用于组合治疗冠状病毒的pld
技术领域
1.本发明涉及使用普立肽(pld)组合治疗冠状病毒感染。
背景技术:
2.冠状病毒(cov)是包膜的单股正链rna病毒,其基因组范围在26.2-31.7kb之间。这个大的、帽状的和多聚腺苷酸化的基因组包含七个常见的冠状病毒基因,其顺序如下:5'-orf1a-orf1b-s-orf3-e-m-n-3'。orf1a/b产生基因组长度mrna(mrna1),其编码多蛋白1a(pp1a)和pp1ab形式的两个重叠的病毒复制酶蛋白。这些多蛋白被病毒编码的蛋白酶酶解成成熟的非结构蛋白(nsp1至nsp16),非结构蛋白组装形成膜相关的病毒复制酶-转录酶复合物(rtc)。基因组的最后三分之一产生亚基因组(sg)mrna,其编码四种结构蛋白,刺突(s)、包膜(e)、膜(m)和核衣壳(n),以及许多辅助蛋白。cov属于niodovirales目的冠状病毒科的冠状病毒亚科。该科包括α-冠状病毒、β-冠状病毒、γ-冠状病毒和δ-冠状病毒四个属。(sars)严重急性呼吸综合征-cov-2和sars-cov属于β-冠状病毒属,约占其基因组的80%。冠状病毒n蛋白在感染细胞内大量产生。n蛋白具有多种功能,包括与病毒rna结合形成螺旋状核衣壳,并在冠状病毒组装中具有结构作用。n蛋白也被认为在病毒复制、转录和翻译中具有作用。
3.冠状病毒(cov)感染各种人类和动物宿主,引起各种疾病,从动物胃肠道感染、脑炎和脱髓鞘,到人类大多数上呼吸道相对轻微的感染。然而,人畜共患冠状病毒、sars-cov、mers-cov和sars-cov2可导致严重疾病和死亡。由sars-cov2引起的疾病被称为2019年冠状病毒病或covid-19。
4.普立肽(pld)是一种环状缩酚酸肽,最初从海洋海鞘白色念珠菌(aplidium albicans)中分离出来。pld也被称为aplidin。pld目前正在进行概念验证研究,以评估covid-19患者的pld的安全性。
5.世卫组织宣布2019-2020冠状病毒疫情为国际公共卫生紧急事件(pheic)。根据世卫组织的数据,截至2021年2月12日,共有107252265例sars-cov-2病例,包括2355339例死亡。对于sars-cov感染,包括sars-cov(引起sars)和sars-cov-2(引起covid-19),没有特定的治疗方法。已经开发了一些疫苗,并自2020年12月起批准用于预防covid-19的个人免疫接种。然而,由于病毒突变、疫苗接种和/或其他因素,患有covid-19的患者的住院率仍然很高。
6.正在对住院患者进行大量药物和不同方案的测试,以确定管理covid-19感染的合适策略。然而,到目前为止,还没有明确的治疗策略。虽然一些药物已显示出疗效,但其他药物没有或在其他方面不适用。因此,对于cov感染的治疗,特别是covid-19的治疗,存在着迫切的未满足的医疗需求。本发明解决了这一需求。
技术实现要素:
7.在一个方面,本发明涉及用于治疗冠状病毒(cov)感染的pld或其药学上可接受的
盐或立体异构体,其中pld或其药学上可接收的盐或立体异构体与一种或多种其他cov抗病毒剂组合使用。
8.令人惊讶地发现,根据本发明的组合保持活性并具有可接受的毒性。
9.在另一方面,提供了一种或多种用于治疗冠状病毒(cov)感染的其他cov抗病毒剂,其中所述一种或更多种其他cov抗病毒剂与pld或其药学上可接受的盐或立体异构体组合使用。
10.在另一方面,提供了一种治疗冠状病毒(cov)感染的方法,其包括向有需要的患者施用pld或其药学上可接受的盐或立体异构体以及一种或多种其他cov抗病毒剂的组合。
11.在另一方面,提供了pld或其药学上可接受的盐或立体异构体在制备用于治疗冠状病毒(cov)感染的药物中的用途,其中在所述治疗中还施用一种或多种其他cov抗病毒剂。
12.在另一方面,提供了一种或多种其他cov抗病毒剂在制备用于治疗冠状病毒(cov)感染的药物中的用途,其中在所述治疗中还施用pld或其药学上可接受的盐或立体异构体。
13.在另一方面,提供了pld或其药学上可接受的盐或立体异构体与一种或多种其他cov抗病毒剂的组合。
14.在另一方面,提供了包括根据本发明的pld的组合,其用作药物。
15.在另一方面,提供了包括根据本发明的pld的组合,其用于治疗冠状病毒(cov)感染。
16.在另一方面,提供了一种药物组合物,其包含药学上可接受的稀释剂和包含根据本发明的pld的组合。
17.在另一方面,提供了一种试剂盒,其包含组合,所述组合包含根据本发明的pld;所述试剂盒可选地进一步包括用于治疗患者的说明书;所述说明书可选地提供用于治疗冠状病毒(cov)感染的根据本发明的组合的使用说明。
18.冠状病毒(cov)感染导致疾病covid-19。因此:
19.在另一方面,提供了用于治疗covid-19的pld或其药学上可接受的盐或立体异构体,其中pld或其药学上可接收的盐或立体异构体与一种或多种其他cov抗病毒剂组合使用。
20.在另一方面,提供了一种或多种用于治疗covid-19的一种或几种其他cov抗病毒剂,其中所述一种或多种其他cov抗病毒药与pld或其药学上可接受的盐或立体异构体组合使用。
21.在另一方面,提供了一种治疗covid-19的方法,其包括向有需要的患者施用pld或其药学上可接受的盐或立体异构体以及一种或多种其他cov抗病毒剂的组合。
22.在另一方面,提供了pld或其药学上可接受的盐或立体异构体在制备用于治疗covid-19的药物中的用途,其中在所述治疗中还施用一种或多种其他cov抗病毒剂。
23.在另一方面,提供了一种或多种其他cov抗病毒剂在制备用于治疗covid-19的药物中的用途,其中在所述治疗中还施用pld或其药学上可接受的盐或立体异构体。
24.在另一方面,提供了pld或其药学上可接受的盐或立体异构体与一种或多种其他cov抗病毒剂的组合。
25.在另一方面,提供了包括根据本发明的pld的组合,其用作药物。
26.在另一方面,提供了包括根据本发明的pld的组合,其用于治疗covid-19。
27.在另一方面,提供了一种药物组合物,其包含药学上可接受的稀释剂和包含根据本发明的pld的组合。
28.在另一方面,提供了一种试剂盒,其包含组合,所述组合包含根据本发明的pld;所述试剂盒可选地进一步包括用于治疗患者的说明书;所述说明书可选地提供用于治疗covid-19的根据本发明的组合的使用说明。
29.以下实施例适用于本发明的所有方面。
30.所述一种或多种其他cov抗病毒剂可选自抑制病毒进入、抑制病毒细胞融合、抑制内吞或抑制病毒复制的药剂。
31.所述一种或多种其他cov抗病毒剂可选自钙蛋白酶、组织蛋白酶或钙蛋白酶/组织蛋白酶抑制剂、rna聚合酶抑制剂、氯氰菊酯介导的内吞的抑制剂、hiv-1蛋白酶抑制剂、丝氨酸蛋白酶抑制剂、tmprss2抑制剂、ifn刺激的抗病毒蛋白、pparα受体激动剂、胆固醇转运蛋白抑制剂、细胞内胆固醇转运抑制剂、神经节苷脂生物合成途径抑制剂、胆固醇消耗剂、糖皮质激素、抑制病毒与宿主细胞膜融合的药剂和jak抑制剂。
32.所述一种或多种其他cov抗病毒剂可选自钙蛋白酶、组织蛋白酶或钙蛋白酶/组织蛋白酶抑制剂、rna聚合酶抑制剂、氯氰菊酯介导的内吞的抑制剂、hiv-1蛋白酶抑制剂、丝氨酸蛋白酶抑制剂、tmprss2抑制剂、ifn刺激的抗病毒蛋白和pparα受体激动剂。
33.所述一种或多种其他cov抗病毒剂可以是组织蛋白酶抑制剂、组织蛋白酶b抑制剂、组组织蛋白酶l抑制剂、钙蛋白酶抑制剂、组合的钙蛋白酶和组织蛋白酶a抑制剂、泛组织蛋白酶b/l抑制剂;其中所述药剂可优选选自mdl28170、e64d或ca-074;优选mdl28170。
34.所述一种或多种其他cov抗病毒剂可以是rna聚合酶抑制剂;其中所述药剂可优选选自瑞德西韦或法匹拉韦;优选瑞德西韦。
35.所述一种或多种其他cov抗病毒剂可以是氯氰菊酯介导的内吞的抑制剂;其中所述药剂可优选选自羟氯喹、氯喹,金刚烷胺或氯丙嗪;优选羟氯喹或氯喹;进一步优选羟氯喹。
36.所述一种或多种其他cov抗病毒剂可以是hiv-1蛋白酶抑制剂;其中,所述药剂可优选地选自甲磺酸奈非那韦水合物、洛匹那韦、替拉那韦、安普那韦或达芦那韦,优选甲磺酸奈非那韦水合物。
37.所述一种或多种其他cov抗病毒剂可以是丝氨酸蛋白酶抑制剂,其中所述剂可以优选地选自卡莫司他、溴己辛、抑肽酶或萘莫司他,优选地,卡莫司他。
38.所述一种或多种其他cov抗病毒剂可以是tmprss2抑制剂;其中所述药剂可优选选自卡莫司他、溴己辛、抑肽酶或萘莫司他,优选卡莫司他。
39.所述一种或多种其他cov抗病毒剂可以是ifn刺激的抗病毒蛋白;其中所述药剂可优选选自干扰素2α或干扰素γ。
40.所述一种或多种其他cov抗病毒剂可以是pparα受体激动剂;其中所述药剂可以优选为非诺贝特。
41.所述一种或多种其他cov抗病毒剂可以抑制胆固醇转运体以抑制细胞内胆固醇转运;其中所述药剂可以优选为伊曲康唑。
42.所述一种或多种其他cov抗病毒剂可以是神经节苷脂生物合成途径抑制剂;其中
所述药剂可以优选为nb-dnj。
43.所述一种或多种其他cov抗病毒剂可以是胆固醇消耗剂;其中所述药剂可以优选为甲基β-环糊精。
44.所述一种或多种其他cov抗病毒剂可以是糖皮质激素;其中所述药剂可以优选为环索奈德。
45.所述一种或多种其他cov抗病毒剂可以是抑制病毒与宿主细胞膜融合的药剂;其中所述药剂可以优选为阿比朵尔。
46.所述一种或多种其他cov抗病毒剂可以是jak抑制剂;其中所述药剂可以优选为托法替尼。
47.pld或其药学上可接受的盐或立体异构体以及一种或多种其他cov抗病毒剂可以同时、顺序或单独施用。
48.pld或其药学上可接受的盐或立体异构体与一种或多种其他cov抗病毒剂的摩尔比可以为1:1000至1000:1、1:700至700:1、2:500至500:1、3:300至300:1、4:100至100:1或1:50至50:1。
49.所述化合物可以是pld。
50.冠状病毒(cov)感染的治疗可用于减少与cov感染相关的并发症,包括住院、icu和死亡。
51.冠状病毒(cov)感染的治疗可用于预防、减少或治疗covid持续性(也称为长时间covid或后covid综合征)。
52.冠状病毒(cov)感染的治疗可用于治疗covid-19引起的肺炎。
53.冠状病毒(cov)感染的治疗可用于降低cov患者的传染性。患者可能是无症状的或不是非常有症状的患者。
54.冠状病毒(cov)感染的治疗可用于减少超级感染者(具有高病毒载量(例如tc《25)的无症状或非非常症状患者)的发生。
55.除了pld或其药学上可接受的盐或立体异构体和一种或多种其他cov抗病毒剂之外,本发明的组合还可以包括皮质类固醇。在这些实施例中,该方案将包括pld或其药学上可接受的盐或立体异构体、一种或多种其他cov抗病毒剂和皮质类固醇。皮质类固醇相对于本发明的组合可以同时、单独或顺序施用。皮质类固醇可以是地塞米松。
56.cov感染可以是轻度感染、和/或中度感染、和/或重度感染。
57.所述cov感染可以是急性cov感染,优选地,其中所述cov感染是急性covid-19感染;和/或可以是持续有症状的cov感染,优选地,其中所述cov感染是持续有症状的covid-19感染;和/或可以是后cov综合征、持续性cov或长cov;优选地,其中所述cov感染是后covid-19综合征、持续性covid或长covid。后cov综合征,持续性cov或长cov可包括心血管、呼吸、胃肠、神经、肌肉骨骼、代谢、肾脏、皮肤、耳鼻咽喉、血液学和自主神经系统;精神问题、全身疼痛、疲劳和/或持续发烧引起的一种或多种症状。
58.所述用途可以是治疗具有cov感染(优选covid-19)症状和体征长达4周;和/或4周至12周;和/或超过12周以上的患者。
59.所述用途可以是预防、减少或治疗持续性covid、长covid或后covid综合征;优选的是,预防、减少或治疗可将患者遭受持续性covid、长covid或后covid综合征症状的可能
性降至最低;和/或降低此类症状的严重程度;进一步优选的是,治疗将cov感染的症状降至最低。
60.该治疗可以降低cov患者的传染性,包括无症状或症状不明显但具有高病毒载量的患者。
61.pld或其药学上可接受的盐或立体异构体可以根据每日一次,给药10天、9天、8天、7天、6天、5天、4天、3天、2天或1天的方案给药;优选2-5天、3-5天、或3、4或5天;最优选3天或5天;最优选3天。
62.pld或其药学上可接受的盐或立体异构体的给药剂量可以为:每天5mg或更少、每天4.5mg或更少、每天4mg或更少、每天3.5mg或更少、每天3mg或更少、每天2.5mg或更少、或每天2mg或更少;0.5mg/天、1mg/天、1.5mg/天、2mg/天、2.5mg/天、3mg/天、3.5mg/天、4mg/天、4.5mg/天、或5mg/天;优选1mg/天、1.5mg/天、2mg/天或2.5mg/天;优选1.5-2.5mg/天;进一步优选1.5mg/天、2mg/天或2.5mg/天。
63.pld或其药学上可接受的盐或立体异构体的总给药剂量可以为:1-50mg、1-40mg、1-30mg、1-20mg、1-15mg、3-15mg、3-12mg、4-12mg、4-10mg、4.5-10mg;4.0mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg;优选4.5mg、5mg、6mg、7.5mg、8mg、9mg、或10mg;更优选4.5-7.5mg/天。总剂量可以分成1、2、3、4、5、6、7、8、9或10天,优选3天或5天;最优选3天。
64.pld或其药学上可接受的盐或立体异构体可以以1.5-2.5mg/天的剂量每日一次给药,持续3天。剂量可以为1.5mg/天。剂量可以为2.5mg/天。
65.pld或其药学上可接受的盐或立体异构体可以是以1.5小时注射给药的pld,每天一次,连续3天。可以1.5小时注射给药1.5mg pld,每天一次,连续3天。可以1.5小时注射给药2mg pld,每天一次,连续3天。可以1.5小时注射给药2.5mg pld,每天一次,连续3天。可以1.5小时注射给药1mg pld,每天一次,连续5天。可以1.5小时注射给药2mg pld,每天一次,连续5天。
66.方案可以是单剂量(1天)的pld或其药学上可接受的盐或立体异构体。pld或其药学上可接受的盐或立体异构体可以1-10mg、4-10mg、4.5-10mg、4mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg;优选4.5mg、5mg、6mg、7.5mg、8mg、9mg或10mg;更优选5-9mg、6.5-8.5mg、7-8mg或7.5mg的单剂量给药。该化合物可以是1.5小时注射、单剂量给药的pld。
67.单剂量方案可与本发明所述的所有疗法一起使用。单剂量方案可用于轻度感染病例。然而,单剂量方案可用于中度和/或重度感染病例。与皮质类固醇的组合使用(包括随后的皮质类固醇给药)可以与单剂量方案一起使用在实施例中。
68.多日方案可以与本发明中提出的所有疗法一起使用。多日方案可用于中度和/或重度感染病例。然而,多日方案也可用于轻度感染病例。
69.另外的皮质类固醇可以在与给药pld或其药学上可接受的盐或立体异构体相同的天数每天给药。可在随后的一天或多天给药皮质类固醇。可以在随后的1、2、3、4、5、6、7、8、9、10天或更多天给药皮质类固醇。当与根据本发明的pld或其药学上可接受的盐或立体异构体在同一天给药时,可以以较高剂量给药皮质类固醇,并且在随后的一天或多天以较低剂量给药。皮质类固醇可以是地塞米松。
70.pld或其药学上可接受的盐或立体异构体可以在给药方案的第1-3天以根据本发明的剂量给药。另外的皮质类固醇可以在给药方案的第1-3天静脉注射给药。此后,皮质类固醇可以从第4天至第10天通过口服给药或iv给药(取决于医生根据患者临床状况和进展的判断)。皮质类固醇可以是地塞米松。剂量可以是第1至3天6.6mg/天iv(例如8mg磷酸地塞米松),然后从第4天到第10天口服或iv地塞米松6mg/天(例如7.2mg磷酸地塞米松或6mg地塞米松碱)。
71.在实施例中,地塞米松是磷酸地塞米松,并且例如在第1天至第3天以8mg/天的剂量iv给药,然后从第4天至第10天口服或iv地塞米松7.2mg/天。
72.pld或其药学上可接受的盐或立体异构体可以注射给药,优选1小时注射、1.5小时注射、2小时注射、3小时注射或更长时间,特别优选1.5小时注射。
73.方案可以包括1.5小时注射给药1.5mg普立肽,每天一次,连续3天;或1.5小时注射给药2mg普立肽,每天一次,连续3天;或1.5小时注射给药2.5mg普立肽,每天一次,连续3天;或1.5小时注射给药1mg普立肽,每天一次,连续5天;或1.5小时注射给药2mg普立肽,每天一次,连续5天。
74.方案可以包括在第1天作为单剂量以1.5小时注射给药7.5mg普立肽。
75.pld或其药学上可接受的盐或立体异构体可以使用负荷剂量和维持剂量给药。
76.根据本发明方案的pld或其药学上可接受的盐或立体异构体组分可以是:
77.第1天2.5mg的负荷剂量,随后几天2mg/天的维持剂量;
78.第1天2.5mg的负荷剂量,随后几天1.5mg/天的维持剂量;
79.第1天2.5mg的负荷剂量,随后几天1mg/天的维持剂量;
80.第1天2.5mg的负荷剂量,随后几天0.5mg/天的维持剂量;
81.第1天2mg的负荷剂量,随后几天1.5mg/天的维持剂量;
82.第1天2mg的负荷剂量,随后几天1mg/天的维持剂量;
83.第1天2mg的负荷剂量,随后几天0.5mg/天的维持剂量;
84.第1天1.5mg的负荷剂量,随后几天1mg/天的维持剂量;
85.第1天1.5mg的负荷剂量,随后几天0.5mg/天的维持剂量;或
86.第1天1mg的负荷剂量,随后几天0.5mg/天的维持剂量。
87.pld或其药学上可接受的盐或立体异构体可以与另外的皮质类固醇组合给药。皮质类固醇可以在与pld给药相同的天数给药。
88.皮质类固醇也可以在随后的一天或更多天给药;例如其中皮质类固醇在第1-3天与pld一起给药,皮质类固醇在第4-10天中的一天或更多天进一步给药。
89.皮质类固醇可以在给药pld的天数里静脉注射给药,但在随后的天数里通过口服或iv给药。
90.皮质类固醇可以是地塞米松。地塞米松可以在给药pld的天数里以6.6mg/天的剂量iv给药。
91.地塞米松可以在随后的天数,优选第4、5、6、7、8、9和10天中的一天或更多天,以6mg/天的剂量口服给药或iv给药。
92.本文定义的地塞米松剂量是指碱重量(base weight)。因此,如果以盐形式使用,可以调节剂量。例如,地塞米松可以是磷酸地塞米松,使得8mg/天磷酸地塞米松相当于
6.6mg地塞米松碱,7.2mg/天磷酸地塞米松相当于6mg地塞米松碱。
93.pld可以在第1天至第3天与iv地塞米松6.6mg/天组合静脉注射(iv)给药1.5mg/天,随后从第4天至第10天口服(po)/iv地塞米松6mg/天(取决于医生根据患者临床状况和进展的判断),并与一种或多种其他cov抗病毒剂一起。
94.pld可以在第1天至第3天与iv地塞米松6.6mg/天组合静脉注射(iv)给药2.0mg/天,随后从第4天至第10天口服(po)/iv地塞米松6mg/天(取决于医生根据患者临床状况和进展的判断),并与一种或多种其他cov抗病毒剂一起。
95.pld可以在第1天至第3天与iv地塞米松6.6mg/天组合静脉注射(iv)给药2.5mg/天,然后从第4天至第10天口服(po)/iv地塞米松6mg/天(取决于医生根据患者临床状况和进展的判断),并与一种或多种其他cov抗病毒剂一起。
96.皮质类固醇可在开始用pld治疗前20至30分钟给药。
97.在根据本发明的方案中,患者可另外接受其他药物,优选在开始用pld或其药学上可接受的盐或立体异构体治疗前20至30分钟:
[0098]-昂丹司琼8mg iv(或等同物);
[0099]-盐酸苯海拉明25mg iv(或等同物);以及
[0100]-雷尼替丁50mg iv(或等同物)。
[0101]
在根据本发明的方案中,在第4天和第5天,患者可po接受每天两次4mg的昂丹司琼(或等同物)。
[0102]
当以单剂量给药pld或其药学上可接受的盐或立体异构体(即在一天内而不是多天方案的一部分)时,患者可在注射普立肽前20-30分钟接受以下预防性药物:
[0103]-盐酸苯海拉明25mg i.v;
[0104]-雷尼替丁50mg i.v;
[0105]-地塞米松6.6mg静脉注射;
[0106]-昂丹司琼8mg i.v,缓慢注射15分钟。
[0107]
可在pld给药后的每12小时口服昂丹司琼4mg,持续3天,以减轻药物引起的恶心和呕吐。如果在早晨给予普立肽,患者可在下午接受第一剂量的昂丹司琼。
附图说明
[0108]
在以下非限制性附图中进一步描述本发明:
[0109]
图1显示了在pld浓度增加的情况下,暴露于固定浓度的sars-cov-2的vero e6细胞的细胞病变效应。
[0110]
图2显示了在pld与所示化合物组合浓度增加的情况下,暴露于固定浓度的sars-cov-2的vero e6细胞的细胞病变效应,所述化合物抑制病毒复制(瑞德西韦和奈非那韦)或病毒进入(羟氯喹和mdl28170)。组合时,以相同浓度加入每种药物。药物的使用浓度范围为0.5nm至10μm。显示了一个代表性实验的变量响应曲线的非线性拟合(平方),不包括具有相关毒性的药物浓度数据。还显示了在没有病毒的情况下暴露于药物浓度降低的vero e6细胞的细胞毒性效应(圆圈)。
[0111]
图3-6显示了在瑞德西韦、羟氯喹、mdl28170和甲磺酸奈非那韦浓度增加的情况下,暴露于固定浓度的sars-cov-2的vero e6细胞的细胞病变效应。
[0112]
图7和8显示了根据本发明的给药计划和施用所预测的总血浆浓度曲线随时间变化。
[0113]
图9显示了根据本发明的进一步给药计划和施用所预测的总血浆浓度曲线随时间变化。
[0114]
图10显示了使用1.5小时输注的单剂量普立肽7.5mg以及第1天至第3天1.5、2.0和2.5mg的总浓度与血浆浓度曲线。
具体实施方式
[0115]
现在将进一步描述本发明。在下面的段落中,更详细地限定本发明的不同方面。如此限定的每个方面可与任何其他一个或多个方面组合,除非明确指示不要这样组合。特别地,指示为优选或有利的任一特征可以与指示为优选或有利的任何其他一个或多个特征组合。
[0116]
在本技术中,使用了多个通用术语和短语,其应当被解释如下。
[0117]
除非另有说明,本文所使用的术语“治疗”是指反转、减弱、减轻或抑制该术语所适用的疾病或病症、或该疾病或病症的一种或多种症状的进展。本文所使用的术语治疗还可包括预防性治疗,即设计为防止疾病发生或使疾病发生的可能性最小化的治疗。
[0118]“治疗”(“treat”、“treating”、以及“treatment”)可指以下一项或多项:1)减少感染细胞的数量;2)减少血清中存在的病毒粒子数量;3)抑制(即在一定程度上减缓,优选停止)病毒复制速率;和4)在某种程度上缓解或减轻与cov感染相关的一种或多种症状。
[0119]
治疗可以是治疗cov感染。治疗可以是治疗sars-cov-2感染。治疗可以是治疗covid-19感染。治疗可以是治疗covid-19。治疗可以是治疗由cov感染引起的疾病。治疗可以是治疗由sars-cov-2感染引起的疾病。治疗可以是治疗由cov感染引起的肺炎。治疗可以是治疗由sars-cov-2感染引起的肺炎。治疗可以是治疗由covid-19感染引起的肺炎。治疗可以是治疗由covid-19引起的肺炎。
[0120]
感染可以是中度感染。感染可以是严重感染。感染可以是轻微的感染。
[0121]
治疗可以减少与cov感染相关的并发症,包括住院、icu和死亡。
[0122]
本发明可用于治疗急性covid-19感染(高达4周的covid-19的体征和症状),治疗(或最小化)正在出现症状的covid-19(从4周到12周的covid-19的体征和症状),或治疗或最小化后covid-19综合征(post-covid-19syndrome)(在感染期间或之后出现的与covid-19一致的症状和体征,持续超过12周,并且不能通过其他诊断来解释。它通常表现为一系列症状,通常重叠,这些症状会随着时间的推移而波动和变化,并可能影响身体的任何系统。后covid-19综合征可在12周前考虑,同时也在评估其他潜在疾病的可能性)。本发明的化合物可以治疗具有covid-19症状和体征长达4周的患者。本发明的化合物可治疗具有covid-19症状和体征4周至12周的患者。本发明的化合物可以治疗具有covid-19症状和体征超过12周的患者。
[0123]
治疗可以是预防、减少或治疗持续性covid(covid persistent)(也称为长covid或后covid综合征)。根据本发明的化合物可使患者遭受持续性covid症状的可能性最小化。或者,本发明的化合物可以降低这些症状的严重性,优选可以使cov感染的症状最小化。
[0124]
后covid综合征可被认为是在感染期间或之后出现的与covid-19一致的持续12周
以上的症状和体征,其不能通过其他诊断来解释。这种病症通常会出现一系列症状,通常是重叠的,这些症状可能会随着时间的推移而改变,并可能影响体内的任何系统。许多患有后covid综合征的人也会经历全身疼痛、疲劳、持续的高温和精神问题。症状包括(但不限于)心血管、呼吸系统、胃肠道、神经系统、肌肉骨骼、代谢、肾脏、皮肤病、耳鼻喉科、血液学和自主神经系统出现的症状,此外还有精神疾病、全身疼痛、疲劳和持续发烧。
[0125]
治疗可以是降低cov患者的传染性。本发明实现了病毒负荷(viral burden)的快速且显著的减少。减少病毒负荷可以降低患者的传染性。这对于无症状或症状不明显但具有高病毒载量的患者(例如,tc《25)特别有益。这样的患者可能是超级感染者或超级传播者。在检测到感染时给药根据本发明的化合物可减少病毒负荷并因此减少患者的感染性。
[0126]
所述治疗可导致病毒载量的减少。这可以表现为在给药后第6天,复制周期阈值(ct)大于30(ct》30)。该治疗可从基线开始减少病毒载量。这可以表现为给药后需要住院治疗的患者百分比的降低。这可以表现为在给药后需要有创机械通气和/或进入icu的患者百分比的降低。这可以表现为出现与持续性疾病有关的后遗症的患者减少。这可以表现为被选择为不良预后标准的分析参数(包括例如淋巴细胞减少、ldh、d-二聚体或pcr)正常化的患者百分比的增加。这可以表现为临床标准正常化(包括例如:头痛、发烧、咳嗽、疲劳、呼吸困难(气短)、关节肌痛或腹泻的症状消失)的患者百分比的增加。
[0127]“患者”包括人类、非人类哺乳动物(如狗、猫、兔子、牛、马、绵羊、山羊、猪、鹿等)和非哺乳动物(如鸟类等)。患者需要住院治疗以管理感染。
[0128]
普立肽(plitidepsin)(pld)是一种环状缩酚酸肽,最初从海洋海鞘白色念珠菌(aplidium albicans)中分离得到。pld也被称为aplidin。在优选实施例中,本发明涉及pld的用途。
[0129]
pld的化学名称为(-)-(3s,6r,7s,10r,11s,15s,17s,20s,25as)-11-羟基-3-(4-甲氧基苄基)-2,6,17-三甲基-15-(1-甲基乙基)-7-[[[(2r)-4-甲基-2-[甲基[[(2s)-1-(2-氧代丙酰基)吡咯烷-2-基]羰基]氨基]戊酰基]氨基]-10-[(1s)-1-甲基丙基]-20-(2-甲基丙基)十四氢-15h-吡咯并[2,1-f]-[1,15,4,7,10,20]二氧杂环戊环三环烷-1,4,8,13,16,18,21(17h)-庚酮((-)-(3s,6r,7s,10r,11s,15s,17s,20s,25as)-11-hydroxy-3-(4-methoxybenzyl)-2,6,17-trimethyl-15-(1-methylethyl)-7-[[(2r)-4-methyl-2-[methyl[[(2s)-1-(2-oxopropanoyl)pyrrolidin-2-yl]carbonyl]amino]pentanoyl]amino]-10-[(1s)-1-methylpropyl]-20-(2-methylpropyl)tetradecahydro-15h-pyrrolo[2,1-f]-[1,15,4,7,10,20]dioxatetrazacyclotricosine-1,4,8,13,16,18,21(17h)-heptone),对应分子式c
57h87
n7o
15
。它的相对分子质量为1110.34g/mol,结构如下:
[0130][0131]
人翻译延伸因子eef1a是真核翻译延伸1复合物(eef1)的一亚基。该复合物在蛋白质合成期间将氨基酰化的trna递送至伸长的核糖体。然而,eef1a不仅是主要的翻译因子,也是最重要的多功能蛋白之一,在新合成的蛋白的质量监测、泛素依赖性降解和促进凋亡中起作用。
[0132]
已经显示出cov的n蛋白,例如sars-cov和tgev(传染性胃肠炎冠状病毒),与真核延伸因子1a(eef1a)直接结合。此外,已显示eef1a的敲低导致病毒数目的显著减少,证明n蛋白与eef1a的相互作用对于病毒复制是必要的。
[0133]
已显示pld以高亲和力和低解离速率结合人翻译延伸因子eef1a。flim-phasor fret实验证明,pld定位在肿瘤细胞中足够接近eef1a,从而提示在活细胞中形成药物-蛋白质复合物。pld抗性细胞系(pld-resistant cell lines)还显示降低的eef1a蛋白水平和这些抗性细胞中eef1a的异位表达恢复了对pld的敏感性,表明eef1a直接参与了pld的作用机制。
[0134]
如上所述,cov的n蛋白也与eef1a结合,并且这种结合对于病毒复制是必需的。此外,n蛋白在cov中高度保守,尤其是sars-cov-2与sars-cov中的n蛋白具有约90%的氨基酸同一性。然而,给药pld和随后的与eef1a的结合阻止了cov n蛋白与eef1a的结合。这反过来又可以防止病毒复制。因此,pld和eef1a之间的相互作用降低了从头合成病毒衣壳的效率,并因此导致病毒载量的降低。
[0135]
除此之外,pld与eef1a的结合阻止了eef1a与其通常的结合配偶体的相互作用。一种这样的结合配偶体是dsrna激活的蛋白激酶(pkr或eif2ak2)。pld与eef1a的结合利用eef1a从复合物中释放pkr,导致pkr的激活。pkr是已知的先天免疫应答的激活剂和在抗病毒免疫应答中的关键角色。具体而言,
[0136]
(i)激活的pkr磷酸化起始因子eif2的α亚基,导致形成无活性的eif2复合物;
[0137]
(ii)激活的pkr诱导iκb降解、nf-κb核转位和nf-κb途径活化。nf-κb是调节负责先天和适应性免疫应答的基因,例如参与t细胞发育、成熟和增殖的基因的主要转录因子;
[0138]
(iii)pkr的激活通过fas聚集和nf-κb转位的机制诱导细胞凋亡,从而消除感染细胞。
[0139]
值得注意的是,cov的蛋白4a通过螯合(sequestration)dsrna强烈抑制pkr的激活。pld绕过这种病毒反应,通过从eef1a复合物释放pkr导致pkr激活,如可以从在没有病毒感染的情况下pkr的激活中看到的。
[0140]
最后,除上述外,pld与eef1a的结合也激活了er应激诱导的未折叠蛋白反应
(upr),这反过来又导致了许多抗病毒反应,包括eif2α的磷酸化。
[0141]
通过这些机制的结合-(i)抑制cov n蛋白/eef1a相互作用;(ii)激活pkr和(iii)激活upr;pld阻止cov复制,并导致宿主反应的激活,从而消除cov。两者都有助于有效的病毒疗法。靶向eef1a的另一个优点是它是人类目标,因此不会像病毒蛋白那样突变以逃避pld。
[0142]
如上所述,在真核细胞中,flim-fret实验证明,普立肽位于足够接近eef1a的位置以提示药物-蛋白质复合物在细胞质中的形成。用14c-普立肽和从兔肌肉纯化的eef1a进行的单独的一组实验表明,普立肽以高亲和力和低解离速率结合eef1a。
[0143]
普立肽对sars-cov-2的体外作用
[0144]
进行了旨在确定普立肽对sars-cov-2的作用的若干体外实验并在本文公开。分别使用用sars-cov-2感染的vero e6细胞和直接定量sars-cov-2核衣壳(n)蛋白(其明确涉及到普立肽诱导的抗病毒活性的机制)的两个研究显示,普立肽在体外是sars-cov-2生长的有效抑制剂,其ic
50
为0.7至3.0nm。在另一项研究中,将人干细胞衍生的肺细胞样细胞预防性地暴露于10nm普立肽1小时,然后用sars-cov-2(4
×
104空斑形成单位)感染。在48小时孵育期后,确定抗病毒和细胞毒性普立肽的效果。结果表明,普立肽完全消除了sars-cov-2的复制,对肺细胞样细胞没有明显的细胞毒性。
[0145]
普立肽对体内sars-cov-2的影响
[0146]
使用先前描述的用sars-cov-2感染的腺病毒介导的hace2的小鼠模型,证明了普立肽在体内有效的抗病毒效果。使用先前描述的表达由用sars-cov-2感染的细胞角蛋白-18基因启动子(k18-hace2)驱动的hace2的转基因小鼠模型,证明了普立肽在体内也有效的抗病毒效果。
[0147]
普立肽对宿主炎症反应的影响
[0148]
与sars-cov类似,sars-cov-2感染也会产生多种细胞因子的高度分泌,随着疾病的发展,血浆水平也会增加,这表明细胞因子的释放与疾病严重程度之间可能存在联系。
[0149]
先天免疫是抵御入侵病原体的第一道防线。在sars-cov-2的情况下,病毒进入宿主上皮细胞是由病毒包膜刺突(s)蛋白和细胞表面受体ace2之间的相互作用介导的。病毒rna,作为病原体相关的分子模式,然后被宿主模式识别受体检测,宿主模式识别受体包括toll样受体家族。然后,toll样受体通过激活转录因子核因子κb(nf-κb)上调抗病毒和促炎介质(antiviral andproinflammatory mediator),如白细胞介素(il)6、il-8和干扰素(ifn)-。在非临床物种和患者中探索sarscov感染的研究中,已经强调了nf-κb对促炎基因表达(特别是在肺中)的重要性。在感染sars cov的小鼠中,nf-κb的药理学抑制导致肺中肿瘤坏死因子α(tnf)、ccl2和cxcl2的较高的存活率和降低的表达。
[0150]
早期的体外研究表明,普立肽诱导肿瘤细胞中nfκb的下调。随后,进行体外和离体研究以评估普立肽对免疫细胞的作用。
[0151]
使用thp-1细胞进行体外研究,thp-1细胞是一种自发永生化的单核细胞样细胞系,来自急性单核细胞白血病儿童患者的外周血,广泛用于研究单核细胞结构和功能。结果表明,所有与病原体相关的分子模式模拟化合物(mimicking化合物)都能诱导在thp-1细胞中产生促炎细胞因子,且添加普立肽可显著减少促炎细胞因子的分泌。
[0152]
离体研究评估了普立肽对小鼠肺中细胞因子il 6、il 10和tnfα表达的影响。结果
显示,来自安慰剂治疗的小鼠的分化(cd)45 细胞群能够在lps-b5刺激下产生il 6、il 10和tnfα。然而,与未刺激的对照组相比,来自用普立肽治疗的小鼠的cd45 细胞未能显示出il 6、il 10和tnfα的显著增加。这些结果表明,体内暴露于普立肽阻止了在从支气管肺泡灌洗液分离的cd45 细胞中增加由lps-b5介导的促炎细胞因子的产生。
[0153]
因此,pld可用于治疗cov感染。
[0154]
术语“cov”感染是指冠状病毒科和正冠状病毒亚科的病毒的任何感染。在一个实施例中,感染来自乙型冠状病毒属中的病毒,乙型冠状病毒属包括乙型冠状病毒1(betacoronavirus 1)、人冠状病毒hku1(human coronavirus hku1)、小鼠冠状病毒(murine coronavirus)、蝙蝠冠状病毒hku5(pipistrellus bat coronavirus hku5)、果蝠冠状病毒hku9(rousettus bat coronavirus hku9)、严重急性呼吸综合征相关冠状病毒(sars-cov)、扁颅蝠冠状病毒hku4(tylonycteris bat coronavirus hku4)、中东呼吸综合征相关冠状病毒、人冠状病毒oc43和刺猬冠状病毒1(ericov)。优选地,所述病毒是sars-cov或sars-cov-2,最优选sars-cov-2。sars-cov-2先前称为2019-ncov,并且这些术语在本文中可互换使用。
[0155]
在本发明的方面,本文定义的组合可用于治疗covid-19。covid-19是由sars-cov-2感染引起的疾病。在本发明方面,本文通过其抗病毒活性定义的组合可以用于最小化或防止covid-19相关肺炎的发展。
[0156]
术语“药学上可接受的盐”是指给予患者后能够(直接或间接)提供本文所述化合物的任何盐。应理解,非药学上可接受的盐也落入本发明的范围内,因为这些盐可用于制备药学上可接受的盐。盐的制备可以通过本领域已知的方法进行。例如,本文提供的化合物的药学上可接受的盐是由包含碱性或酸性部分的母体化合物通过常规化学方法合成的。通常,这样的盐例如通过使这些化合物的游离酸或碱形式与化学计量量的合适的碱或酸在水中或在有机溶剂中或在两者的混合物中反应来制备。通常,优选非水介质如醚、乙酸乙酯、乙醇、异丙醇或乙腈。酸加成盐的示例包括无机酸加成盐(如盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、硝酸盐、磷酸盐)和有机酸加成盐(如乙酸盐、三氟乙酸盐、马来酸盐、富马酸盐、柠檬酸盐、草酸盐、琥珀酸盐、酒石酸盐、苹果酸盐、扁桃酸盐、甲磺酸盐和对甲苯磺酸盐)。碱加成盐的示例包括无机盐(如钠盐、钾盐、钙盐和铵盐),以及有机碱盐(如乙二胺、乙醇胺、n,n-二烷基乙醇胺(n,n-dialkylenethanolamine)、三乙醇胺和碱性氨基酸盐)。
[0157]
本发明的化合物(包括pld)可以是作为游离化合物或溶剂合物(例如水合物、醇化物,特别是甲醇合物)的结晶形式,这两种形式都在本发明的范围内。溶剂化方法通常是本领域已知的。本发明的化合物可以具有不同的多晶型形式,并且本发明意欲包括所有这些形式。
[0158]
本文所提及的任何化合物旨在代表这样的特定化合物以及某些变体或形式。特别地,本文提及的化合物可具有不对称中心,因此以不同的对映异构体或非对映异构体形式存在。因此,本文提及的任何给定的化合物旨在代表外消旋物、一种或多种对映异构体形式、一种或多种非对映异构体形式及其混合物中的任何一种。同样,关于双键的立体异构或几何异构也是可能的,因此在某些情况下,分子可以(e)-异构体或(z)-异构体(反式和顺式异构体)存在。如果分子含有几个双键,每个双键都会有自己的立体异构体,这可能与分子中其他双键的立体异构体相同或不同。此外,本文提及的化合物可作为阻转异构体存在。本
文所提及的化合物的所有立体异构体(包括对映体、非对映体、几何异构体和阻转异构体)及其混合物都被认为在本发明的范围内。
[0159]
本发明涉及pld与其他一种或多种药剂的组合。
[0160]
组合疗法可能比单一疗法有优势。使用组合疗法可以避免或尽量减少耐药病毒的出现。组合治疗可以增强抗病毒活性。这些效应增加了改善临床结果的机会。在实施例中,可以选择药剂来处理病毒生命周期的不同步骤。在一个实施例中,抑制病毒进入或病毒细胞融合的药剂可与作用于病毒进入后的药剂组合。或者,这些药剂可以处理相同的步骤——例如,与pld组合的药剂可以仅针对病毒进入或仅针对病毒进入后。
[0161]
在一个实施例中,组合剂具有抗炎性质。
[0162]“组合剂”是指本文定义的与pld组合使用的一种或多种其他药剂。
[0163]
本发明的组合涉及pld的抗病毒用途,而不是pld也被批准的肿瘤适应症。
[0164]
下列药剂已被确定为组合药剂。
[0165]
在一个方面,组合剂抑制病毒进入或病毒细胞融合。sars-cov-2有两种进入途径(1)通过与质膜融合(“早期途径”)或(2)通过内吞和细胞组织蛋白酶,后者随后切割病毒尖峰蛋白,触发融合途径,并将cov基因组(“晚期途径”)释放到细胞胞质中。在一个实施例中,可以使用一种以上的组合剂来阻断早期和晚期途径。
[0166]
因此,在一个实施例中,组合剂抑制宿主细胞质膜和病毒之间的质膜融合。这将阻止病毒基因组进入细胞内部并最终产生新的病毒颗粒。冠状病毒膜融合发生在病毒s蛋白受体结合后,使得病毒膜和宿主细胞膜彼此接近。然而,s蛋白必须首先被蛋白酶(预处理)切割,才能具有融合能力。在一个实施例中,组合剂是外源性或跨膜蛋白酶抑制剂,更优选丝氨酸蛋白酶抑制剂或丝氨酸蛋白酶。优选地,蛋白酶抑制剂防止或减少病毒s蛋白的切割。
[0167]
在一个实施例中,所述组合剂选自阿比朵尔、抑肽酶、hai-1、hai-2、sfti-1、ε-氨基己酸、4-(2-氨基乙基)苯磺酰氟、对氨基苯甲脒、卡莫司他(例如甲磺酸卡莫司他)、萘莫司他(如甲磺酸萘莫司他)、h-d-htyr-ala-4-amba、bapa、cvs-3983、in-1、wx-uk1、mesupron、mi-432、mi-462、溴己辛、mi-1148、pf-429242、达诺普瑞、西米普瑞、洛米布韦、达克拉他韦、替拉普利韦、波塞普利韦、纳拉普利韦、曲格列汀、阿格列汀、利格列汀、西格列汀、沙格列汀、维拉格列汀、阿尔维列他汀和加贝酯中的任何一种。
[0168]
在另一个实施例中,组合剂是ii型跨膜丝氨酸蛋白酶(ttsp)抑制剂。ttsp可以是可溶性蛋白酶,例如tmprss11a。在另一个实施例中,组合剂是tmprss2抑制剂。在优选实施例中,所述药剂是卡莫司他(甲磺酸)、溴己辛(盐酸盐)、抑肽酶或萘莫司他。
[0169]
在另一优选实施例中,组合剂是卡莫司他。在另一个实施例中,组合剂是阿比朵尔。
[0170]
膜融合还取决于病毒和/或宿主细胞膜的脂质含量。在另一个实施例中,组合剂可从质膜消耗胆固醇和/或破坏脂质筏的形成。因此,在一个实施例中,组合剂是胆固醇消耗剂。尤其是,破坏筏的形成可能会阻止病毒对接和细胞进入。在一个实施例中,所述药剂选自氟伐他汀、阿托伐他汀,洛伐他汀、普伐他汀、辛伐他丁、吉非罗齐、甲基β-环糊精、β-环葡精、拉美布、二聚体、hpβcd和三聚体中的任何一种。在优选实施例中,所述药剂是甲基β-环糊精,其从质膜消耗胆固醇并干扰脂质筏的形成,脂质筏可包含sars-cov-2对接膜蛋白
ace2。
[0171]
在另一个实施例中,组合剂可以抑制胆固醇转运体以抑制细胞内胆固醇转运。在优选实施例中,所述药剂是伊曲康唑。
[0172]
在另一个实施例中,组合剂可以防止或抑制神经节苷脂生物合成以防止病毒结合。在一个实施例中,组合剂是神经节苷脂生物合成途径抑制剂。神经节苷脂生物合成途径抑制剂抑制介导鞘磷脂生物合成的酶(如丝氨酸棕榈酰转移酶、鞘磷脂合成酶和cer合成酶),导致病毒感染性降低。在一个实施例中,所述药剂选自isp-1、杨梅素、na255、d609、spk-601、ms-209、n-丁醛氧基吉霉素(nb-dnj)和d,l-苏氨酸-1-苯基-2-癸酰基氨基-3-吗啉-1-丙醇(pdmp)中的任何一种。在优选实施例中,所述药剂是nb-dnj,其是gm1神经节苷脂途径的抑制剂。
[0173]
在另一个实施例中,组合剂抑制内吞作用,更优选地,抑制氯氰菊酯介导的内吞作用。在一个实施例中,组合剂是氯氰菊酯介导的内吞的抑制剂。氯氰菊酯介导的内吞的抑制剂以多种方式干扰氯氰菊酯介导的内吞过程。例如,药剂可以将网格蛋白和衔接蛋白2(ap2)从细胞表面转移到细胞内的内体;阻止病毒颗粒进入包被的凹坑结构;影响氯氰菊酯包被的囊泡的功能(例如,通过提高内体ph,从而干扰病毒融合过程);影响网格蛋白包被囊泡的形成;干扰质子梯度;阻断dynamin(例如dynamin i)的gtpase活性;干扰网格蛋白盒配体与网格蛋白n端结构域的结合(从而停止网格蛋白包被的凹坑动力学,从而干扰网格蛋白介导的内吞作用)。
[0174]
在一个实施例中,组合剂选自氯丙嗪、单丹酰卡达瓦林、金刚烷胺、金刚乙胺、氧化苯胂、羟氯喹、氯喹、莫能菌素、吩噻嗪、dynasore、dynoles、dyngoes和pitstop(例如pitstop1和2)中的任何一种。在优选实施例中,所述药剂是羟氯喹、氯喹,金刚烷胺或氯丙嗪。在另一优选实施例中,组合剂是羟氯喹或氯喹或者氯喹衍生物,其提高内体ph并干扰病毒融合过程。在另一优选实施例中,组合剂是羟氯喹。
[0175]
在一个实施例中,组合剂通过干扰ap2相关蛋白激酶1(aak1)减少病毒进入,aak1是上皮细胞中氯氰菊酯介导的内吞的关键调节因子。aak1的抑制已被证明可减少病毒感染。已知的aak1抑制剂包括jak抑制剂。因此,在一个实施例中,组合剂是jak抑制剂。在一个实施例中,所述组合剂选自托法替尼、cyt387、巴利替尼、鲁索替尼、tg101348、莱司他尼、azd1480、r348、vx-509、glpg0634、gsk2586184、ac-430、帕西替尼和bms-911543中的任何一种。在优选实施例中,该药剂是对aak1具有特别强亲和力的托法替尼。
[0176]
在另一个实施例中,组合剂是蛋白酶抑制剂,更优选半胱氨酸蛋白酶抑制剂。蛋白酶抑制剂在进入宿主细胞期间干扰病毒的蛋白水解过程,特别是s蛋白。在优选实施例中,所述蛋白酶是内体蛋白酶,特别是内体酶,优选由内体环境的低ph激活的内体蛋白酶。更优选地,组合剂是组织蛋白酶抑制剂。或者,抑制剂是钙蛋白酶抑制剂。
[0177]
在一个实施例中,组合剂是组织蛋白酶抑制剂、组织蛋白酶b抑制剂、组组织蛋白酶l抑制剂、钙蛋白酶抑制剂、组合的钙蛋白酶和组织蛋白酶a抑制剂、泛组织蛋白酶b/l抑制剂。在一个实施例中,所述药剂选自奥当卡替、组织蛋白酶抑制剂1、e64、e64d(阿洛司他丁)、mg-132、pd151746、亮肽、z-fa-fmk、洛司他丁酸、ca-074、mdl28170、西美普瑞、博卡普瑞、纳拉普瑞、mg-132calpeptin、mdl27170(钙蛋白酶抑制剂iii)、钙蛋白酶抑制物vi、钙蛋白酶抑制物i、mg-115、钙蛋白酶抑制剂ii、钙蛋白酶抑制剂xii、psi、gc-376和芦平曲韦中
的任何一种。在一个优选的实施例中,该药剂是mdl28170(其是钙蛋白酶和组织蛋白酶b的组合抑制剂)、e64d(其是一旦病毒内化于内切体中就在下游起作用的泛组织蛋白酶b/l抑制剂)或ca-074(其是组织蛋白酶b抑制剂)。在优选实施例中,组合剂是mdl28170。
[0178]
病毒进入后,病毒rna被释放到细胞质中,在细胞质中其基因组被翻译和切割以产生新的病毒颗粒。因此,在另一方面,所述组合剂抑制病毒感染的步骤,例如病毒复制,所述步骤是病毒后细胞进入。
[0179]
在一个实施例中,组合剂是rna聚合酶抑制剂。rna聚合酶抑制剂阻止病毒基因组的复制。一些rna聚合酶抑制剂可作为前药,并在体内磷酸化。这些rna聚合酶抑制剂(或其磷酸化类似物)作为rna聚合酶的竞争性、非竞争性、无竞争性或自杀性抑制剂。在一个实施例中,所述药剂选自索非布韦、索非布韦 利巴韦林、索非布韦 利巴韦林 聚乙二醇干扰素α、索非布韦 莱迪帕韦(哈沃尼)、索非布韦 西美普韦、索非布韦 达克拉他韦、奥姆比塔韦 达萨布韦 帕利他韦 利托那韦(viekira-pak)、达萨布维、利巴韦韦、法匹拉韦、瑞德西韦、贝拉布韦、德洛布韦、菲利布韦、拉德布韦和塞特罗布韦中的任何一种。在优选实施例中,所述组合剂是瑞德西韦或法匹拉韦。在另一个优选实施例中,所述药剂是瑞德西韦。
[0180]
在一个实施例中,组合剂是hiv-1蛋白酶抑制剂。hiv-1蛋白酶抑制剂干扰由hiv-1病毒基因组编码的gag和gag-pol多蛋白前体的切割。这种干扰通过阻止成熟活性蛋白如蛋白酶、逆转录酶(p51)、核糖核酸酶h(p15)和整合酶的产生来限制病毒成熟。在一个实施例中,组合剂选自替拉那韦、达芦那韦、达芦那韦 科比司他、安瑞那韦、福沙普雷那韦、洛匹那韦、洛匹那韦-利托那韦、阿塔扎那韦、阿他那韦 科比司他、萨奎那韦、印地那韦、利托那维、奈非那韦(例如甲磺酸奈非那韦水合物)、spi-256和gs 8374中的任何一种。在一个优选实施例中,该剂为甲磺酸奈非那韦水合物、洛匹纳韦、替拉那韦、安瑞那韦或达芦那韦。在另一优选实施例中,组合剂是甲磺酸奈非那韦水合物。
[0181]
在一个实施例中,组合剂是ifn刺激的抗病毒蛋白。干扰素是一个多基因诱导细胞因子家族,具有抗病毒活性。干扰素分为两类:i型干扰素(也称为病毒性干扰素),包括干扰素-α(白细胞)、干扰素-β(成纤维细胞)和干扰素-ω;和ii型ifn(也称为免疫ifn或ifn-γ)。干扰素靶向单个细胞内干扰素诱导的蛋白,如:pkr激酶,其通过蛋白合成起始因子eif-2α的磷酸化抑制翻译起始;介导rna降解的oas合成酶家族和rna酶l核酸酶;mx蛋白gtpase家族,其似乎靶向病毒核衣壳并抑制rna合成;和adar,其通过腺苷脱氨基编辑双链rna以产生肌苷。干扰素诱导的mhc i类和ii类抗原以及一氧化氮合酶的表达也有助于在整个动物体内观察到的抗病毒反应。在一个实施例中,所述药剂是干扰素-α,例如干扰素-α2a(roferon a)、干扰素-α2b(内含子a)、共有干扰素-αcon(infergen)、人白细胞衍生的干扰素-αn3(alferon n)、人淋巴母细胞衍生的ifn-αn1(wellferon)、聚乙二醇-干扰素-α2a(pegasys)和聚乙二醇干扰素-α2b(聚乙二醇内含子);干扰素-β,如avonex和betaseron;γ,如活化免疫。在优选实施例中,组合剂是干扰素2-α或干扰素γ。
[0182]
在一个实施例中,所述组合剂是tlr 7激动剂,优选地为维托莫德。
[0183]
在一个实施例中,组合剂是pparα受体激动剂。pparα受体激动剂具有抗病毒和抗炎特性。在一个实施例中,组合剂选自吉非罗齐、氯贝特、wy14643、油酰乙醇胺、棕榈酰乙醇酰胺、非诺贝特、苯扎贝特和培马贝特中的任一种。在优选实施例中,所述药剂是非诺贝特。
[0184]
在一个实施例中,组合剂抑制冠状病毒复制复合物的活性。在一个实施例中,组合
剂抑制病毒蛋白酶mpro,也称为3c样蛋白酶(3clpro)。在一个实施例中,蛋白酶被钙蛋白酶抑制剂抑制。
[0185]
在一个实施例中,组合剂是糖皮质激素。糖皮质激素已显示抑制sars-cov-2的病毒复制。在一个实施例中,所述组合剂选自对甲松、泼尼松、曲安奈德、美松松松、安昔奈德、氟甲龙、倍氯米松(例如二丙酸倍氯米酮)、倍他米松(如磷酸倍他米酮)、强的松(例如醋酸泼尼松酮)、利美索龙、利美索酮、利美索酮、氯丙酸倍氯米松、利美松(例如,氯倍他醇(例如丙酸氯倍他索)、氟氯西奈、环索奈德、氟泼尼定(例如醋酸氟泼尼定)、氟考通酮、二氟考通隆、地塞米松(例如异烟酸地塞米松)、氟替卡松(例如糠酸氟替卡森)、美强的松、去甲可的松、可的唑、氯吡诺、莫米松(例如呋酸莫米松)、可的松,泼尼松龙(例如磷酸泼尼松隆、半琥珀酸泼尼松松)、甲基泼尼松酮(例如半琥珀酸酯甲基泼尼松)、氯可的龙(例如醋酸氯可的松)、美伦格斯特洛(例如醋酸美伦格斯特洛)和卤代米松。在优选的实施例中,组合剂是环索奈德。为免生疑问,本发明允许除了本发明的组合药剂之外,还额外施用皮质类固醇(优选地塞米松)。如果组合剂是糖皮质激素(例如环索尼),也可以如本文所定义的那样施用额外的糖皮质激素,优选地塞米松。
[0186]
在一个实施例中,组合药剂是抑制病毒复制的药剂。
[0187]
在一个实施例中,组合剂是抑制病毒进入的药剂。
[0188]
在一个优选实施例中,pld与一种或多种选自抑制病毒进入、抑制病毒细胞融合、抑制内吞或抑制病毒复制的其他药剂组合。特别地,pld或其药学上可接受的盐或立体异构体与一种或多种其他cov抗病毒剂组合使用,所述cov抗病毒剂选自calpain、组织蛋白酶或钙蛋白酶/组织蛋白酶抑制剂、rna聚合酶抑制剂、氯氰菊酯介导的内吞的抑制剂、hiv-1蛋白酶抑制剂、丝氨酸蛋白酶抑制剂、tmprss2抑制剂、ifn刺激的抗病毒蛋白、ifn-,pparα受体激动剂、胆固醇转运蛋白抑制剂、细胞内胆固醇转运抑制剂、神经节苷脂生物合成途径抑制剂、胆固醇消耗剂、糖皮质激素、抑制病毒与宿主细胞膜融合的药剂和jak抑制剂。
[0189]
在另一优选实施例中,pld与一种或多种选自钙蛋白酶、组织蛋白酶或钙蛋白酶/组织蛋白酶抑制剂、rna聚合酶抑制剂、氯氰菊酯介导的内吞的抑制剂、hiv-1蛋白酶抑制剂、丝氨酸蛋白酶抑制剂、tmprss2抑制剂、ifn刺激的抗病毒蛋白和pparα受体激动剂的其他药剂组合。具体的药剂可能包括mdl 28170、瑞德西韦、羟氯喹、甲磺酸奈非那韦水合物、氯喹和干扰素2α,干扰素-γ和非诺贝特。
[0190]
在本发明组合的优选实施例中,pld或其药学上可接受的盐或立体异构体与所述组合中的其他药剂的摩尔比为1:1000至1000:1,或1:700至700:1,1:500至500:1,2:300至300:1,3:100至100:1,或者1:50至50:1。
[0191]
本发明的化合物可用于具有生物/药理学活性的药物组合物中,用于治疗上述感染和相关病症。这些药物组合物包含本发明的化合物和药学上可接受的载体。术语“载体(carrier)”是指与活性成分一起给药的稀释剂、佐剂、赋形剂或载体(vehicle)。e.w.martin的“remington’s pharmaceutical sciences”,1995,中有描述合适的药物载体。药物组合物的示例包括用于口服、局部或胃肠外给药的任何固体(片剂、丸剂、胶囊剂、颗粒剂等)或液体(溶液、混悬剂、乳剂等)组合物。含有本发明化合物的药物组合物可以通过脂质体或纳米球包封、以缓释制剂的方式或通过其他标准递送方式递送。
[0192]
pld的示例性组合物是用于注射的溶液的粉末形式。例如wo9942125中描述的组合
物。例如包括水溶性材料,其次是混合溶剂的重构溶液的本发明化合物的冻干制剂。具体的例子是pld和甘露醇和混合溶剂的重构溶液(例如peg-35蓖麻油、乙醇和注射用水)的冻干制剂。每个小瓶例如可以包含2mg的pld。重构后,每ml重构溶液可包含:0.5mg pld、158mg peg-35蓖麻油和0.15ml/ml乙醇。
[0193]
组合中其他活性剂(即非pld)的示例性组成将取决于所讨论的活性剂。
[0194]
在实施例中,提供了一种试剂盒,其包括组合,所述组合包括根据本发明的pld和可选的用于治疗患者的说明书。通常地,试剂盒可以包括pld或其药学上可接受的盐或立体异构体和其他组合剂以及用于治疗患者的说明书。每种活性剂可以提供在合适的容器中。试剂盒还可以包括输送系统。试剂盒可以包括pld以及用于与根据本发明的一种或多种其他药剂组合使用的说明书。试剂盒可以包括根据本发明的一种或多种其他药剂以及用于与pld组合使用的说明书。
[0195]
说明书可建议将根据本发明的pld与根据本发明用于治疗cov感染或covid-19的其他药剂组合施用。试剂盒可指示同时、顺序或单独施用这些组合。
[0196]
可基于多种因素选择特定组合,包括所使用的特定化合物的活性、所使用的具体制剂、应用模式、年龄、体重、一般健康、性别、饮食、给药时间、排泄率、药物组合、反应敏感度、确定的或潜在的抗性以及正在治疗的特定疾病或病症的严重程度。
[0197]
本发明的组合可适于同时、顺序或单独施用。
[0198]
任何特定试剂的特定剂量和治疗方案可以变化,并且将取决于多种因素,包括所使用的特定化合物的活性、所使用的特定制剂、应用模式、年龄、体重、综合健康、性别、饮食、给药时间、排泄率、药物组合、反应敏感性、确定的或潜在的抗性和正在治疗的特定疾病或病症的严重性。
[0199]
在本发明的实施例中,组合药剂(即除pld以外的药剂)的给药方案将根据该药剂的适当给药方案进行给药。
[0200]
在本发明的实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次剂量的给药方案给药。
[0201]
在进一步的实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日剂量给药10天、9天、8天、7天、6天、5天、4天、3天、2天或1天的给药方案给药。优选的方案是2-5天,或3-5天,或3、4或5天,最优选3天或5天。
[0202]
剂量可为每天5mg或以下、每天4.5mg或以下、每天4mg或以下、每天3.5mg或以下、每天3mg或以下、每天2.5mg或以下或2mg或以下。
[0203]
具体剂量包括0.5mg/天、1mg/天、1.5mg/天、2mg/天、2.5mg/天、3mg/天、3.5mg/天、4mg/天、4.5mg/天或5mg/天。优选的剂量为1mg/天、1.5mg/天、2mg/天和2.5mg/天。
[0204]
在进一步的实施例中,pld或其药学上可接受的盐或立体异构体可以根据1-50mg、1-40mg、1-30mg、1-20mg、1-15mg、3-15mg、3-12mg、4-12mg、4-10mg或4.5-10mg的总剂量给药。总剂量可为4mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg。优选的总剂量为4.5mg、5mg、6mg、7.5mg、8mg、9mg或10mg。总剂量可以分成1、2、3、4、5、6、7、8、9或10天,优选3天或5天。
[0205]
在一个具体实施例中,pld或其药学上可接受的盐或立体异构体可根据每日一次,每天2.5mg或更少的剂量,给药5天的给药方案给药。
[0206]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可根据每日一次,每天2mg或更少的剂量,给药5天的给药方案给药。
[0207]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天1.5mg或更少的剂量,给药3天的给药方案给药。
[0208]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可根据每日一次,每天2mg或更少的剂量,给药3天的给药方案给药。
[0209]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天2.5mg或更少的剂量,给药3天的给药方案给药。
[0210]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天1.5mg的剂量,给药3天的给药方案给药。
[0211]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天2.0mg的剂量,给药3天的给药方案给药。
[0212]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天2.5mg的剂量,给药3天的给药方案给药。
[0213]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可以根据每日一次,每天1.5至2.5mg的剂量,给药3天的给药方案给药。
[0214]
另一种方案是在第1天单剂量给药pld或其药学上可接受的盐或立体异构体。单剂量方案可能特别适合治疗:轻度感染;减少与cov感染相关的并发症(包括住院、icu和死亡);预防、减少、避免或治疗持续性covid、长covid、后covid综合征;和/或降低cov患者的传染性。单次剂量的pld可以是1-10mg、4-10mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg;优选4.5mg、5mg、6mg、7.5mg、8mg、9mg、9mg或10mg;更优选5-9mg、6.5-8.5mg、7-8mg或7.5mg。
[0215]
在另一个实施例中,pld或其药学上可接受的盐或立体异构体可根据本发明给药,其中本发明的化合物与皮质类固醇一起给药。优选地,皮质类固醇是地塞米松。
[0216]
皮质类固醇可以与pld或其药学上可接受的盐或立体异构体一起每日给药。给药可以是按次序的、同时的或连续的。皮质类固醇可在给药pld或其药学上可接受的盐或立体异构体后的几天内进一步给药。例如,对于3天给药方案,皮质类固醇可以在第1-3天给药,然后每天进一步给药3、4、5、6、7、8、9或10天或更多天。
[0217]
在一个具体实施例中,皮质类固醇可以在第1-3天以静脉注射给药,然后在第6-10天以口服给药。在另一个实施例中,皮质类固醇的剂量在与pld或其药学上可接受的盐或立体异构体的共同给药阶段期间可以更高,并且在随后的天数里降低。
[0218]
具体剂量时间表包括:
[0219]
·
在第1天至第3天静脉注射(iv)1.5mg/天的pld以及6.6mg/天的盐酸地塞米松,然后从第4天到第10天口服(po)/iv6 mg/天的地塞米松直至第10天(取决于医生根据患者临床状况和病情进展的判断)。
[0220]
·
在第1天至第3天静脉注射(iv)2.0mg/天的pld并静脉注射6.6mg/天的地塞米松,然后从第4天到第10天口服(po)/iv6 mg/天的地塞米松直至第10天(取决于医生根据患者临床状况和病情进展的判断)。
[0221]
·
在第1天至第3天静脉注射(iv)2.5mg/天的pld并静脉注射6.6mg/天的地塞米
松,然后从第4天到第10天口服(po)/iv 6mg/天的地塞米松直至第10天(取决于医生根据患者临床状况和病情进展的判断)。
[0222]
在一个实施例中,为了避免与给药相关的输液反应,患者可以在开始注射pld或其药学上可接受的盐或立体异构体之前20至30分钟接受以下药物:
[0223]
·
昂丹司琼8mg iv(或等同物)
[0224]
·
盐酸苯海拉明25mg iv(或等同物)
[0225]
·
雷尼替丁50mg iv(或等同物)
[0226]
·
地塞米松6.6mg静脉注射(包括在上述时间表中)
[0227]
另外,在第4天和第5天,用本发明化合物治疗的患者可po接受每天两次的昂丹司琼4mg。
[0228]
地塞米松、昂丹司琼和雷尼替丁的剂量在此以碱形式为基础进行定义。盐酸苯海拉明的剂量以盐酸盐为基础给出。本发明化合物的剂量基于碱形式给出。
[0229]
日剂量可以注射给药。注射可以是1小时注射、1.5小时注射、2小时注射、3小时注射或更长时间。优选地,注射为1.5小时。
[0230]
在某些实施例中,可根据使用负荷剂量和维持剂量的方案来给药剂量。根据本发明的负荷/维持剂量包括:
[0231]
第1天2.5mg的负荷剂量,随后几天2mg/天的维持剂量;
[0232]
第1天2.5mg的负荷剂量,随后几天1.5mg/天的维持剂量;
[0233]
第1天2.5mg的负荷剂量,随后几天1mg/天的维持剂量;
[0234]
第1天2.5mg的负荷剂量,随后几天0.5mg/天的维持剂量;
[0235]
第1天2mg的负荷剂量,随后几天1.5mg/天的维持剂量;
[0236]
第1天2mg的负荷剂量,随后几天1mg/天的维持剂量;
[0237]
第1天2mg的负荷剂量,随后几天0.5mg/天的维持剂量;
[0238]
第1天1.5mg的负荷剂量,随后几天1mg/天的维持剂量;
[0239]
第1天1.5mg的负荷剂量,随后几天0.5mg/天的维持剂量;以及
[0240]
第1天1mg的负荷剂量,随后几天0.5mg/天的维持剂量。
[0241]
根据另一实施例,日剂量可在方案的最后一天或多天减少。
[0242]
根据另一实施方式,如果日剂量为2mg,则剂量可在第4天和第5天减少至1mg。
[0243]
具体的治疗方案包括:
[0244]-1mg pld,注射1.5小时,每天一次,连续5天。(总剂量5mg);
[0245]-2mg pld,注射1.5小时,每天一次,连续5天。根据研究人员的判断,第4天和第5天的剂量可以减少到1mg/天(总剂量8-10mg);
[0246]-1.5mg pld,注射1.5小时,每天一次,连续3天。(总剂量4.5mg);
[0247]-2mg pld,注射1.5小时,每天一次,连续3天。(总剂量6mg);以及
[0248]-2.5mg pld,注射1.5小时,每天一次,连续3天。(总剂量7.5mg)。
[0249]
组合中的pld成分的单剂量方案包括:
[0250]
·
给药pld,注射1.5小时,第1天一次,剂量为1-10mg、4-10mg、4.5-10mg、4mg、4.5mg、5mg、5.5mg、6mg、6.5mg、7mg、7.5mg、8mg、8.5mg、9mg、9.5mg或10mg,优选4.5mg、5mg、6mg、7.5mg、8mg、9mg或10mg,更优选5-9mg、6.5-8.5mg、7-8mg或最优选7.5mg。
[0251]
·
单剂量方案可在注射pld前20-30分钟进一步包括以下预防性药物:
[0252]-静脉注射盐酸苯海拉明25mg,
[0253]-静脉注射雷尼替丁50mg,
[0254]-静脉注射地塞米松6.6mg,
[0255]-静脉注射昂丹司琼8mg,缓慢注射15分钟。
[0256]
·
服用pld后,可每12小时口服昂丹司琼4mg,持续3天,以减轻药物引起的恶心和呕吐。
[0257]
如果在早晨给予pld,患者可以在下午接受第一剂量的昂丹司琼。
[0258]
根据另外的实施例,可以基于临床参数和/或患者特征选择患者用于使用pld的治疗。合适的参数可以是本技术中公开的测量。
[0259]
如上所述,组合药剂(即除pld以外的药剂)的给药方案将根据该药剂的适当给药方案进行给药。
[0260]
上述方案和剂量可适用于如本文所定义的治疗方法和用途。
[0261]
为了提供更简洁的描述,本文给出的一些定量表达式没有限定术语“约”。应理解,无论是否明确使用术语“约”,本文给出的每一量均意指实际给定值,且其还意指将基于本领域技术人员合理地推断的对所述给定值的近似值,包括所述给定值的实验和/或测量条件导致的等效物和近似物。
[0262]
虽然上述公开提供了对包含在本发明范围内的主题(包括制造和使用本发明的方法及其最佳模式)的概述,但是提供以下实施例以进一步使本领域技术人员能够实践本发明并提供完整的书面描述。然而,本领域技术人员将认识到,这些实施例的细节不应理解为对本发明的限制,本发明的范围应从本公开的权利要求及其等同物中理解。鉴于本公开,本发明的各种进一步的方面和实施例对于本领域技术人员将是显而易见的。
[0263]
实施例
[0264]
pld可以根据wo 02/02596或如文献中其他地方公开的那样制备。
[0265]
材料和方法
[0266]
细胞培养。vero e6细胞(atcc crl-1586)在dulbecco改良的eagle培养基(dmem;lonza)中培养,该培养基补充有5%胎牛血清(fcs;euroclone)、100u/ml青霉素、100μg/ml链霉素和2mm谷氨酰胺(均来自thermofisher scientific)。
[0267]
病毒分离、滴定和测序。
[0268]
sars-cov-2病毒是从一名89岁男性患者的鼻咽拭子中分离出来的,该患者获得知情同意,并在样本采集前用倍泰龙和羟氯喹治疗2天。将拭子收集在3ml培养基(deltaswab vicum)中以降低粘度,并在-80℃下储存直至使用。vero e6细胞在1.5x106个细胞的细胞培养烧瓶(25cm2)上培养过夜,然后在37℃和5%co2下接种1ml处理过的样品1小时。之后,供应4ml补充2%fcs的dmem,并将细胞孵育48小时。收集上清液,以200x g离心10分钟以去除细胞碎片,并在-80℃下储存。每天评估细胞的细胞病变效应,并使用sars-cov-2upe、rdrp和n分析对上清液进行病毒rna提取和特异性rt-qpcr(corman等人,2020)。将病毒进行两次传代,制备收集vero e6上清液的病毒原液。
[0269]
使用indimag病原体试剂盒(indical biosciences)直接从病毒库中提取病毒rna,并根据制造商的方案使用具有低聚dt和随机六聚体的primescript
tm
rt试剂盒
(takara)转录为cdna。使用swift扩增子sars-cov-2组(swift biosciences)进行dna文库制备。测序就绪文库,然后加载到illumina miseq平台和300bp配对末端测序试剂盒上。对序列读取进行质量过滤,并使用trimmomatic修剪适配器引物序列。使用cutadapt去除扩增引物序列(martin,2011)。然后使用bowtie2工具(langmead,b.和salzberg,s,2012)针对冠状病毒参考(nc_045512.2)绘制测序读数。使用samtools从18x1800x879平均覆盖率的结果比对中调用一致基因组序列(li等人,2009)。基因组序列保存在gisaid储存库中(http://gisaid.org)具有登录id epi_isl_510689。
[0270]
抗病毒剂和化合物。pld的使用浓度范围为以1/5系列稀释从100μm至0.0512nm和以1/3稀释度从10μm至0.5nm进行测定。当pld和其他药物合并时,每种药物以1:1的摩尔比添加,浓度范围为以1/5系列稀释从100μm至0.0512nm和以1/3稀释度从10μm至0.5nm进行测定。
[0271]
抗病毒活性。向vero e6细胞中加入浓度升高的抗病毒化合物以及10
1.8
tcid
50
/ml的sars-cov-2,该浓度达到50%的细胞病变效果。未暴露的细胞用作感染的阴性对照。为了检测任何药物相关的细胞毒性效应,vero e6细胞在药物浓度增加的情况下同样培养,但不存在病毒。病毒或药物的细胞病变或细胞毒性效应在感染后3天使用细胞滴度glo发光细胞存活率测定法(promega)测定。在fluoroscan ascent fl光度计(thermofisher scientific)中测量发光。
[0272]
ic
50
计算和统计分析。将化合物或其混合物的响应曲线调整为非线性拟合回归模型,用具有可变斜率的四参数logistic曲线计算。未暴露于病毒的细胞用作感染的阴性对照,并设置为100%的存活率,并用于标准化数据和计算细胞病变效应的百分比。通过单样本t检验评估与100%的统计差异。所有分析和图形均使用graphpad prism v8.0b软件生成。
[0273]
研究了pld对sars-cov-2的抗病毒活性。pld还与其他抗病毒剂组合使用。
[0274]
据信,sars-cov-2进入需要病毒结合和通过与细胞受体ace2和细胞蛋白酶的相互作用激活尖峰蛋白,这一机制有利于通过内吞作用进行病毒内化。已经证明,干扰这些初始过程中的任何一个都可以减少sars-cov-2的进入和传染性。此外,sars-cov-2可通过内吞作用进入细胞,并在内切体中积累,细胞组织蛋白酶也可引发棘突蛋白,并有利于切割后的病毒融合。
[0275]
因此,进行了实验以确定pld与进入抑制剂化合物的组合的活性,其可能在病毒进入之前通过损害病毒细胞融合而产生影响。
[0276]
此外,当sars-cov-2与质膜或内质体膜融合时,它触发病毒rna释放到细胞质中,在那里多蛋白被蛋白酶翻译和切割。这导致rna复制酶-转录酶复合物的形成,该复合物通过复制和转录驱动负链rna的产生。负链rna驱动正rna基因组的转录和病毒核蛋白的翻译,这些核蛋白在细胞质的病毒衣壳中组装。这些衣壳随后发芽进入内质网高尔基体腔,病毒最终通过胞吐释放到细胞外空间。这导致了病毒循环的多个步骤,这些步骤可能容易被不同的抗病毒化合物与pld组合靶向。因此,进行实验以确定pld与进入后抑制剂组合的活性。
[0277]
进一步的抗病毒抑制剂具有不同或未知的作用机制。因此,进行了实验以确定pld组合的活性以及具有未知作用机制的抑制剂的抗病毒活性。
[0278]
实施例1-对暴露于pld及其与多喹(dolquine)、瑞德西韦、mdl28170和甲磺酸奈非
那韦水合物的组合的vero e6细胞的细胞病变作用
[0279]
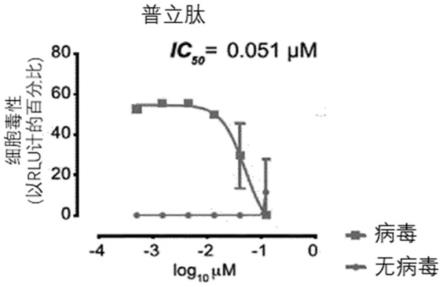
pld的抗病毒活性如图1所示,图1显示了在pld浓度增加的情况下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应。pld的使用浓度为以1/3稀释度从10μm至0.5nm。显示了一个具有两个重复的代表性实验的变量响应曲线的非线性拟合(平方)。显示了该图的特定ic
50
值。还显示了在没有病毒的情况下暴露于浓度增加的药物的vero e6细胞的细胞毒性效应(圆圈)。
[0280]
将恒定浓度的sars-cov-2临床分离物(id epi_isl_418268)与增加浓度的pld混合并添加到vero e6细胞中。为了控制药物诱导的细胞毒性,在不存在sars-cov-2的情况下,还将vero e6与增加浓度的pld一起培养。在未观察到药物细胞毒性作用的浓度下(圆形),pld能够抑制病毒诱导的细胞病变效应(方形)。在两个重复的实验中,pld的平均ic
50
值和sd分别为0.051
±
0.02μm。
[0281]
pld与其他活性抗病毒剂组合试验。组合时,以相同浓度以1:1摩尔比添加每种药物。pld和羟氯喹的组合显示ic
50
为0.011μm。该实验的结果如图2所示。重要的是,未发现pld活性降低。同样重要的是,未见毒性增加。
[0282]
pld和瑞德西韦的组合显示ic
50
为0.07μm。同样重要的是,未发现pld活性降低。同样重要的是,未发现毒性增加。该实验的结果如图2所示。
[0283]
pld和mdl28170的组合显示ic
50
为0.021μm。同样重要的是,未发现pld活性降低。同样重要的是,未发现毒性增加。该实验的结果如图2所示。
[0284]
pld和甲磺酸奈非那韦水合物的组合显示ic
50
为0.015μm。同样重要的是,未发现pld活性降低。同样重要的是,未发现毒性增加。该实验的结果如图2所示。
[0285]
图3-6显示了瑞德西韦、羟氯喹、mdl28170和甲磺酸奈非那韦水合物的活性。
[0286]
实施例2-羟氯喹对sars-cov-2的抗病毒活性
[0287]
羟氯喹的抗病毒活性如图3所示,该图显示了在羟氯喹(多喹)浓度增加的情况下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应。使用浓度范围为100μm至0.0512nm的羟氯喹。显示了一个具有两个重复的代表性实验的变量响应曲线的非线性拟合(红色方块),排除了具有相关毒性的药物浓度数据。显示了该图的特定ic
50
值。还显示了在没有病毒的情况下暴露于浓度增加的药物的vero e6细胞的细胞毒性效应(灰色圆圈)。
[0288]
羟氯喹(多喹)对sars-cov-2诱导的vero e6细胞细胞毒性的抑制作用如图3所示。将恒定浓度的sars-cov-2临床分离物(id epi_isl_418268)与浓度增加的羟氯喹混合并添加到vero e8细胞中。为了控制药物诱导的细胞毒性,在不存在sars-cov-2的情况下,还将vero e6与越来越高浓度的羟氯喹一起培养。在未观察到药物细胞毒性作用的浓度下(灰色圆圈),羟氯喹能够抑制病毒诱导的细胞病变效应(红色方块)。该药物的平均ic
50
值和sd来自至少三个独立实验,每个重复两次,为9.3
±
11.1μm。
[0289]
实施例3-在浓度增加的瑞德西韦的存在下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应
[0290]
图4显示了在浓度增加的瑞德西韦的存在下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应。药物使用浓度范围为100μm至0.0512nm。显示了一个具有两个重复的代表性实验的变量响应曲线的非线性拟合(红色方块),排除了具有相关毒性的药物浓度数据。显示了该图的特定ic
50
值。还显示了在没有病毒的情况下暴露于浓度增加的药物的
vero e6细胞的细胞毒性效应(灰色圆圈)。
[0291]
组合用药的浓度范围为100μm至0.0512nm。在未观察到药物细胞毒性的浓度下,瑞德西韦被证实具有体外抑制sars-cov-2诱导的vero e6细胞病变作用的能力。该药物的平均ic
50
值和sd来自至少三个独立实验,每个重复两次,为2.16
±
4.1μm。
[0292]
实施例4-在浓度增加的mdl28170的情况下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应
[0293]
图4显示了在浓度增加的mdl 28170钙蛋白酶抑制剂iii的情况下,暴露于固定浓度sars-cov-2的vero e6细胞的细胞病变效应。该药物的浓度范围为100μm至0.0512nm。显示了一个具有两个重复的代表性实验的变量响应曲线的非线性拟合(红色方块),排除了具有相关毒性的药物浓度数据。显示了该图的特定ic
50
值。还显示了在没有病毒的情况下暴露于药物浓度降低的vero e6细胞的细胞毒性效应(灰色圆圈)。
[0294]
mdl 28170被证实在体外具有抑制sars-cov-2诱导的vero e6细胞病变作用的能力,其浓度未观察到药物的细胞毒性。该药物的平均ic
50
值和sd来自至少两个独立实验,每个重复两次,为0.14
±
0.06μm。
[0295]
实施例5-暴露于甲磺酸奈非那韦水合物的vero e6细胞的细胞病变效应
[0296]
暴露于固定浓度sars-cov-2的vero e6细胞在抗hiv-1、甲磺酸奈非那韦水合物的蛋白酶抑制剂浓度增加的情况下的细胞毒性效应。结果如图6所示。药物使用浓度范围为100μm至0.0512nm。显示了一个具有两个重复的代表性实验的变量响应曲线的非线性拟合(红色方块),排除了具有相关毒性的药物浓度数据。显示了该图的特定ic
50
值。还显示了在没有病毒的情况下暴露于浓度增加的药物的vero e6细胞的细胞毒性效应(灰色圆圈)。
[0297]
甲磺酸奈非那韦水合物被证实在体外具有抑制sars-cov-2诱导的vero e6细胞病变作用的能力,其浓度未观察到该药物的细胞毒性。
[0298]
这些数据表明,pld可以与其他药剂组合施用,而不会降低pld活性。这些数据还表明,pld可与其他药剂组合施用,而不会对毒性产生不利影响。这些重要发现强调了将pld与其他药剂结合以最小化或避免选择耐药病毒的可能性。
[0299]
已证明pld与一种或多种其他cov抗病毒剂组合有效,所述其他cov抗病毒剂选自抑制病毒进入、抑制病毒细胞融合、抑制内吞或抑制病毒复制的药剂。特别地,一种或多种其他cov抗病毒剂选自钙蛋白酶、组织蛋白酶或钙蛋白酶/组织蛋白酶抑制剂、rna聚合酶抑制剂、氯氰菊酯介导的内吞的抑制剂、hiv-1蛋白酶抑制剂、丝氨酸蛋白酶抑制剂、tmprss2抑制剂、ifn刺激的抗病毒蛋白、pparα受体激动剂、胆固醇转运体抑制剂、细胞内胆固醇转运抑制剂、神经节苷脂生物合成途径抑制剂、胆固醇消耗剂、糖皮质激素、抑制病毒与宿主细胞膜融合的药剂和jak抑制剂。
[0300]
本文描述的具有抗病毒活性的抗病毒剂处理病毒生命周期的不同步骤,因此可以考虑与pld结合使用。这些组合保持效力并具有可接受的细胞毒性,可用于最小化或避免耐药病毒的出现。
[0301]
pld组合保留功效并保留可接受的细胞毒性。本文所公开的组合可用作抗sars-cov-2和covid-19的抗病毒组合剂。
[0302]
实施例6
–
pld的血浆模型
[0303]
图7显示了每日剂量(d1-d5)为1.0mg和2.0mg后,总血浆普立肽浓度模型随时间变
treatments.lancet infect.dis.20,400
–
402.
[0325]
tu,y.-f.,chien,c.-s.,yarmishyn,a.a.,lin,y.-y.,luo,y.-h.,lin,y.-t.,lai,w.-y.,yang,d.-m.,chou,s.-j.,yang,y.-p.,等人(2020).a review of sars-cov-2and the ongoing clinical trials.int.j.mol.sci.21,2657.
[0326]
wang,y.,zhang,d.,du,g.,du,r.,zhao,j.,jin,y.,fu,s.,gao,l.,cheng,z.,lu,q.,等人(2020).remdesivir in adults with severe covid-19:a randomised,double-blind,placebo-controlled,multicentre trial.the lancet s0140673620310229.
[0327]
williamson,b.,feldmann,f.,schwarz,b.,meade-white,k.,porter,d.,schulz,j.,van doremalen,n.,leighton,i.,yinda,c.k.,perez-perez,l.,等人(2020).clinical benefit of remdesivir in rhesus macaques infected with sars-cov-2(microbiology).
[0328]
wu,c.,liu,y.,yang,y.,zhang,p.,zhong,w.,wang,y.,wang,q.,xu,y.,li,m.,li,x.,等人(2020).analysis of therapeutic targets for sars-cov-2and discovery of potential drugs by computational methods.acta pharm.sin.b 10,766
–
788.
[0329]
zhou,y.和simmons,g.(2012).development of novel entry inhibitors targeting emerging viruses.expert rev.anti infect.ther.10,1129
–
1138。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。