1.本发明属于基于三人核心家系的ngs测序的生物信息数据分析领域,涉及三人核心家系全外显子组测序(trio-wes)和三人核心家系全基因组测序(trio-wgs)的数据分析,尤其涉及一种基于二代测序数据的孟德尔遗传错误分析方法。
背景技术:
2.基于三人核心家系的二代测序方法已普遍用于遗传病的筛查和辅助诊断。单亲源二体(uniparental disomy,upd)是一种与表观遗传相关的疾病,指来自父母一方的染色体片段被另一方的同源部分取代,或一个个体的两条同源染色体都来自同一亲本,前者称为节段性单亲源二体。单亲源二体可分为单亲同二体(isodisomy upd(iupd),来自同一亲体的同一染色体)和单亲源异二体(heterodisomy upd(hupd),分别来自同一亲体的两条同源染色体)。
3.upd是一种临床上重要的疾病,在生长衰竭和内分泌异常的病因中占一定比例。最近的研究表明,在3500例活产婴儿中,约存在1例upd。
4.全外显子组测序(wes)和全基因组测序(wgs)已经广泛地应用于遗传疾病的筛查和诊断方面,三人核心家系的检测方法结合遗传模式分析可提升新发突变和符合杂合突变的检出率,但对于分析出来的点突变数据用于额外的变异类型(如upd及其遗传亲本来源)的分析缺乏根据,像15号染色体upd不同亲本来源可能跟不同的疾病相关,母源缺失导致angleman-syndrome,父源缺失导致prader-willi syndrome。对于应用trio-wes或trio-wgs的不符合孟德尔遗传的位点数目和分布在亲缘关系不符、是否近亲结婚、是否有样本混淆和样本污染等质量控制方面也缺乏方法依据。
5.目前用于分析roh的方法主要是基于单核苷酸多态性阵列(snp-array,cma)分析拷贝数缺失或拷贝数中性roh,但目前的方法不能准确区分亲本来源。如果要区分亲本来源,通常还得做甲基化分析,如snp-array检测15号染色体的情况,通常还需对15q11.2区域做先证者和父母的ms-mlpa以确定亲本来源。基于ngs分析roh的分析工具automap,也仅用于单样本分析iupd,不能分析hupd,且无法做更多的分析和质控。目前亲缘关系是否相符、是否有样本污染或样本混淆一般通过核心家系(trio)的str检测来比对。
技术实现要素:
6.为了克服现有技术的不足,考虑到基于trio-wes或trio-wgs的分析方法可根据先证者是否有连续的不符合孟德尔遗传的位点来分析upd;结合特殊类型的不符合孟德尔遗传的基因型可用于准确推断upd的亲本来源,区分iupd和hupd;对于整个基因组范围内出现有很多大段的roh且占全基因组范围一定比例的可用于推断先证者的是否存在血亲同源(ibd)的情况;对于全基因组范围均匀分布的不符合孟德尔遗传的位点及其比例可用于提示是否存在亲缘关系不符、样本混淆和样本污染、产前羊水或流产物样本母源污染,及异体骨髓移植或器官移植等情况。本发明提出了一种基于二代测序数据的孟德尔遗传错误分析
方法。
7.本发明基于二代测序数据的孟德尔遗传错误分析方法,包括如下步骤:
8.1)将三人核心家系的双端150bp读长的二代测序原始序列比对到参考基因组,查找出三个样本全基因组范围内的点突变,包括单核苷酸多态性(snp)和插入缺失(indel);
9.2)对超过2种基因型的突变拆分成多个双等位基因突变;
10.3)对低质量和位于明确致病性良性或人群多态性cnv区域的snp和indel位点进行过滤;
11.4)标记基因型;
12.根据不同情况,将突变位点分别标记为denovo、iupdpat、iupdmat、updpat、updmat、duo-del;将所有denovo、iupdpat、iupdmat、updpat、updmat、duo-del位点都标记为mie(mendelian inheritance error);
13.5)将denovo、iupdpat、iupdmat、mie、updpat、updmat、duo-del赋予1、2、3、4、5、6、7数值以便于数据可视化,在全基因组范围内做分布图,用cbs方法将连续符合某种特征的点连成片段,输出片段分布,标注上已知的upd疾病区域;
14.6)不同情况的判定:
15.6.1如果基因组范围内存在某段与已知upd疾病区域有重叠的upd片段,结合表型分析提示upd相关疾病可能;有可能单独存在iupd或单独hupd,也有可能iupd和hupd同时存在,可结合标记类型判定亲本来源;
16.6.2如果在基因组范围内存在多段iupd,且占到基因组范围内一定比例,则提示存在近亲婚配的可能;
17.6.3如果基因组范围内较均匀分布着mie的点,但又不那么密集的情况,则可能存在样本污染或者产前样本的母源污染,对于非产前样本提示肿瘤发生可能;
18.6.4如果全基因组范围内较均匀分布着mie的点且密集程度高,mie位点多的情况,则提示可能存在样本混淆或者亲缘关系不符合的情况,或可能存在异体器官/骨髓移植。
19.根据本发明的优选方案,所述步骤1)中的二代测序的方法为trio-wes或trio-wgs。
20.根据本发明的优选方案,所述步骤1)中,查找出三个样本全基因组范围内的点突变,具体为:基于gatk best practice(http://gatk.broadinstitute.org/hc/en-us/sections/360007226651-best-practices-workfl ows)分析出每个样本点突变gvcf格式文件,然后用joint-calling分析方法合并成一个vcf文件;
21.根据样本家系关系ped文件(https://www.mv.helsinki.fi/home/tsjuntun/autogscan/pedigreefile.html),将生成的vcf文件按三人核心家系分成多个家系vcf文件,过滤掉每个家系vcf文件中的三个样本都未检出或都为纯合野生型的位点。
22.根据本发明的优选方案,所述步骤3)对位点进行过滤,具体为:
23.过滤掉除1-22和x、y染色体以外的contig上的位点,过滤掉质量值低于30、最大基因分型质量小于20、最大测序深度小于8、最大突变等位基因频率小于0.2、平均测序深度小于5、任意一个样本测序深度为0或未检出的位点。
24.过滤掉人群频率》1%的且致病性为明确良性或人群多态性的cnv区域的位点。
25.5.根据权利要求1所述的基于二代测序数据的孟德尔遗传错误分析方法,其特征
在于,所述步骤4),具体为:
26.4.1将父亲和母亲都为纯合野生型、先证者为杂合突变或纯合突变型的位点标记为denovo;
27.4.2将父亲是纯合野生型、母亲是杂合突变型、先证者是纯合突变型或者父亲是纯合突变型、母亲是杂合突变型、先证者是纯合野生型的位点标记为iupdmat;
28.4.3将母亲是纯合野生型、父亲是杂合突变型、先证者是纯合突变型或者母亲是纯合突变型、父亲是杂合突变型、先证者是纯合野生型的位点标记为iupdpat;
29.4.4将母亲是纯合野生型,父亲是杂合或者纯合突变型、先证者是纯合突变型的位点标记为updpat;
30.4.5将父亲是纯合野生型,母亲是杂合或者纯合突变型、先证者是纯合突变型的位点标记为updmat;
31.4.6将父亲和母亲都为纯合突变型、先证者为杂合突变或纯合野生型的位点标记为duo-del;
32.4.7将所有denovo、iupdpat、iupdmat、updpat、updmat、duo-del位点都标记为mie。
33.本发明主要解决了trio-wes或trio-wgs二代测序数据用于分析upd(包括iupd和hupd)及其亲本来源问题,无需再借助额外的ms-mlpa等甲基化检测项目就可确定亲本来源,同时结合实际情况用于提示亲缘关系不符、是否近亲结婚、是否有样本混淆和样本污染、产前羊水或流产物样本是否存在母源污染,是否存在骨髓移植或器官移植等质量控制情况,可作为二代测序过程中一项常规的质量控制项目,对于二代测序过程中隐含的信息可做进一步的核实和确定,最终提高二代测序样本检测的阳性率。
34.本方法是基于二代测序突变的生物信息学分析,不涉及实验层面的修改或优化,所提示的信息,均需用金标准的检验方法做进一步验证,不代表最终结果。
附图说明
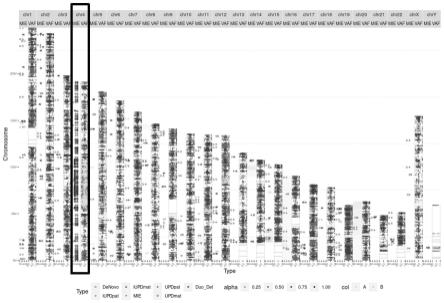
35.图1表示先证者存在严重异常临床表型且经affymetrix cyto750k snparray验证的拷贝数正常但存在大段roh区域样本和父母全血样本的mie分析结果;
36.图2表示存先证者在严重异常临床表型且经affymetrix cyto750k snparray验证的存在大片段缺失的样本和父母全血样本的mie分析结果
37.图3为图2所示样本的cnv(copy number variation)和vaf分析结果;
38.图4为通过临床随访明确其祖父母间为近亲关系的三人核心家系全血样本的mie分析结果;
39.图5为一对表型正常夫妻和另一个没任何亲缘关系的正常人组成的假三人家系样本的全基因组mie分析结果。
40.图6为先证者来自于经str检测验证存在母源污染的产前羊水及其父母全血样本的mie分析结果;
41.图7为先证者为男性的正常表型样本和其父母正常的的全血样本的mie分析结果;
42.图8位300例(100个trio核心家系)的所有染色体上mie位点总数的箱线图统计分布。
具体实施方式
43.下面结合具体实施方式对本发明做进一步阐述和说明。所述实施例仅是本公开内容的示范且不圈定限制范围。本发明中各个实施方式的技术特征在没有相互冲突的前提下,均可进行相应组合。
44.1.本实施例将300例(100个trio核心家系)由华大基因t7型号测序仪的双末端150bp测序模式输出的trio-wes数据比对到人参考基因组(参考基因组版本为hg19)上;(大部分是正常样本,其中异常的样本有:1.先证者来自于经str检测验证存在母源污染的产前羊水及其父母全血样本,2.先证者存在严重异常临床表型且经affymetrix cyto750k snparray验证的拷贝数正常但存在大段roh区域样本和父母全血样本,3.存在严重异常临床表型且经affymetrix cyto750ksnparray验证的存在大片段缺失的样本和父母全血样本,4.通过临床随访明确其祖父母间为近亲关系的三人核心家系全血样本,5.一对表型正常夫妻和另一个没任何亲缘关系的正常人组成的假三人家系样本);
45.2.基于gatk best practice(http://gatk.broadinstitute.org/hc/en-us/sections/360007226651-best-practices-workflows)分析出每个样本点突变gvcf格式文件,然后用joint-calling分析方法合并成一个vcf文件;
46.3.根据样本家系关系ped文件(https://www.mv.helsinki.fi/home/tsjuntun/autogscan/pedigreefile.html),我们将步骤2生成的vcf按三人核心家系分成多个家系vcf文件,过滤掉每个家系vcf文件中的三个样本都未检出或都为纯合野生型的位点;
47.4.对每个家系vcf文件中超过2种基因型的突变拆分成多个双等位基因突变;
48.5.将每个家系vcf文件过滤掉除1-22和x、y染色体以外的contig上的位点,过滤掉质量值低于30、最大基因分型质量小于20、最大测序深度小于8、最大突变等位基因频率小于0.2、平均测序深度小于5、任意一个样本测序深度为0或未检出的位点;过滤掉位于人群频率》1%的且致病性为明确良性或人群多态性的cnv区域的位点;
49.6.将每个家系vcf文件标记上各种位点进行分类,对不符合孟德尔遗传的位点分别进行分类;
50.其中,将父亲和母亲都为纯合野生型、先证者为杂合突变或纯合突变型的位点标记为denovo;
51.将父亲是纯合野生型、母亲是杂合突变型、先证者是纯合突变型或者父亲是纯合突变型、母亲是杂合突变型、先证者是纯合野生型的位点标记为iupdmat;
52.将母亲是纯合野生型、父亲是杂合突变型、先证者是纯合突变型或者母亲是纯合突变型、父亲是杂合突变型、先证者是纯合野生型的位点标记为iupdpat;
53.将母亲是纯合野生型,父亲是杂合或者纯合突变型、先证者是纯合突变型的位点标记为updpat;
54.将父亲是纯合野生型,母亲是杂合或者纯合突变型、先证者是纯合突变型的位点标记为updmat;
55.将父亲和母亲都为纯合突变型、先证者为杂合突变或纯合野生型的位点标记为duo-del;
56.将所有denovo、iupdpat、iupdmat、updpat、updmat、duo-del位点都标记为mie。
57.7.对每个家系vcf文件将denovo、iupdpat、iupdmat、mie、updpat、updmat、duo-del
赋予1、2、3、4、5、6、7数值,在全基因组范围内做分布图,用cbs(circular binary segmentation)方法将连续符合某种特征的点连成片段,输出片段分布,标注上已知的upd疾病区域;
58.8.如没有过多mie位点的家系,本实施例不做进一步分析。
59.如果基因组范围内存在某段与已知upd疾病区域有重叠的upd片段,那么则结合表型,提示upd相关疾病可能,即有可能单独存在iupd或单独hupd,也有可能iupd和hupd同时存在,可结合标记类型判定亲本来源;
60.如果在基因组范围内存在多段iupd,且占到基因组范围内一定比例,那么则提示可能存在近亲婚配可能;
61.如果基因组范围内较均匀分布着mie的点,但又不那么密集的情况,则可能存在样本污染或者产前样本的母源污染,对于非产前样本可能提示肿瘤发生;
62.如果全基因组范围内较均匀分布着mie的点且密集程度高,mie位点多的情况,则可能存在样本混淆或者亲缘关系不符合的情况,也有可能存在异体器官(骨髓)移植。
63.图1-8为具体案例情况。其中,图1、2、4、5、6、7为6个trio(三人核心家系)的mie分析及其先证者的vaf分布图,denovo、iupdpat、iupdmat、mie、updpat、updmat、duo-del分别用正方形、圆形、正三角形、加号、叉、菱形、倒三角形表示,对应的数值为1、2、3、4、5、6、7,vaf是variant allele fraction的简写,其定义为突变型基因型的测序深度除以此位点的总测序深度,纯合野生型对应的理论值为0,杂合突变型对应的理论值为0.5,纯合突变型对应的理论值为1。vaf部分的灰色区域采用automap(https://github.com/mquinodo/automap)软件计算的可能存在大段roh区域。
64.图1表示先证者存在严重异常临床表型且经affymetrix cyto750k snparray验证的拷贝数正常但存在大段roh区域(chr4:54441842-109664820)样本和父母全血样本,其mie分析表明chr4:54441842-109664820区域为iupdmat,整条4号染色体为updmat;
65.图2表示存先证者在严重异常临床表型且经affymetrix cyto750k snparray验证的存在大片段缺失(chr7:114333664-128000091)的样本和父母全血样本,其mie分析表明chr7:114333664-128000091区域为iupdmat和updmat,表示父源基因型缺失;
66.图3为图2所示样本的cnv(copy number variation)和vaf分析,拷贝数变异分析表明,chr7:114333664-128000091区域存在1个拷贝的缺失,且vaf分析提示此区域为roh区域,结合图2mie的分析结论我们可推测次样本存在13.7mb大小的父源拷贝数缺失;
67.图4为通过临床随访明确其祖父母间为近亲关系的三人核心家系全血样本,vaf分析发现chr11:67395456-84878075、chr11:40106074-48346729、chr11:48388067-56143225、chr19:23676134-30018369、chr13:102235519-107145829等大段的纯合区域,但在这些区域的mie分布都正常;
68.图5为一对表型正常夫妻和另一个没任何亲缘关系的正常人组成的假三人家系样本,全基因组mie分析发现所有染色体都存在数量非常多的mie位点,vaf分布正常,表明家系关系不符。
69.图6为先证者来自于经str检测验证存在母源污染的产前羊水及其父母全血样本,mie分析发现其总的mie位点较正常样本稍多。
70.图7为先证者为男性的正常表型样本和其父母正常的的全血样本的mie分析结果,
mie位点较少,且vaf分布正常。
71.图8为300例(100个trio核心家系)的所有染色体上mie位点总数的箱线图统计分布。
72.表1:本发明特定mie基因型比较及分类
[0073][0074]
表1为本发明特定mie基因型比较及分类,结合图表中的结果可知,trio-wgs或trio-wgs数据通过本发明方法的标记,可确定拷贝数中性或拷贝数缺失情况的loh(loss of heterozygous)的亲本来源。性染色体拷贝数异常的情况,也可通过此分析方法确定是哪种配子(精子或卵子)出现了异常。
[0075]
trio-wgs或trio-wgs数据通过本发明方法的标记,可以用于分析亲本是否存在近亲结婚、样本混淆、家系不符、产前样本母源污染等情况、以及受检者是否存在异体器官或骨髓移植情况。
[0076]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。