1.本发明涉及有机合成技术领域,特别涉及一种四吲哚甲烷化合物及其制备方法与应用。
背景技术:
2.吲哚类化合物是一大类具有苯并五元氮杂环结构单元的化合物,在自然界中广泛存在。吲哚类化合物是一种重要的杂环衍生物生物碱,其在生物体内显现出了丰富多彩的生物活性作用。到19世纪末,就已经有部分吲哚化合物被应用于染料行业。自20世纪30年代起,科研人员发现吲哚具有的良好生物活性,诸多以吲哚结构作为核心药效基团生物碱相继出现。目前,含有吲哚结构的药物已大量上市,对治疗人类的疾病起到了非常重要的作用;同时,吲哚类化合物已广泛应用于医药、农药、香料、染料以及其他精细化工产品的中间体等领域。
3.近年来,随着有关生物碱的深入研究,有一部分四吲哚甲烷类物质被相继发现。alice团队从海水里分离的反硝化假弧菌中发现了一种新型的四吲哚化合物,该化合物对l929和a549细胞进行的活性测试表现出较好的生物毒性。wang团队合成出四吲哚甲烷类化合物sk228,其对乳腺癌细胞的调控作用和机制进行了探讨,经过动物研究,sk228可以有效抑制癌细胞的生长和转移。
技术实现要素:
4.发明目的:本发明提供了一种四吲哚甲烷化合物,该类化合物具有较好的生物活性,尤其是抗肿瘤活性,可为抗肿瘤药物筛选提供化合物库,并提供了该类化合物的制备方法。
5.技术方案:本发明一种四吲哚甲烷化合物,其结构如式(i)所示:
[0006][0007]
其中,r为卤素或c1~c3烷氧基,r位于吲哚环的4位或6位。
[0008]
优选地,r为f、cl、br或甲氧基。
[0009]
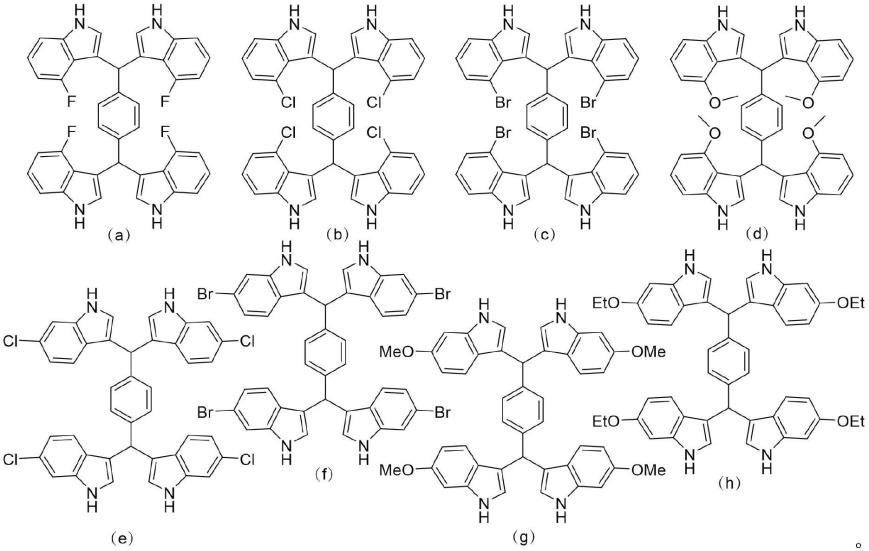
优选地,四吲哚甲烷化合物结构如下所示:
[0010][0011]
本发明所述一种四吲哚甲烷化合物的制备方法,操作步骤为:
[0012][0013]
将化合物1和化合物2置于圆底烧瓶中,加入溶剂溶解,加入催化剂,充入氮气保护,在一定温度下反应一段时间,反应完毕后,加水,用乙酸乙酯萃取,有机相用硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得化合物3。其中,r位于吲哚环的4位或6位。
[0014]
优选地,所述溶剂为甲醇、乙醇、乙腈或四氢呋喃。
[0015]
优选地,所述催化剂为盐酸、碘、溴化铜或三氟甲基磺酸铋。
[0016]
优选地,所述化合物1和化合物2的摩尔比为1:4~1:4.5。
[0017]
优选地,所述反应温度为0℃~80℃,所述反应时间为0.5~2h。
[0018]
优选地,所述化合物2为4-氟吲哚、4-氯吲哚、4-溴吲哚、4-甲氧基吲哚、6-氯吲哚、6-溴吲哚、6-甲氧基吲哚或6-乙氧基吲哚。
[0019]
本发明还提供了上述一种四吲哚甲烷化合物在制备抗肿瘤药物中的应用。
[0020]
有益效果:本发明的一种四吲哚甲烷化合物,其对a549癌细胞的半数抑制浓度(ic50)介于7.5~12.5μmol
·
l-1,优于标准抗癌药物顺铂对a549细胞的ic50值(36μmol
·
l-1),具有较好的抗肿瘤效果。
附图说明
[0021]
下面对说明书附图所表达的内容做简要说明:
[0022]
图1为本发明实施例1制备的四吲哚甲烷化合物的质谱图;
[0023]
图2为本发明实施例2制备的四吲哚甲烷化合物的质谱图;
[0024]
图3为本发明实施例3制备的四吲哚甲烷化合物的质谱图;
[0025]
图4为本发明实施例4制备的四吲哚甲烷化合物的质谱图;
[0026]
图5为本发明实施例5制备的四吲哚甲烷化合物的质谱图;
[0027]
图6为本发明实施例6制备的四吲哚甲烷化合物的质谱图;
[0028]
图7为本发明实施例7制备的四吲哚甲烷化合物的质谱图;
[0029]
图8为本发明实施例1制备的四吲哚甲烷化合物的1h nmr图;
[0030]
图9为本发明实施例2制备的四吲哚甲烷化合物的1h nmr图;
[0031]
图10为本发明实施例3制备的四吲哚甲烷化合物的1h nmr图;
[0032]
图11为本发明实施例4制备的四吲哚甲烷化合物的1h nmr图;
[0033]
图12为本发明实施例5制备的四吲哚甲烷化合物的1h nmr图;
[0034]
图13为本发明实施例6制备的四吲哚甲烷化合物的1h nmr图;
[0035]
图14为本发明实施例7制备的四吲哚甲烷化合物的1h nmr图。
具体实施方式
[0036]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0037]
实施例1:将1mmol原料1和4mmol 4-氟吲哚置于50ml圆底烧瓶中,加入20ml甲醇溶解,加入0.1ml盐酸,充入氮气保护,0℃下反应2h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物a,收率65%。esi-ms m/z:661.19(m na)
,如图1所示。1h nmr(300mhz,dmso-d6)δ11.06(s,4h),7.16(d,j=7.5hz,8h),6.99(d,j=5.1hz,4h),6.61(s,8h),6.13(s,2h),如图8所示。
[0038]
实施例2:将1mmol原料1和4.5mmol 4-氯吲哚置于50ml圆底烧瓶中,加入20ml乙腈溶解,加入0.1mmol碘,充入氮气保护,60℃下反应0.5h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物b,收率82%。esi-ms m/z:727.07(m na)
,如图2所示。1h nmr(400mhz,dmso-d6)δ11.12(s,4h),7.32(d,j=8.1hz,4h),7.10(s,4h),7.01(t,j=7.8hz,4h),6.90(d,j=8.1hz,6h),6.53(s,4h),如图9所示。
[0039]
实施例3:将1mmol原料1和4.2mmol 4-溴吲哚置于50ml圆底烧瓶中,加入20ml四氢呋喃溶解,加入0.1mmol溴化铜,充入氮气保护,50℃下反应1h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物c,收率80%。esi-ms m/z:904.87(m na)
,如图3所示。1h nmr(500mhz,dmso-d6)δ11.10(s,4h),7.38(d,j=8.7hz,4h),7.10(d,j=7.6hz,4h),7.05(d,j=10.3hz,6h),6.95(s,4h),6.51(s,4h),如图10所示。
[0040]
实施例4:将1mmol原料1和4.2mmol 4-甲氧基吲哚置于50ml圆底烧瓶中,加入20ml乙醇溶解,加入0.1mmol三氟甲基磺酸铋,充入氮气保护,80℃下反应0.5h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物d,收率85%。esi-ms m/z:709.27(m na)
,如图4所示。1h nmr(500mhz,dmso-d6)δ10.61(s,4h),7.10(s,4h),6.91(s,8h),6.62(s,2h),6.50(s,4h),6.36(d,j=6.3hz,4h),3.59(s,12h),如图11所示。
[0041]
实施例5:将1mmol原料1和4.5mmol 6-氯吲哚置于50ml圆底烧瓶中,加入20ml乙腈溶解,加入0.1mmol三氟甲基磺酸铋,充入氮气保护,60℃下反应1h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物e,收率88%。esi-ms m/z:727.07(m na)
,如图5所示。1h nmr(400mhz,dmso-d6)δ10.96(s,4h),7.36(d,j=8.1hz,4h),7.48(s,4h),7.21(t,j=7.9hz,4h),6.87(d,j=7.5hz,4h),6.84(s,4h),5.78(s,2h),如图12所示。
[0042]
实施例6:将1mmol原料1和4.5mmol 6-溴吲哚置于50ml圆底烧瓶中,加入20ml乙醇溶解,加入0.1mmol碘,充入氮气保护,80℃下反应0.5h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物f,收率85%。esi-ms m/z:904.87(m na)
,如图6所示。1h nmr(500mhz,dmso-d6)δ10.96(s,4h),7.54(s,4h),7.25(s,4h),7.16(d,j=8.5hz,4h),6.99(d,j=8.4hz,4h),6.85(s,4h),5.79(s,2h),如图13所示。
[0043]
实施例7:将1mmol原料1和4.5mmol 6-甲氧基吲哚置于50ml圆底烧瓶中,加入20ml四氢呋喃溶解,加入0.1ml盐酸,充入氮气保护,50℃下反应2h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物g,收率80%。esi-ms m/z:709.27(m na)
,如图7所示。1h nmr(500mhz,dmso-d6)δ10.56(s,4h),7.24(s,4h),7.10(d,j=8.7hz,4h),6.85(s,4h),6.67(s,4h),6.52(d,j=8.7hz,4h),5.69(s,2h),3.75(s,12h),如图14所示。
[0044]
实施例8:将1mmol原料1和4.5mmol 6-乙氧基吲哚置于50ml圆底烧瓶中,加入20ml甲醇溶解,加入0.1mmol溴化铜,充入氮气保护,60℃下反应2h,反应完毕后,加20ml蒸馏水,分别用10ml乙酸乙酯萃取3次,有机相用无水硫酸镁干燥后减压蒸除溶剂,硅胶柱层析得目标产物h,收率82%。esi-ms m/z:765.25(m na)
。
[0045]
合成产物对人肺癌细胞抑制率测试
[0046]
取对数生长期的a549肿瘤细胞,胰酶消化后用含10%胎牛血清的dmem/f12培养基调整细胞密度5
×
104个/ml,接种至96孔培养板中,每孔含100μl细胞悬液,继续培养24h后,吸弃培养液,更换含不同药物浓度及溶剂对照的空培,每个浓度设4个复孔。继续培养24h后,每孔加入100μl mtt(0.5g
·
l-1
),放入培养箱继续孵育4h,吸弃上清,每孔加入150μl dmso,振荡10min,溶解结晶,用酶标免疫检测仪测定490nm波长处的吸光度(a)。实验重复3次,取平均值。以溶剂处理为对照组,由a计算细胞存活率,细胞存活率(%)=a用药/a对照
×
100%。采用spss17.0软件计算对细胞的半数抑制浓度(ic
50
),结果如表1所示。
[0047]
表1实施例1-8的半数抑制浓度ic
50
(μmol
·
l-1
,n=3)
[0048]
编号实施例1实施例2实施例3实施例4实施例5实施例6实施例7实施例8ic
50
7.5010.239.8612.507.988.5411.4310.35
[0049]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。