示反应完成。将溶液用盐水洗涤并用ea萃取、经na2so4干燥。通过硅胶柱(pe:etoac=10:1)纯化粗产物以产生产物。化学式:c
27h25
fn4o
2.
计算值(m h
):456.52,实验值:457.1。
[0555]
步骤3:2-(4-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)乙酸甲酯
[0556]
在室温下向2-{4-[2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶-2-基}乙酸甲酯(300mg,0.6557mmol)于meoh(10ml)中的溶液中添加pd/c(150mg)。将混合物搅拌6h。过滤反应并在真空下浓缩滤液。通过通用制备型hplc纯化残余物以产生产物。化学式:c
21h21
fn4o
2.
计算值(m h
):380.42,实验值:381.1。
[0557]1h nmr(400mhz,dmso)δ8.41(d,j=4.0hz,1h),7.41
–
7.34(m,2h),7.18(t,j=8.9hz,2h),7.12-7.09(m,1h),7.02(dd,j=5.1,1.5hz,1h),4.19(t,j=4.0hz,2h),3.80
(s,2h),3.65(s,2h),3.60(s,3h),2.92(t,j=5.4hz,2h),2.41(s,3h)。
[0558]
实施例43.制备化合物2-55
[0559]
如图43中所示,具体合成步骤如下:
[0560]
步骤1:1-(4-溴吡啶-2-基)丙-2-酮
[0561]
向4-溴-2-甲基吡啶(3g,17.4mmol)于thf(30ml)中的溶液中添加lda(13ml,26mmol,2n)。将反应混合物在-78℃下搅拌1h。添加n-甲氧基-n-甲基乙酰胺(2.69g,26mmol)。将反应混合物在25℃下搅拌15h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(10:1)洗脱以产生产物。化学式:计算值(m h
)c8h8brno:214.06,实验值:214.0。
[0562]
步骤2:1-(4-(5-苄基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)丙-2-酮
[0563]
向2-(4-氟苯基)-5-(1-甲基苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪(6g,13.9mmol)于二恶烷(30ml)和水(5ml)中的溶液中添加1-(4-溴吡啶-2-基)丙-2-酮(2g,9.3mmol)、碳酸钾(2.57g,18.6mmol)和四(三苯基膦)钯(1g,0.9mmol)。将反应混合物在100℃下搅拌18h。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用pe/etoac(1:1)洗脱以产生产物。化学式:计算值(m h
)c
27h25
fn4o:440.52,实验值:441.1。
[0564]
步骤3:1-(4-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)吡啶-2-基)丙-2-醇
[0565]
向1-{4-[2-(4-氟苯基)-5-(1-甲基苯基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶-2-基}丙-2-酮(1200mg)于meoh(30ml)中的溶液中添加pd/c(100mg)。将反应混合物在25℃下搅拌15h。过滤溶液、收集滤液。将反应混合物在压力下浓缩以产生产物。化学式:计算值(m h
)c
20h21
fn4o:352.41,实验值:353.8。
[0566]
步骤4:1-(2-(4-氟苯基)-3-(2-(2-羟基丙基)吡啶-4-基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0567]
在0℃下向1-{4-[2-(4-氟苯基)-4h,5h,6h,7h-吡唑并[1,5-a]吡嗪-3-基]吡啶-2-基}丙-2-醇(300mg,0.85mmol)和nahco3(143mg,1.7mmol)于thf(10ml)和水(10ml)中的溶液中添加乙酸乙酰酯(174mg,1.7mmol)。将反应混合物在25℃下搅拌5h。用thf(10ml*2)萃取残余物。将反应混合物在压力下浓缩。通过通用制备型hplc纯化残余物以产生产物。
[0568]1h nmr(400mhz,meod)δ8.40(d,j=5.3hz,1h),8.32(s,1h),7.41(dd,j=8.7,5.4hz,2h),7.18
–
7.00(m,4h),4.30(dt,j=34.8,5.2hz,2h),4.10(ddd,j=20.0,12.4,5.7hz,3h),2.82(qd,j=13.3,6.5hz,2h),2.21(d,j=35.0hz,3h),1.21
–
1.11(m,3h)。
[0569]
实施例44.制备化合物2-56
[0570]
如图44中所示,具体合成步骤如下:
[0571]
步骤1:7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2,3-二氢呋喃并[3,2-b]吡啶
[0572]
向3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(100mg,0.34mmol)于二恶烷(5ml)和水(0.5ml)中的溶液中添加7-(3,3,4,4-四甲基环戊硼烷-1-基)-2,3-二氢呋喃并[3,2-b]吡啶(85.11mg,0.35mmol)、pd(dppf)cl2(10mg,10%w/w))和k3po4(144.34mg,
0.68mmol)。将混合物在n2气氛下在90℃下搅拌4h。反应混合物用水稀释并将水相用ea萃取两次,将有机相合并并在压力下浓缩。通过制备型tlc纯化残余物以产生奶白色固体产物(77.92mg,68.6%)。
[0573]
化学式:计算值(m h
):c
19h17
fn4o:336.37,实验值:337。
[0574]
步骤2:1-(3-(2,3-二氢呋喃并[3,2-b]吡啶-7-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0575]
在0℃下向7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2,3-二氢呋喃并[3,2-b]吡啶(35mg,0.10mmol)于dcm(65ml)中的溶液中添加tea(20.24mg,0.20mmol),然后滴加乙酰氯(8.64mg,0.11mmol)。将混合物在25℃下搅拌2h。将反应混合物用水稀释并将水相用dcm萃取两次,将有机相合并并在压力下浓缩。通过通用制备型hplc纯化残余物以产生白色固体产物(30.95mg,78.6%)。
[0576]
化学式:计算值(m h
):c
21h19
fn4o2,378.41,实验值:339。
[0577]
实施例45.制备化合物2-57
[0578]
如图45中所示,具体合成步骤如下:
[0579]
步骤1:1-(3-(2,3-二氢呋喃并[3,2-b]吡啶-7-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0580]
在室温下在氮气氛下向7-溴-2,3-二氢呋喃并[3,2-b]吡啶(68mg,0.34mmol,1.0当量)和2-(4-氟苯基)-5-甲基-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(121mg,0.34mmol,1.0当量)于甲苯(15ml)中的搅拌混合物中添加cs2co3(230mg,0.69mmol,2.0当量)和pd(dppf)cl
2 ch2cl2(28mg,0.034mmol,0.1当量)。将所得混合物在氮气氛下在100℃下搅拌2h。将所得混合物在减压下浓缩。将残余物通过硅胶柱色谱纯化、用pe/ea(1:2)洗脱以提供浅棕色固体状的1-(3-(2,3-二氢呋喃并[3,2-b]吡啶-7-基)-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮(36mg,28%)。
[0581]
实施例46.制备化合物2-58
[0582]
如图46中所示,具体合成步骤如下:
[0583]
步骤1:1-(3-溴-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮
[0584]
在0℃下向7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基-2,3-二氢异恶唑并[4,5-b]吡啶(150mg,0.51mmol)于dcm(10ml)中的溶液中添加tea(103.21mg,1.02mmol),然后滴加乙酰氯(40.04mg,0.51mmol)。将混合物在25℃下搅拌2h。将反应混合物用水稀释并将水相用dcm萃取两次,将有机相合并并在压力下浓缩。通过制备型tlc纯化残余物以产生奶白色固体产物(147.14mg,85.9%)。
[0585]
化学式:计算值(m h
)c
14h13
brfn3o:338.18,实验值:339。
[0586]
步骤2:7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基-2,3-二氢异恶唑并[4,5-b]吡啶
[0587]
向1-(3-溴-2-(4-氟苯基)-6,7-二氢吡唑并[1,5-a]吡嗪-5(4h)-基)乙-1-酮(35mg,0.10mmol)于二恶烷(3ml)和水(0.5ml)中的溶液中添加2-甲基-7-(3,3,4,4-四甲基环戊硼烷-1-基)-2,3-二氢异恶唑并[4,5-b]吡啶(28.40mg,0.11mmol)、pd(dppf)cl2(3.5mg,10%w/w)和k3po4(42.45mg,0.20mmol)。将混合物在n2气氛下在90℃下搅拌4h。将反应混合物用水稀释并将水相用ea萃取两次,将有机相合并并在压力下浓缩。通过通用制备
型hplc纯化残余物以产生白色固体产物(15.27mg,37.5%)。
[0588]
化学式:计算值(m h
)c
21h20
fn5o2:393.42,实验值:394。
[0589]
实施例47.制备化合物2-59
[0590]
如图47中所示,具体合成步骤如下:
[0591]
步骤1:7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基-2,3-二氢异恶唑并[4,5-b]吡啶
[0592]
向3-溴-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(100mg,0.34mmol)于二恶烷(5ml)和水(0.5ml)中的溶液中添加2-甲基-7-(3,3,4,4-四甲基环戊硼烷-1-基)-2,3-二氢异恶唑并[4,5-b]吡啶(87.78mg,0.34mmol)、pd(dppf)cl2(10mg,10%w/w))和k3po4(144.34mg,0.68mmol)。将混合物在n2气氛下在85℃下搅拌6h。将反应混合物用水稀释并将水相用ea萃取两次,将有机相合并并在压力下浓缩。通过制备型tlc纯化残余物以产生灰色固体产物(85.31mg,71.9%)。
[0593]
化学式:计算值(m h
)c
19h18
fn5o:351.39,实验值:352。
[0594]
步骤2:7-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基-2,3-二氢异恶唑并[4,5-b]吡啶
[0595]
向7-(2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)-2-甲基-2,3-二氢异恶唑并[4,5-b]吡啶(50mg,0.14mmol)于thf(65ml)中的溶液中添加多聚甲醛(4.50mg,0.15mmol),并将混合物在25℃下搅拌30min。然后在0℃下添加氰基硼氢化钠()17.60mg,0.28mmol)。将混合物在25℃下搅拌2h。将反应用水淬灭并用ea萃取两次。将有机相合并并在压力下浓缩。通过通用制备型hplc纯化残余物以产生白色固体产物(24.91mg,47.9%)。
[0596]
化学式:计算值(m h
)c
20h20
fn5o:365.41,实验值:366。
[0597]
实施例48.制备化合物2-60
[0598]
如图48中所示,具体合成步骤如下:
[0599]
步骤1:7-(2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)呋喃并[3,2-b]吡啶-2(3h)-酮
[0600]
向3-溴-2-(4-氟苯基)-5-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪(30mg,0.10mmol)于二恶烷(3ml)和水(0.3ml)中的溶液中添加7-(3,3,4,4-四甲基环戊硼烷-1-基)呋喃并[3,2-b]吡啶-2(3h)-酮(28.29mg,0.11mmol)、pd(dppf)cl2(3mg,10%w/w)和k3po4(42.45mg,0.20mmol)。将混合物在n2气氛下在90℃下搅拌4h。将反应混合物用水稀释并将水相用ea萃取两次,将有机相合并并在压力下浓缩。通过制备型tlc纯化残余物以产生黄色固体产物(16.14mg,45.8%)。
[0601]
化学式:计算值(m h )c
20h17
fn4o2:364.38,实验值:365。
[0602]
实施例49.制备化合物2-61
[0603]
如图49中所示,具体合成步骤如下:
[0604]
步骤1:7-(5-乙酰基-2-(4-氟苯基)-4,5,6,7-四氢吡唑并[1,5-a]吡嗪-3-基)呋喃并[3,2-b]吡啶-2(3h)-酮
[0605]
向1-[2-(4-氟苯基)-3-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)-4h,6h,7h-吡唑并[1,5-a]吡嗪-5-基]乙酮(100mg,0.259mmol)于1,4-二恶烷/h2o=10:1(30ml)中的溶液中添加7-溴呋喃并[3,2-b]吡啶-2(3h)-酮(83.23mg,0.389mmol)、na2co3(82.546mg,
0.778mmol)、pd(dppf)cl2(37.951mg,0.0519mmol)。将反应混合物在100℃下搅拌16h。通过lcms检测到反应完成。过滤溶液、收集滤液。将反应混合物在压力下浓缩。将粗物质加入硅胶柱并用dcm/meoh(10:1)洗脱。化学式:计算值(m h
)c
21h17
fn4o3:392.3,实验值:393.0。
[0606]
实施例50.生物活性
[0607]
在1μl的ck1δ抑制剂(例如,本技术的化合物)或4%dmso(例如,作为对照)存在下,用含有50mm tris、10mm mgcl2、1mm二硫苏糖醇、含10μm atp的100μg/ml bsa、2nm野生型ck1δ和42μm肽底物plsrtlpsvaslpgl(flotow等,1990)的缓冲剂(40μl,ph 7.5)进行ck1δ激酶分析。将反应混合物在25℃下培育85min;如关于kinase-glo分析(promega)所述进行检测。在perkin elmer envision酶标仪(perkinelmer,waltham,ma)上测量发光输出。
[0608]
将bmal1-dluc或per2-dluc u2os细胞悬浮于培养基(补充有10%胎牛血清、0.29mg/ml l-谷氨酰胺、100单位/ml青霉素和100mg/ml链霉素的dmem)中并以每孔200μl(10,000个细胞)涂在96孔白色实心底培养板上。2天后,每孔分配100μl外植体(explant)培养基(补充有2%b27、10mm hepes、0.38mg/ml碳酸氢钠、0.29mg/ml l-谷氨酰胺、100单位/ml青霉素、100mg/ml链霉素、0.1mg/ml庆大霉素和1mm荧光素的dmem,ph7.2),接着施用1μl本技术的化合物(溶解于dmso中;在dmso中的最终浓度为0.7%)。用光学透明膜覆盖培养板并设置到酶标仪(infinite m200,tecan)。每1h记录一次发光值,持续3至4天。通过曲线拟合程序cellularhythm或multicycle(actimetrics)从发光节律中获得周期参数,两种程序产生类似的结果。
[0609]
ck1δ抑制结果(ic50)汇总在表5中。
[0610]
表5
[0611]
表5中的“*”表示该值超过测量范围,例如该值》100000。
[0612]
ck1δ抑制结果(ec50)汇总在表6中。
[0613]
表6
[0614]
表6中的“*”表示该值超过测量范围,例如该值》30。
[0615]
实施例51.药物转运分析
[0616]
制备caco-2细胞
[0617]
分别向transwell插片(insert)和储存器(reservoir)的每个孔中加入50μl和25ml细胞培养基。然后将hts transwell培养板在37℃、5%co2下培育1小时,之后进行细胞接种。
[0618]
用培养基将caco-2细胞稀释至6.86
×
105个细胞/ml,并将50μl细胞悬浮液分配至96孔hts transwell培养板的过滤孔中。将细胞在37℃、5%co2、95%相对湿度的细胞培养箱中培养14至18天。从初始种植后不超过24小时开始,每隔一天替换一次细胞培养基。
[0619]
制备储备溶液
[0620]
在dmso中制备10mm测试化合物储备溶液。在dmso中制备浓度为10mm的阳性对照储备溶液。在此分析中,使用地高辛(digoxin)和普萘洛尔(propranolol)作为对照化合物。
[0621]
评估细胞单层完整性
[0622]
从储存器和每个transwell插片中去除培养基并用经过预热的新鲜培养基替换。
[0623]
使用millicell上皮电压电阻测量系统(millipore,usa)测量跨单层的跨上皮电阻(teer)。
[0624]
测量结束,即将培养板返回培养箱中。
[0625]
根据下式计算teer值:teer测量值(ohm)*膜面积(cm2)=teer值(ohm
·
cm2)teer值应大于230ohm
·
cm2,表示合格的caco-2单层。
[0626]
分析程序
[0627]
将caco-2培养板从培养箱中去除,并用经过预热的hbss(10mm hepes,ph 7.4)洗涤两次,然后在37℃下培育30分钟。
[0628]
将对照化合物和测试化合物的储备溶液在dmso中稀释以得到1mm溶液,然后用hbss(10mm hepes,ph 7.4)稀释以得到5μm工作溶液。培育体系中dmso的最终浓度为0.5%。
[0629]
为了确定从顶端到基底外侧方向的药物转运速率,将125μl的对照化合物与测试化合物的5μm工作溶液加入transwell插片(顶端室)中,并将50μl样品(d0样品)立即从顶端室转移到新的96孔培养板。用235μl hbss(10mm hepes,ph 7.4)填充接收培养板的孔(基底外侧室)。
[0630]
为了确定从基底外侧到顶端方向的药物转运速率,将285μl的对照化合物与测试化合物的5μm工作溶液加入接收培养板的孔(基底外侧室)中,并将50μl样品(d0样品)立即从基底外侧室转移到新的96孔培养板。用75μl hbss(10mm hepes,ph 7.4)填充transwell插片的孔(顶端室)。进行一式两份分析。
[0631]
将培养板在37℃下培育2小时。
[0632]
培育结束后,将来自供体侧(对于从顶端流向基底外侧(ap
→
bl)而言为顶端室,且对于从基底外侧流向顶端(bl
→
ap)而言为基底外侧室)和接收侧(对于从顶端流向基底外侧(ap
→
bl)而言为基底外侧室,且对于从基底外侧流向顶端(bl
→
ap)而言为顶端室)的50μl样品转移到新的96孔培养板的孔中,接着添加4体积的含有适当内标物(is)的冷甲醇。使样品涡旋5分钟,然后以3,220g离心40分钟。将一份100μl上清液与适当体积的超纯水混合,然后进行lc-ms/ms分析。
[0633]
为了确定在2小时转运期之后的荧光黄泄漏结果,在水中制备荧光黄储备溶液,并用hbss(10mm hepes,ph 7.4)稀释达到100μm的最终浓度。向每个transwell插片(顶端室)
中加入100μl荧光黄溶液,接着用300μl hbss(10mm hepes,ph 7.4)填充接收培养板的孔(基底外侧室)。将这些培养板在37℃下培育30分钟。(使用基底外侧通道孔)从顶端孔和基底外侧孔直接去除80μl样品,并转移至新的96孔培养板的孔中。在485nm激发和530nm发射的荧光酶标仪中测量荧光黄荧光信号(用以监控单层完整性)。
[0634]
数据分析
[0635]
可使用下式计算caco-2药物转运分析的表观渗透系数(p
app
),单位为厘米/秒:
[0636]
p
app
=(va×
[药物]
接收体
)/(面积
×
时间
×
[药物]
初始,供体
)
[0637]
其中va为接收孔的体积(ml),面积为膜的表面积(对于transwell-96孔通透性支持物而言为0.143cm2)且时间为总的转运时间(以秒计)。
[0638]
将使用下式来确定外排率:
[0639]
外排率=p
app(b-a)
/p
app(a-b)
[0640]
其中p
app(b-a)
表示从基底外侧到顶端方向的表观渗透系数,且p
app(a-b)
表示从顶端到基底外侧方向的表观渗透系数。
[0641]
可使用下式来确定回收率:
[0642]
回收率%=(va×
[药物]
接收体
vd×
[药物]
供体
)/(vd×
[药物]
初始,供体
)
[0643]
其中va为接收孔中的体积(以ml计)(对于从顶端流向基底外侧(ap
→
bl)而言为0.235ml且对于从基底外侧流向顶端(bl
→
ap)而言为0.075ml)、vd为供体孔中的体积(以ml计)(对于从顶端流向基底外侧(ap
→
bl)而言为0.075ml且对于从基底外侧流向顶端(bl
→
ap)而言为0.235ml)。
[0644]
可使用下式计算荧光黄泄漏率,单位以百分比(%)计:
[0645]
%ly泄漏率=100
×
[ly]
接收体
/([ly]
供体
[ly]
接收体
)
[0646]
ly泄漏率《1%为可接受的,表示合格的caco-2单层。
[0647]
p
app(b-a)
、p
app(a-b)
和外排率汇总在表7中。
[0648]
表7化合物p
app(a-b)
(10-6
,cm/s)p
app(b-a)
(10-6
,cm/s)外排率地高辛0.3016.9856.52普萘洛尔26.5513.630.512-438.530.83.62-5019.2517.320.92-5222.7623.871.05
[0649]
实施例52.内在清除率分析
[0650]
1.根据表8制备主溶液。
[0651]
表8试剂储备浓度体积最终浓度磷酸盐缓冲液200mm200μl100mm超纯h2o-106μl-mgcl2溶液50mm40μl5mm
[0652]
2.如下进行三次独立实验。a)使用nadph:将10μl的20mg/ml肝微粒体和40μl的10mm nadph加入培育。微粒体和nadph的最终浓度分别为0.5mg/ml和1mm。b)不使用nadph:
将10μl的20mg/ml肝微粒体和40μl的超纯h2o加入培育。微粒体的最终浓度为0.5mg/ml。c)不含nadph的热灭活微粒体:将10μl的20mg/ml热灭活肝微粒体和40μl的超纯h2o加入培育。微粒体的最终浓度为0.5mg/ml。
[0653]
3.反应从添加4μl的200μm测试化合物溶液或最终浓度为2μm的对照化合物溶液开始,并在37℃下进行。
[0654]
4.在0、15、30、45和60min时从反应溶液中取50μl等分试样。通过添加4体积含内标物(100nm阿普唑仑(alprazolam)、200nm拉贝洛尔(labetalol)、200nm咖啡因和2μm酪洛芬)的冷乙腈终止反应。将样品以3,220g离心40分钟。将一份100μl上清液与100μl的超纯h2o混合,然后进行lc-ms/ms分析。
[0655]
5.数据分析
[0656]
使用microsoft excel进行所有计算。
[0657]
由提取的离子色谱图确定峰面积。通过对母体药物剩余百分比的自然对数与孵育时间的曲线作线性回归来确定斜率值k。
[0658]
活体外半衰期(活体外t
1/2
)由斜率值来确定:
[0659]
活体外t
1/2
=-(0.693/k)
[0660]
使用下式将活体外t
1/2
(min)转化为活体外内在清除率(活体外cl
int
,单位为μl/min/mg蛋白)(两次测定的平均值):
[0661]
活体外cl
int
=(0.693*培育体积(μl))/(活体外t
1/2
*蛋白的量(mg))
[0662]
使用下式将活体外t
1/2
(min)转化为放大未结合内在清除率(放大cl
int
,单位为ml/min/kg)(两次测定的平均值):
[0663]
肝微粒体中内在清除率预测值的比例因子汇总在表9中。
[0664]
表9
[0665]
a.iwatsubo等,davies和morris,1993,10(7)第1093-1095页。
[0666]
b.barter等,2007,curr drug metab,8(1),第33-45页;iwatsubo等,1997,jpet,283第462-469页。
[0667]
papp(b-a)、papp(a-b)和外排率汇总在表10中。
[0668]
表10
[0669]
尽管在本文中已示出和描述了本技术的优选实施例,但是对于本领域技术人员显而易见的是,这些实施例仅作为示例提供。本发明并不意在受说明书中提供的具体示例的限制。尽管已参考前述说明书描述了本发明,但本文中对实施例的描述和说明并不意味着被解释为具有限制意义。在不背离本发明的情况下,本领域技术人员现在将想到多种变化、改变和替换。此外,应了解,本发明的所有方面不限于本文所阐述的具体描述、配置或相对比例,其取决于各种条件和变量。应了解,在实施本发明时可以采用对本文描述的本技术实施例的各种替代方案。因此,预期本发明还应涵盖任何这样的替代、修改、变化或等效物。以下权利要求书旨在定义本技术的范畴,并且这些权利要求范畴内的方法和结构及其等效物由此被涵盖。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。