1.本发明涉及电化学能量储存领域,并且更特别地涉及表现出高能量和高功率密度两者的金属离子电容器(metal ion capacitor,mic)。
背景技术:
2.近年来,大量研究集中在电化学能量储存上以响应对高功率的高能量密度传输日益增长的需求。一方面,诸如锂离子电池(lithium ion battery,lib)的电池依赖于可以提供非常高的能量密度的法拉第反应,然而,所述电池受到功率和可循环性的限制。另一方面,超级电容器借助于容抗来存储电荷,从而以能量密度为代价实现高的功率和长的可循环性。最近,金属离子电容器(mic),特别是基于锂离子的金属离子电容器,已作为一种解决方案出现,其潜力在于通过使用与电容器级电极配对的电池级电极的组合将上述技术相结合而在高功率密度和长的可循环性下提供高能量密度。在过去几年中开发的不同锂离子电容器(lithium ion capacitor,lic)中,基于双碳的lic很有前景,甚至在市场上存在商业产品(jm energy)。然而,这种系统的密集商业化因循环期间的电荷(e-)和活性锂(li )的不可逆损失影响库仑效率和能量效率而受到阻碍。目前为止已确定的主要机制对应于在第一次充电期间在负电极处形成固体电解质界面(sei)。因此,对该li (和电子)损失进行补偿的所谓的预锂化步骤对于实现活性材料的充分利用是必要的。这是在制造高效率的lib和lic期间考虑的主要问题之一。值得注意的是,虽然它们对于lib和lic两者而言是常见的,但是预锂化对于lic甚至更关键,因为任何附加的li 离子源应当足够大以便不仅解决sei形成,而且还(在充电时)使负电极锂化。因此,lic中碳的预锂化仍然是主要的技术障碍。因此,商业lic到目前为止都是基于由石墨制成的负极,尽管其动力学缓慢,但是其呈现出与其他碳质材料如硬碳相比更低的第一次循环不可逆性,并且能够实现更容易的预锂化解决方案。
3.多年来,已经使用若干个技术对lic中的石墨进行预锂化。fuji co.提出了目前在工业中使用的方法jm energy):其使用锂箔作为第三电极以对石墨负极电极进行预锂化,并且该lic结构产生具有三个电极(正极、负极和牺牲锂金属电极)的电化学能量储存装置。这种三电极结构的lic对于正极和负极两者都需要使用网格型集电器,以便能够将锂离子传输到正极和负极中并且在正极与负极之间传输,这使得电池设计复杂并且制造昂贵。此外,这种三电极结构的lic重新引入了与早期基于使用金属锂的锂离子电池有关的危害,例如短路和热失控的风险。或者,电化学预锂化可以在单独的电池中完成。然而,电化学预锂化过程通常需要在惰性气氛下将经预锂化的负电极重新组装到lic电池中的步骤,这会使成本增长并且降低了以商业方式使用该方法的可能性。
4.因此,近年来已经开发了替代的具有成本效益的预锂化策略。最广泛推广的方法之一包括使用复合正极。将锂化金属氧化物并入至正电极并且与活性炭组合作为不可逆锂源。已经提出了诸如li2moo3、li5feo6或li2ruo3的金属氧化物,实现了石墨电极的完全预锂
化程度。尽管如此,该方法表现出作为用于预锂化的可行解决方案而针对其实现方式应当解决的一些缺点。首先,这些金属氧化物中的大多数的锂脱嵌电位(extraction potential)高于4.7v vs li/li
,促进了不利地影响后lic可循环性的电解质分解。其次,分解的金属氧化物作为死料残留在电池中,对能量密度不利。最后,金属氧化物的高成本对整体最终产品的价格具有负面影响。为了解决这些问题,最近采用了来自锂离子电池(lib)技术的牺牲盐构思。将可以不可逆地被氧化(即,锂提取)的在其结构中并入有锂的羰基类有机化合物与正电极中的活性炭混合。这是有协同作用的混合物,因为这些有机化合物是高度绝缘的并且需要大量的导电碳以分解——lib技术中的缺点,所述缺陷在lic中由于活性材料已经是高导电的碳而不存在。jezowski等人(safe and recyclable lithium-ion capacitors using sacrificial organic lithium salt,nature materials,2017,1-7)通过合成可以将石墨电极完全预锂化的具有3.5v vs li/li
的脱锂电位和365mah g-1
的高理论容量的3,4-二羟基苄腈二锂盐首次开发了这种方法。所产生的氧化醌是可溶的,从致了穿梭机制(shuttle mechanism)。此外,所开发的lic受碍于使用石墨而缺乏高功率。
5.由于硬碳的第一次循环不可逆性较高,因此硬碳已被排除在上述的预锂化解决方案之外,并且受到的关注要少得多。鉴于硬碳的不可逆容量较高,其要求非常高的预锂化程度,因此,基于硬碳的负极的预锂化一直通过使用提供几乎无限供应的锂离子的锂金属来解决。在这方面,zheng等人率先通过使用不同形式的金属锂对硬碳进行预锂化。首先,使用稳定化锂金属粉末(stabilized lithium metal powder,slmp)成功地对硬碳进行预锂化。然而,尽管锂被稳定化,但是其仍然具有高反应性,并且出现了与工业规模制造不兼容的安全问题。同一团队还开发了一种替代策略——使用与电解质直接接触的金属锂条。再次成功地对硬碳进行了预锂化,但是金属锂的使用以及该方法的复杂性阻碍了其工业化。参见zheng等人,constructing high energy and power densities li-ion capacitors using li thin film for pre-lithiation,journal of the electrochemical society,2017,164,a2164-a2170。
6.因此,仍然需要开发新的电化学能量储存解决方案来克服上述系统的局限性的。
技术实现要素:
7.本发明通过提供具有出色功率容量的基于硬碳负极的金属离子电容器(mic)解决了现有技术的局限性。特别地,发明人发现,可以将牺牲盐作为金属离子源添加至正电极中的活性炭(activated carbon,ac)中,以有效地补偿硬碳(hard carbon,hc)负极的高不可逆容量,从而允许在负极与正极之间实现1:1的优越质量平衡。有利地,该方法的优异性能不仅在锂离子电容器(lic)中,而且在诸如钠离子电容器和钾离子电容器的其他金属离子电容器中都得到了成功证明。此外,本文中描述的mic与工业化且易于扩展的制造工艺兼容,这使得能够使用hc作为mic中的负电极,从而增加能量并且允许实现比基于石墨的电容器的功率更高的功率的目标。
8.在一个方面中,本发明涉及金属离子电容器,所述金属离子电容器包括:
[0009]-包含硬碳的负电极,
[0010]-包含活性炭和牺牲盐的正电极,以及
[0011]-定位在两个电极之间的分隔件;
[0012]
其中,牺牲盐选自方酸盐、草酸盐、酮基丙二酸盐和二酮基琥珀酸盐或者其组合。
[0013]
在操作中,可以将电解溶液并入整个正极、负极和分隔件。电解溶液包含溶解在溶剂中的电解质材料(溶质)。
[0014]
在另一个方面中,本发明涉及用于制造如本文所述的金属离子电容器的方法,所述方法包括由生物质废弃物制备负硬碳电极和/或正电极的活性炭。
[0015]
在另一个方面中,本发明涉及牺牲盐作为金属离子源用于对金属离子电容器的负电极进行预掺杂以及补偿用于在负电极上形成固体电解质界面(sei)的对金属离子的需求的用途,其中所述牺牲盐与活性炭在正电极中组合,牺牲盐选自方酸盐、草酸盐、酮基丙二酸盐和二酮基琥珀酸盐或其组合,并且其中所述负电极包含硬碳。
[0016]
在下文中在详细描述中和在权利要求书中还另外限定了这些方面及其优选实施方案。
附图说明
[0017]
为了更好地理解本发明、本发明的目的和优点,以下附图附于说明书,在附图中中描绘了下述内容:
[0018]
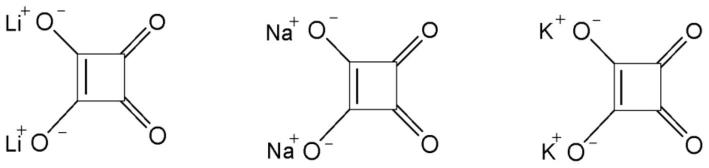
图1示出了所合成的方酸盐(实验)的x射线图案:li2c4o4、na2c4o4和k2c4o4。对于方酸锂和方酸钠,还示出了来自无机晶体结构数据库(icds)的x射线图案。
[0019]
图2示出了如实施例3至5中所述的,与super c65以60:30比例混合的li2c4o4、na2c4o4和k2c4o4的第1次、第2次和第5次循环伏安法(cyclic voltammetry,cv)以及充电/放电循环,其示出了鉴于第5次循环中残留的可忽略电流,由所有方酸盐表现出的其分解电位和高不可逆性。
[0020]
图3示出了如实施例3至5所述的,具有5:5的c65导电碳和pvdf粘合剂的与活性炭以40:50的比例混合的li2c4o4、na2c4o4和k2c4o4的第1次、第2次、第5次和第10次cv以及充电/放电循环。在第一次循环之后,残留容量仅归因于ac的贡献。
[0021]
图4示出了如实施例3中所述的,对于li/li
而言,橄榄核产生的硬碳的倍率性能测试和库仑效率。
[0022]
图5示出了如实施例3中所述的,对于li/li
而言,橄榄核产生的硬碳与可商购的石墨相比的倍率性能测试和库仑效率。
[0023]
图6示出了如实施例3中所述的,为了选择可能的最佳质量平衡,橄榄核产生的硬碳和与方酸锂混合的活性碳的容量比较。
[0024]
图7示出了如实施例3中所述的,在c/10下进行的lic的第一次充电以及hc和ac 盐的电位变化。使用1比1的质量平衡组装lic,并且在c/10下进行充电。
[0025]
图8示出了如实施例3中所述的,考虑到从几分钟到几秒钟的不同放电时间,在不同的施加电流下在2v与4v之间运行的lic的电压曲线。还示出了hc和ac 盐的电位变化。
[0026]
图9示出了如实施例3中所述的,为了比较将本文报告的lic与基于对称的橄榄核产生的ac的edlc进行比较的ragone图。
[0027]
图10示出了如实施例4中所述的,为了选择可能的最佳质量平衡,橄榄核产生的硬碳和与方酸钠混合的活性碳的容量比较。
[0028]
图11示出了如实施例4中所述的,在c/10下进行的nic的第一次充电以及hc和ac
盐的电位变化。
[0029]
图12示出了如实施例4中所述的,考虑到从几分钟到几秒钟的不同放电时间,在不同的施加电流下在2v与4v之间运行的nic的电压曲线。还示出了hc和ac 盐的电位变化。
[0030]
图13示出了如实施例4中所述的,为了比较目的将本文报告的nic与基于对称的橄榄核产生的ac的edlc进行比较的ragone图。
[0031]
图14示出了如实施例5中所述的,为了选择可能的最佳质量平衡,橄榄核产生的硬碳和与方酸钾混合的活性碳的容量比较。
[0032]
图15示出了如实施例5中所述的,在c/10下进行的kic的第一次充电以及hc和ac 盐的电位变化。
具体实施方式
[0033]
当前的碱金属离子电容器技术,例如锂离子电容器(lic),基于双碳电极系统,石墨是负电极中最常用的材料。对于lic,与锂离子电池(lib)相比需要更高的预锂化程度,因为正电极不是锂源并且所有锂必须来自预锂化源。因此,当前使用石墨作为负电极,原因是石墨的不可逆容量较低,仅为约5%,从而使预锂化步骤中所需的锂量最小化。然而,石墨缓慢的动力学在功率方面限制了lic技术,并且非常期望在负电极中使用更大功率的材料。此外,该技术应当不仅适用于lic,而且适用于其他金属离子电容器,例如钠离子电容器和钾离子电容器。
[0034]
本发明中提供的解决方案是金属离子电容器,所述金属离子电容器包括:
[0035]-至少一个包含硬碳(hc)的负极,
[0036]-至少一个包含活性炭(ac)和牺牲盐的正极,所述牺牲盐选自方酸盐、草酸盐、酮基丙二酸盐和二酮基琥珀酸盐或者其组合,以及
[0037]-介于两个电极之间的分隔件。
[0038]
电极——负极和正极——优选地浸入或以其他方式定位在电解溶液中,其中分隔件介于它们之间。
[0039]
负极由硬碳制成。如本文所使用的,hc材料优选地具有小于约500m2/g,例如小于约100m2/g的比表面积。在实施方案中,用于形成负极的hc材料是不可石墨化的,并且可以优选地具有小于约100微米,例如小于约100微米、约10微米、约5微米、或者小于或等于约1微米的平均颗粒尺寸。在一个更优选的实施方案中,hc材料具有约0.1微米至约5微米,例如约0.5微米至约2微米、或者更具体地约1微米的平均颗粒尺寸。出乎意料的是,即使使用较低颗粒尺寸也可以实现良好的性能。包含hc的负极的厚度可以在例如约25微米至约600微米的范围内。
[0040]
正极由包含活性炭和牺牲盐的复合材料制成。如本文中所使用的,ac材料优选地具有大于约500m2/g的比表面积。在实施方案中,用于形成正极的ac材料可以优选地具有小于100微米,例如小于约100微米、约10微米或约5微米的平均颗粒尺寸。包含ac/牺牲盐的正极的厚度可以在例如约25微米至约600微米的范围内。
[0041]
根据本发明,分别用作负极和正极的hc和ac可以由多种前体例如蔗糖、纤维素、聚乙烯(pvc)、糠醇合成,或者甚至更好地,由可持续资源如回收的生物质合成。在优选实施方案中,这些碳材料由生物质废弃物例如椰子壳、花生壳、果皮、橄榄核等制备。当然,可以采
用两种或更多种前体的组合来制备本发明的碳。在一个特别优选的实施方案中,hc和ac由回收的橄榄核合成。
[0042]
通常,hc的制备包括:在惰性气氛(例如,氩流)下,在高温,通常约600℃或更高(例如,约600℃至1800℃、约650℃至1500℃、约700℃至1200℃、或约750℃至1000℃)下,将碳前体加热至少约30分钟(例如,约30分钟至240分钟)以使所述前体热解。如果为固体,可以在热解之前将碳前体破碎。在某些实例中,使温度逐渐增加(例如,升温速率为约2℃分钟-1
至10℃分钟-1
)直至达到热解温度。
[0043]
根据一个特定的实施方案,hc由生物质废弃物(例如,橄榄核)通过包括下述的方法来制备:以约3℃分钟-1
至8℃分钟-1
的升温速率将生物质废弃物,优选地先前破碎的生物质废弃物加热至约600℃至1800℃(例如,约750℃至1000℃)的预定温度,并且进一步将其保持约1.5小时至2.5小时。在一个更特定的实施方案中,将生物质废弃物破碎并且装载到炉(例如,管式炉)中以通过以下过程进行热解过程:在氩流(例如,约50ml分钟-1
至250ml分钟-1
)下以约4℃分钟-1
至6℃分钟-1
的升温速率加热至约750℃至1000℃(例如约800℃)的预定温度,并且进一步将其保持约1.5小时至2.5小时。
[0044]
为了调节颗粒尺寸并定制hc用于高功率应用,然后可以对hc进行机械研磨和碾碎。在特定实施方案中,在热解之后,然后以研钵/研杵对hc进行手动粗研磨,之后在行星式磨机中对hc进行球磨。碾碎可以进行约60分钟至120分钟。
[0045]
活性炭(ac)可以由先前获得的硬碳(hc)(没有进行机械研磨和碾碎)来制备。通常,为了制备活性炭(ac),可以将先前获得的硬炭(hc)(在没有研磨和球磨步骤的情况下)与氢氧化物例如氢氧化钾以不同的质量比,通常以约1:1或更大(例如约1:10、约1:8、约1:6、约1:4或约1:2)物理混合。然后可以通过在惰性气氛(例如,ar流)下加热直至约600℃至800℃(例如,约650℃、约700℃、约750℃)的温度来使与氢氧化物混合的hc活化。在实施方案中,将材料混合物放置在舟皿中,并且通过以下使其活化:在惰性气氛(例如,约50ml分钟-1
至250ml分钟-1
的ar流)下加热直至约600℃至800℃(例如,约650℃、约700℃、约750℃)的温度,优选地在炉(例如,管式炉)内的水平不锈钢管内部。在某些情况下,使温度逐渐增加(例如,升温速率为约2℃分钟-1
至10℃分钟-1
)。在限定温度下的保持时间通常可以设置为约30分钟至约5小时(例如,约1小时至2小时、约2小时至3小时、约3小时至4小时、约4小时至5小时)。在活化之后,可以对微孔ac进行冲洗直到中性ph并将其干燥。例如,可以用酸和水的稀释溶液对微孔ac进行冲洗直到达到中性ph,然后在约100℃至150℃下将其干燥,优选地在真空下干燥。
[0046]
根据一个特定实施方案,ac通过将先前获得的hc与氢氧化钾以约1:5至1:7的质量比物理混合来制备。将混合材料放置在舟皿(例如,舟皿)中,并且通过在管式炉内的水平不锈钢管内部在约50ml分钟-1
至250ml分钟-1
的ar流下加热直至约650℃至750℃使混合材料活化。使温度以约3℃分钟-1
至8℃分钟-1
的升温速率逐渐增加,并且在限定温度下的保持时间设置为约1.5小时至2.5小时。在活化之后,用盐酸和水的稀释溶液对微孔ac进行冲洗直到达到中性ph,然后在真空下在约120℃下将其干燥。
[0047]
在本发明的金属离子电容器的正极中ac与牺牲盐组合。牺牲盐充当金属离子源,用于对负电极进行预掺杂以及补偿用于在负电极上形成固体电解质界面(sei)的对金属离子的需求。更特别地,术语“牺牲盐”是指能够在第一次充电期间分解释放金属离子使得金
属离子可以被充入负电极中并且使由于sei形成导致的活性金属离子损失得到补偿的金属盐。也就是说,牺牲盐能够向hc的结构供应金属离子,并且能够通过提供在负电极的表面处形成钝化层所必需的金属离子来补偿hc的高不可逆容量。
[0048]
本发明中有用的牺牲盐包括方酸盐、草酸盐、酮基丙二酸盐和二酮基琥珀酸盐或者其组合。牺牲盐的金属优选地选自 1电荷的金属离子,例如碱金属。 1电荷的金属离子例如碱金属离子(例如,li、na和k)的这些盐的化学式如下:方酸盐(m2c4o4)、草酸盐(m2c2o4)、酮基丙二酸盐(m2c3o5)和二酮基琥珀酸盐(m2c4o6),其中m为 1电荷的金属离子。所有这些盐都转换为气态产物co和co2,这不产生残留物并因此可以在电池形成循环之后被去除。
[0049]
根据特定实施方案,牺牲盐选自方酸盐和酮基丙二酸盐或者其组合。在更特定的实施方案中,牺牲盐为式m2c4o4的方酸盐,其中m选自li、na和k。下面描述了li2c4o4、na2c4o4和k2c4o4的化学结构及其分解反应。
[0050][0051]
a)li2c4o4→
2co2 2c 2e- 2li
[0052]
b)na2c4o4→
2co2 2c 2e- 2na
[0053]
c)k2c4o4→
2co2 2c 2e- 2k
[0054]
复合正极可以由合适比例的牺牲盐与活性炭的混合物形成。在实施方案中,复合电极中的牺牲盐的量可以在约10重量%至约60重量%(例如,约10重量%、约20重量%、约30重量%、约40重量%、约50重量%或约60重量%)的范围内。在一个更特定的实施方案中,复合电极中的牺牲盐的量为约40重量%。在实施方案中,复合电极中的ac的量可以在约10重量%至约60重量%(例如,约10重量%、约20重量%、约30重量%、约40重量%、约50重量%或约60重量%)的范围内。在一个更特定的实施方案中,复合电极中的ac的量为约50重量%。在一个甚至更特定的实施方案中,复合正极包含约40重量%的牺牲盐和约50重量%的ac。
[0055]
正极和负极可以通过将相关组分的浆料混合物流延(例如,流延成型(tape casting))来形成。正极浆料可以包含活性炭和任选的粘合剂,负极浆料可以包含硬碳、任选的粘合剂和任选的导电碳源如石墨或炭黑。示例粘合剂包括但不限于聚偏二氟乙烯(pvdf)和聚四氟乙烯(ptfe)。导电碳的具体实例包括但不限于超c45和super c65。
[0056]
对于正极和负极中的每一者,并入到电极中的粘合剂的量可以在总的电极组合物的约0重量%至约20重量%,例如约5重量%至约10重量%的范围内。对于负极,并入到电极中的导电碳的量可以在总的负极组合物的约0重量%至约10重量%,例如约5重量%的范围内。
[0057]
在一个特定实施方案中,复合正极包含约40重量%的牺牲盐、约50重量%的ac、约5重量%的c65和约5重量%的pvdf。
[0058]
在一些实施方案中,包含与牺牲盐混合的活性炭的复合正极可以为多孔的,而硬碳负极可以为多孔的或无孔的并且可以例如对于液体(包括用于形成电解溶液的溶剂)是
不可渗透的。正极和负极可以附接至相应的正集电器和负集电器。集电器可以包括金属箔如铝箔或铜箔。
[0059]
正极、负极、分隔件和集电器在组装时可以被统称为电极组。在一些实施方案中,电极组可以基本上由正极、负极和分隔件组成,或者基本上由正极、负极、分隔件和相应的集电器组成。
[0060]
可以在正极与负极之间并入液体电解溶液,使得电解溶液渗透分隔件。电解溶液可以包含溶解在合适溶剂中的电解质材料(溶质)。电解质材料可以为能够在电化学装置中起作用的任何材料。在实施方案中,本发明涉及锂离子电容器,其中电解质材料为锂盐,即络合锂盐,例如lipf6、libf4、liclo4、liasf6、lif3so3、li[(cf3so2)2n]或li[(fso2)2n]以及其混合物。在实施方案中,本发明涉及钠离子电容器,其中电解质材料为钠盐,即络合钠盐,例如napf6、nabf4、naclo4、naasf6、naf3so3、na[(cf3so2)2n]或na[(fso2)2n]以及其混合物。在实施方案中,本发明涉及钾离子电容器,其中电解质材料为钾盐,即络合钾盐,例如kpf6、kbf4、kclo4、kasf6、kf3so3、k[(cf3so2)2n]或k[(fso2)2n]以及其混合物。用于形成电解溶液的示例溶剂包括有机溶剂或有机溶剂的混合物,例如碳酸二甲酯(dmc)、丙酸甲酯(mp)、碳酸亚乙酯(ec)、碳酸丙二醇酯(pc)、碳酸二乙酯(dec)、碳酸甲乙酯(emc)或其混合物,以及适合于其中电荷载流子为碱金属离子如锂离子、钠离子或钾离子的电解质中使用的其他溶剂。在一些实施方案中,溶剂可以能够溶解电解质材料。
[0061]
诸如络合锂盐、络合钠盐或络合钾盐的络合盐是这样的任何离子化合物:其包含诸如锂、钠或钾的金属,以及本身不电离并且可溶于有机溶剂中的另外的金属、准金属或非金属原子。例如,lipf6包含锂和磷作为金属原子,但是磷本身不电离。相反,磷作为pf
6-离子电离。在另一个实施例中,libf4包含锂金属和准金属硼。虽然锂电离(li
),但是硼本身不电离,而是作为bf
4-离子电离。在又一个实例中,liclo4包含锂金属和非金属原子氯和氧。非金属原子作为高氯酸根离子(clo
4-)电离。溶剂可以为在电化学能量储存装置中使用的任何合适的溶剂。
[0062]
在一个特定实施方案中,电解溶液为mpf6在ec:pc中的溶液,其中m优选为li、na或k。
[0063]
电解质材料和牺牲盐必须基于相同的金属离子。在一个特定实施方案中,本发明涉及碱离子电容器,其包含作为正电极添加剂的碱金属牺牲盐和基于碱金属盐的电解质。在一个更特定的实施方案中,本发明涉及锂离子电容器,其包含作为正电极添加剂的锂牺牲盐和基于锂盐的电解质。在一个特定实施方案中,本发明涉及钠离子电容器,其包含作为正电极添加剂的钠牺牲盐和基于钠盐的电解质。在一个特定实施方案中,本发明涉及钾离子电容器,其包含作为正电极添加剂的钾牺牲盐和基于钾盐的电解质。
[0064]
分隔件或离子传导膜可以介于负极与正极之间。分隔件提供离子导电性,同时确保相对电极之间的有效分隔。
[0065]
基本上,可以使用两种类型的分隔件:多孔分隔件,其中电解质在合适溶剂中的溶液填充分隔件的孔隙;或者无孔分隔件,其通常为纯固体聚合物电解质(即,溶解在用作固体溶剂的的高分子量聚醚主体例如peo和ppo中的电解质)或者凝胶聚合物电解质体系(其将能够在聚合物主基体和电解质中形成稳定凝胶的增塑剂或溶剂并入到聚合物基体中)。
[0066]
在一个特定实施方案中,分隔件由玻璃纤维材料制成。
[0067]
一种形成金属离子电容器的方法包括:组装电极组,所述电极组包括复合正极、负极和设置在负极与正极之间的分隔件;然后向组合件中添加电解溶液。
[0068]
根据特定实施方案,正电极和负电极为层合的或卷绕的。
[0069]
本发明的金属离子电容器可以作为用于电动汽车、混合电动汽车等的驱动或辅助储存装置而得到实际应用。此外,本发明的金属离子电容器适合作为用于诸如太阳能发电和风力发电的各种发电系统的储存装置,以及适合作为用于家用电子设备的储存装置等。
[0070]
本领域技术人员知道,与测量有关的数值会受到测量误差的影响,这限制了数值的准确性。当将诸如“约”或“大约”的术语应用于特定值(例如“约200℃”或“大约200℃”)或者应用于范围(例如“约x至大约y”)时,该值或范围被解释为与用于测量该值或范围的方法一样准确。除非另有明确说明,否则可以应用科学和技术文献中的一般约定,使得数值的最后一位优选地指示测量值的精度。因此,除非给出其他误差容限,否则最大容限优选地通过对最后一个小数位应用四舍五入约定来确定。例如,值3.5优选地具有3.45至3.54的误差容限,并且2%至10%的范围优选地覆盖1.5%至10.4%的范围。指定值的所述变化是本领域技术人员所理解的并且在本发明的范围内。此外,为了提供更简洁的描述,本文中给出的一些定量表达没有使用术语“约”来限定。应当理解,无论是否明确使用术语“约”,本文中给出的每个量都旨在指实际的给定值,并且还旨在指基于本领域普通技术人员可合理推断的这样的给定值的近似值,包括由于这样的给定值的实验条件和/或测量条件而产生的等效值和近似值。
[0071]
浓度、量和其他数值数据在本文中可以以范围格式表示或呈现。要理解的是,这样的范围格式仅为了方便和简洁而使用,并因此应当被灵活地解释成不仅包括明确叙述为范围限制的数值,而且还包括涵盖在该范围内的所有单独的数值或子范围,如同每个数值和子范围被明确地叙述一样。作为例示,“约1%至约5%”的数值范围应当被解释为不仅包括明确叙述的约1%至约5%的值,而且还包括在该指定范围内的单独值和子范围。因此,该数值范围中包括诸如2、3和4的单独值,以及例如1至3、2至4和3至5等的子范围。这种相同的原理适用于列举仅一个数值的范围。
[0072]
应当理解,本公开内容的范围包括本文中公开的实施方案的所有可能的组合。
[0073]
以下实施例仅是本发明的某些实施方案的举例说明,并且不能被认为以任何方式限制本发明。
[0074]
实施例
[0075]
实施例1
–
牺牲盐的制备
[0076]
使用4-二羟基-3-环丁烯-1,2-二酮和相应的金属碳酸盐(即li2co3、na2co3或k2co3)作为起始材料合成牺牲盐方酸二锂(li2c4o4)、方酸二钠(na2c4o4)和方酸二钾(k2c4o4)。取1:1的混合物并且将其溶解在去离子水中,随后搅拌过夜。在使用前通过使用旋转蒸发仪除去去离子水并在真空下在50℃下干燥12小时。
[0077]
图1描绘了牺牲盐的xrd图案。
[0078]
实施例2
–
电极的制备:hc和复合ac/牺牲盐
[0079]
将橄榄核破碎并且装载在管式炉中以通过以下过程进行热解过程:在100ml分钟-1
的ar流下以5℃分钟-1的升温速率加热至800℃的预定温度,并且进一步将其保持2小时(活化产率为约25%)以获得硬碳。为了调整颗粒尺寸并且定制hc用于高功率应用,然后将hc在
硬质瓷研钵/研杵中手动地粗研磨,之后在pulverisette 5(fritsch international,德国)中使用罐和由氧化锆制成的球以1:30hc:球质量比进行球磨。碾碎进行90分钟。
[0080]
为了制备活性炭(ac),将先前获得的硬炭(hc)(在没有研磨和球磨步骤的情况下)与氢氧化钾以1/6的质量比物理混合。将与koh混合的材料放置在舟皿中,并且通过在管式炉内的水平不锈钢管内在ar流(100ml分钟-1
)下加热直至700℃的温度使所述材料活化。加热升温速率为5℃分钟-1
,并且在限定温度下的保持时间为2小时。在活化之后,使用盐酸和水的稀释溶液对微孔ac进行冲洗直到达到中性ph,然后在真空下在120℃下对微孔ac进行干燥(活化产率为约75%)。
[0081]
将ac与实施例1的牺牲盐(li2c4o4、na2c4o4和k2c4o4)中的每一者以及导电碳(c65)和粘合剂(pvdf)混合以制备三种复合材料,所述三种复合材料在以下实施例中用作正极,连同合成的hc用作负极。
[0082]
实施例3
–
锂离子电容器
[0083]
在锂离子电容器中研究了用于负电极预锂化的方法,所述锂离子电容器包括构建在气密swagelok电池中的浸入在1m lifp
6 ec:dmc中的用纤维玻璃分隔的基于硬碳的负电极以及基于活性炭和li2c4o4的正电极。图1a描绘了合成的锂牺牲盐的xrd图案(实验,上)以及在无机晶体结构数据库(icds,下)中注册的锂牺牲盐的xrd图案。
[0084]
图2描绘了半电池配置中合成盐的电化学性能,以确定分解电位和实验容量。由于盐本身是非导电的有机化合物,因此将所述盐与导电碳(super c65、imerys石墨&碳)混合以获得在盐上的碳涂层,以使其能够分解。电位窗口对于相应的m/m
设置在2伏与5伏之间,以确保盐的完全分解。图2a示出了对于在1m lifp
6 ec:dmc电解质中的方酸锂的情况。第一次cv循环示出了在3.8v与4.5v vs.li /li之间的宽峰,其中最大值集中在4.2v处。第二次循环仍示出了宽但强度小得多的峰,其描述了在第一次循环之后仍有一些盐残留在电极中。即使如此,在随后的循环中不存在峰表明所有的锂都已经不可逆地脱嵌。恒电流(ga)充电/放电测量在2v与4.2v之间(由cv确定的电位窗口)进行。在图2d中示出了比容量vs.li
/li电位。在c/10下直至4.2v的第一次充电(c是li2c4o4的理论容量,对应于425.48mah g-1
)示出了在3.8v与4v vs.li
/li之间的大的台阶,由此获得了375mah g-1
的不可逆比容量,而在接下来的循环中容量几乎可以忽略,对应于super c65。
[0085]
图3示出了用于开发正电极的与ac组合的不同牺牲盐,下文被称为“ac 盐”。图3a示出了与ac、导电碳(c65)和粘合剂(pvdf)分别以40重量%、50重量%、5重量%和5重量%的重量比混合的li2c4o4。cv示出了在2v与4.2v vs.li
/li之间在0.1mv秒-1
下记录的伏安图。高电位由于超出该高电位的电解质的不稳定性而受到限制。在第一次负极扫描中,在3.8v与4.2v vs.li
/li之间的宽峰描述了盐的不可逆氧化反应。在接下来的循环中,由于残留在电极中的盐而出现了强度更小的峰。到第10次循环时,仅仅可以看到ac的典型矩形伏安图,从而证明了ac 盐的完全分解,同时ac的贡献保持不变。还探索了ga充电/放电。图3d示出了在c/10(c是li2c4o4的理论比容量)下复合材料的第一次充电步骤,示出了相对于盐质量的480mah g-1
的不可逆比容量。该容量高于盐的理论容量归因于ac的贡献。另外,在2v与4v vs.li
/li之间探索了新的正电极的倍率性能,并且将其与参考ac(没有牺牲盐,ac
参考
)比较以发现残留产物对电化学性能的可能影响。
[0086]
图4示出了对要在lic的组合件上使用的硬碳材料进行的倍率性能测试。示出了在
不同c倍率下的ga充电/放电测量,其中c对应于lic6的理论容量,372mah g-1
。在c/10下的第一次充电步骤中,hc输出了1063mah g-1,在1063mah g-1中,461mah g-1是可逆的,表现出43.4%的ce。对hc观察到的高不可逆容量归因于hc的较低颗粒尺寸以及使电极面积增加的无序性质。在5次循环之后,容量稳定在394mah g-1
。对于hc在2c倍率下的后续充电/放电输出了260mah g-1
并且在高功率需求倍率下保持优异的比容量值,即,在10c下215mah g-1
、在50c下120mah g-1
和在100c下70mah g-1
。
[0087]
图5示出了与类似的石墨电极相比,对要在lic的组合件上使用的硬碳材料进行的倍率性能测试。虽然倍率性能直至2c充电放电倍率都非常相似,但是在5c下开始出现差异,而在10c下时已经明显看到hc是其石墨对应物至少2.5倍好,同时当所应用的c倍率增加时该差异增大。因此,证明了对于大功率lic使用hc优于石墨的便利性。另一方面,hc的第一ce为42%,而石墨的第一ce为72%。
[0088]
为了获得电极的最大容量输出并且能够成功地解决第一锂化步骤,需要平衡正电极与负电极的活性质量,因为两个电极中储存的电荷必须相等(m c δv =m-c-δv-)。在图6中,比较了在不同电流密度下两个电极的比容量。因此,对于lic的开发选择1:1(hc:ac 盐)的质量比。
[0089]
图7示出了基于hc的lic的第一恒电流锂化。首先,在对于hc质量的c/10下对lic(hc:ac 盐)进行充电直至在对于hc质量的c/10下固定的4.2v电压。同时,li
从正电极中脱嵌并且插入到hc的微观结构中,允许达到330mv vs.li
/li。在图8中对后面循环和系统以及电极性能进行了评估,其中示出了从电池可经得起的放电时间(30分钟)到受限于edlc的放电时间(15秒)进行的在不同电流密度下的ac(红色虚线)、hc(绿色点划线)和整个lic(黑色直线)的不同充电/放电曲线。在低电流密度下,在约30分钟的放电时间内,ac呈现出表明其电容性储存机制的对称形状。ac在2在2.8v至4.3v vs.li
/li内变化,而hc在860mv至380mv vs.li
/li内变化,呈现出松散(sloppy)曲线。在约2.5分钟的放电时间内,所施加的电流密度进一步增加至0.5a g-1
,两个体系保持相似的行为,然而,负电极的电位窗口略微变宽。然而,hc仍然在远离电镀电位的安全电位下运行。增加所施加的电流密度并且朝通常对于edlc而受限的放电时间行进,hc在仅15秒的放电时间内表现出色的性能。
[0090]
能量密度值和功率密度值涉及两个电极的活性质量(active mass,am)的总和(hc的mg和ac的mg)并且在图9的ragone图中报告。对于两个装置都实现了在不同功率密度下的高能量密度值。然而,考虑到高功率应用,基于hc的lic实现的超快响应解决了基于石墨的lic的能量特征。与对称的基于橄榄核产生的ac的常规edlc相比,在不同功率密度要求下观察到能量密度2倍至3倍的增加。
[0091]
实施例4
–
钠离子电容器
[0092]
在钠离子电容器中研究了用于负电极预钠化(presodiation)的方法,所述钠离子电容器包括构建在气密swagelok电池中的浸入在1m nafp6ec:pc中的用纤维玻璃分隔的基于硬碳的负电极以及基于活性炭和na2c4o4的正电极。图1b描绘了合成的钠牺牲盐的xrd图案(实验,上)以及在无机晶体结构数据库(icds,下)中注册的钠牺牲盐的xrd图案。
[0093]
如上所述,图2描绘了半电池配置中合成盐的电化学性能,以确定分解电位和实验容量。图2b示出了在1m nafp
6 ec:pc中的方酸钠的情况。第一次循环示出了在3.5v与4.15v vs.na
/na之间的宽峰,其中发生了na2c4o4的分解。第二次循环示出了宽但强度小得多的
峰,这归因于在第一次循环之后残留在电极中的盐。即使如此,在随后的循环中不存在峰表明所有的钠都已经不可逆地分解。图2e示出了在2v至4.2v vs.na
/na的电位窗口内进行的ga充电/放电测量。在c/10下直至4.2v的第一次充电(c是na2c4o4的理论容量,对应于339.268mah g-1
)示出了在3.7v与4.2v vs.na /na之间的松散曲线,由此获得了275mah g-1
的不可逆比容量。在接下来的循环中,容量几乎为零,对应于super c65。
[0094]
图3b示出了与ac、导电碳(c65)和粘合剂(pvdf)分别以40重量%、50重量%、5重量%和5重量%的重量比混合的na2c4o4。cv示出了在2v与4.2v vs.li
/li之间在0.1mv秒-1
下记录的伏安图。高电位由于超出该高电位的电解质的不稳定性而受到限制。在第一次负极扫描中,在3.6v与4v vs.li
/li之间的宽峰描述了盐的不可逆氧化反应。在接下来的循环中,由于残留在电极中的盐而出现了强度更小的峰。到第10次循环时,仅仅可以看到ac的典型矩形伏安图,从而证明了ac 盐的完全分解,同时ac的贡献保持不变。还探索了ga充电/放电。图3e示出了在c/10(c是na2c4o4的理论比容量)下复合材料的第一次充电步骤,示出了相对于盐质量的350mah g-1
的不可逆比容量。该容量高于对于参考盐测量的情况所观察到的容量归因于ac的贡献。
[0095]
为了获得电极的最大容量输出并且能够成功地解决第一钠化步骤,需要平衡正电极与负电极的活性质量,因为两个电极中储存的电荷必须相等(m
c
δv
=m-c-δv-)。为了设置装置的正确质量比,在图10中汇总了hc和ac na2c4o4复合材料的倍率性能。在这种情况下,由于hc在不同电流密度下的松散容量值,为了避免在高电流密度下的进一步钠电镀,选择1:1的质量比。
[0096]
图11示出了在对于hc质量的c/10下直至固定的4.2v的第一次充电步骤。在该步骤中,当对整个装置充电时,正电极的基于钠的牺牲盐在3.5v至4v vs.na
/na电位下被氧化,同时脱嵌的na 插入到hc的微观结构中并且实现了180mv的截止电位。
[0097]
在预钠化步骤完成之后,对nic追踪在不同电流密度下在2v至4v的电池电压下的ga充电/放电测量。图12示出了ac(红色虚线)、hc(绿色点划线)和整个nic(黑色直线)在不同电流密度下的不同充电/放电曲线。在所有的充电/放电曲线中,ac电位窗口在3v至4v vs.na
/na内变化,而hc电位窗口在1v至约300mv vs.na
/na内变化,避免了朝负电极的表面的任何钠电镀。在图13中的ragone图中报道了能量密度值和功率密度值——涉及预钠化nic的两个电极的活性质量(am)(hc的mg和ac的mg)的总和。在低功率密度下,在约30分钟的放电时间内,所开发的预钠化nic可以输出与其lic对应物一样多的能量密度,该能量密度为约72wh kg-1
am
。同时,增加所施加的电流密度并且行进至约10分钟的放电时间,能量密度仍然保持在58wh kg-1
am
。然而,由于在高倍率下na 的扩散受阻,nic输出比所开发的lic更低的比能值。然而,nic仍然能够提供比其edlc对应物更高的能量密度值,高达2000wh kg-1
am
的功率密度——在约1分钟的放电时间内——而没有任何钠电镀。
[0098]
实施例5
–
钾离子电容器
[0099]
图1c描绘了钾牺牲盐的xrd图案。
[0100]
如上所述,图2描绘了半电池配置中合成盐的电化学性能,以确定分解电位和实验容量。图2c示出了在1m kpf
6 ec:dmc中的方酸钾的情况。第一次循环示出了在3.5v与4.5v vs.k
/k之间的宽峰,其中发生了k2c4o4的分解。电流的进一步增加归因于电解质分解。在随后的循环中,3.5v与4.5v之间的峰消失,但是超过4.5v的峰证明了电解质分解。图2f示出了
在2v至4.5v vs.k
/k的电位窗口内进行的ga充电/放电测量。在c/10下直至4.5v的第一次充电(c是k2c4o4的理论容量,对应于282mah g-1
)示出了在3.6v与4.5v vs.k
/k之间的松散曲线,由此获得了225mah g-1
的不可逆比容量。在接下来的循环中,容量几乎为零,仅对应于super c65。
[0101]
图3c示出了与ac、导电碳(c65)和粘合剂(pvdf)分别以40重量%、50重量%、5重量%和5重量%的重量比混合的k2c4o4。cv示出了在2v与4.2v vs.li
/li之间在0.1mv秒-1
下记录的伏安图。高电位由于超出该高电位的电解质的不稳定性而受到限制。在第一次负极扫描中,在3.6v与4.2v vs.li
/li之间的宽峰描述了盐的不可逆氧化反应。在接下来的循环中,由于残留在电极中的盐而出现了强度更小的峰。到第10次循环时,仅仅可以看到ac的典型矩形伏安图,从而证明了ac 盐的完全分解,同时ac的贡献保持不变。还探索了ga充电/放电。图3f示出了在c/10(c是k2c4o4的理论比容量)下复合材料的第一次充电步骤,示出了相对于盐质量的250mah g-1
的不可逆比容量。该容量高于对于参考盐测量的情况所观察到的容量归因于ac的贡献。
[0102]
为了获得电极的最大容量输出并且能够成功地解决第一预钾化(prepotassiation)步骤,需要平衡正电极与负电极的活性质量,因为两个电极中储存的电荷必须相等(m
c
δv
=m-c-δv-)。为了设置装置的正确质量比,在图14中汇总了hc和ac k2c4o4复合材料的倍率性能。在这种情况下,由于hc在不同电流密度下的松散容量值,为了避免在高电流密度下的进一步钾电镀,选择1:1的质量比。
[0103]
图15示出了在对于hc质量的c/10下直至固定的4.2v的第一次充电步骤。在该步骤中,当对整个装置充电时,正电极的基于钾的牺牲盐在3.5v至4.2v vs.k
/k的电位下被氧化,同时脱嵌的k
插入到hc的微观结构中并且实现600mv的截止电位。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。