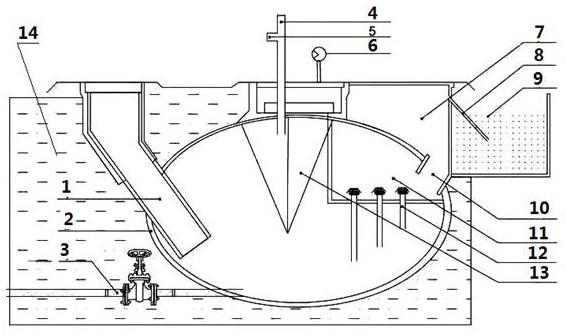
1.本发明属于烯烃聚合催化剂技术领域,尤其涉及一种非对称芳基桥连茂金属化合物及其应用,具体涉及一类非对称芳基桥连茂金属化合物,以及该类化合物在催化烯烃聚合,特别是乙烯与α-烯烃共聚中的应用。
背景技术:
2.近年来,高分子材料因较高的性价比及其优越的物理机械性能,日益渗透到人们日常生产生活的各个方面,如常见的塑料、橡胶、纤维、涂料及粘合剂、高分子共混和复合材料、功能高分子材料等等。聚烯烃材料更是在众多高分子材料中表现突出,俨然成为当今社会生产生活不可或缺的重要组成部分。为了满足国内外市场对聚烯烃材料日新月异的各种需求,众多企业及科研人员致力于研发新型烯烃聚合催化剂,用以制备新型聚烯烃材料,进而促进了烯烃聚合催化剂的进一步发展。
3.茂金属催化剂作为在齐格勒-纳塔催化剂基础上发展起来的一类新型烯烃聚合催化剂,凭借其特有的催化性能,在聚烯烃材料的工业化生产进程中起到了至关重要的作用,在未来的市场中拥有广阔的应用前景。20世纪50年代初,natta、breslow等人首先发现环戊二烯钛化合物(cp2ticl2)与alet2cl协同作用下可以催化乙烯聚合。70年代初,reichert等人发现cp2tietcl/aletcl2体系可以实现乙烯聚合,且聚合体系中因少量水的存在,而使聚合活性急剧上升,同时聚合物相对分子量也有大幅度增加。1989年,美国dow公司和exxon公司先后相隔两周各自提出限定几何构型(cgc)催化剂的专利申请。1991年,美国exxon公司首次成功将茂金属催化剂体系用于聚乙烯的工业化生产。2009年,日本sumitomo公司研发并报道了多种新型半夹心型phenics催化剂,可以高活性催化乙烯与1-己烯共聚,制备高插入率的共聚物。韩国sk公司在其基础上,进一步研究了不同基团对phenics催化剂催化性能的影响,并得到一系列高插入率的产品。
4.随着各种新型茂金属催化剂在不断研制并应用,普遍的弊端也初见端倪,如很多催化共聚得到的产物分子量与插入率不可兼顾。为此,研制新型高效催化剂用以高活性、高插入率催化乙烯/α-烯烃共聚,制备高分子量mpe产品,成为目前工业上烯烃聚合研究领域的重要方向。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种非对称芳基桥连茂金属化合物及其应用,本发明提供的非对称芳基桥连茂金属化合物为一种高活性、高稳定性的烯烃聚合催化剂,可以高效应用于催化乙烯/α-烯烃共聚、制备高分子量的聚烯烃材料。
6.本发明提供了一种非对称芳基桥连茂金属化合物,具有式i结构:
[0007][0008]
式i中,r1选自氢原子、c1~c
20
烷基、c3~c
20
的芳基或取代的芳基;
[0009]
r2选自氢原子、c1~c
20
烷基、c3~c
20
的取代或未取代的芳基;
[0010]
r3选自氢原子、c1~c
20
的烷基、c3~c
20
的芳基或取代的芳基;
[0011]
x选自卤素、烷基、芳基或取代的芳基;
[0012]
m选自第四副族过渡金属元素。
[0013]
在本发明中,所述r1中c1~c
20
烷基优选为c1~c
15
烷基,更优选为c1~c
10
烷基,更优选为c1~c5烷基,最优选为甲基、乙基、异丙基、叔丁基;所述r1中c3~c
20
的芳基或取代的芳基优选由2~4个环烷基或取代的环烷基连接形成,更优选选自两两相连的c3~c
10
的多元环或取代的多元环,所述多元环优选选自五元环或取代的五元环、六元环或取代的六元环中的一种或几种,更优选为两两相连的五元环和六元环。
[0014]
在本发明中,所述r2中c1~c
20
烷基优选为c1~c
15
烷基,更优选为c1~c
10
烷基,更优选为c1~c5烷基,最优选为甲基、丙基、异丙基、正丁基、异丁基、叔丁基;所述r2中c3~c
20
的取代或未取代的芳基优选选自c3~c
20
多元环或取代的多元环、c3~c
20
的稠环或取代的稠环,更优选选自苯基或取代的苯基、苄基或取代的苄基、苯并基或取代的苯并基、茚满基或取代的茚满基,最优选选自苯基、苄基、苯并环戊烷基、茚满基。
[0015]
在本发明中,所述r3中c1~c
20
烷基优选为c1~c
15
烷基,更优选为c1~c
10
烷基,更优选为c1~c5烷基,最优选选自甲基、乙基、异丙基、叔丁基。
[0016]
在本发明中,所述x中的卤素优选为氯;所述x中的烷基优选选自c1~c5的烷基,更优选为甲基;所述x中的芳基或取代的芳基优选选自苄基或取代的苄基,更优选为苄基。
[0017]
在本发明中,所述m优选选自钛、锆或铪。
[0018]
在本发明中,所述r1优选选自氢原子、甲基、乙基、异丙基、叔丁基、两两相连的五元环和六元环;所述r2优选选自氢原子、甲基、丙基、异丙基、正丁基、异丁基、叔丁基、苯基、苄基、苯并环戊烷基、茚满基;所述r3优选选自氢原子、甲基、乙基、异丙基、叔丁基;所述x优选选自氯、甲基。
[0019]
在本发明中,所述式i优选选自式c1~式c11中的一种:
[0020]
[0021][0022]
式c1~式c11中未给出的端基均为甲基。
[0023]
本发明对所述非对称芳基桥连茂金属化合物的制备方法没有特殊的限制,本领域技术人员根据化合物结构按照本领域熟知的各种合成方法进行制备即可。在本发明中,所
述非对称芳基桥连茂金属化合物的制备方法优选包括:
[0024]
将配体和正丁基锂溶液进行第一反应,得到配体的锂盐;
[0025]
将所述配体的锂盐和化合物mx4的溶液进行第二反应,得到非对称芳基桥连茂金属化合物(x为卤素)。
[0026]
在本发明中,所述配体优选具有式l所示的结构:
[0027][0028]
式l中的r1、r2和r3的选择基团与上述技术方案所述式i中的一致。
[0029]
在本发明中,所述配体优选选自式l1~式l10结构化合物中的一种:
[0030]
22℃,最优选为-20℃;所述第三反应的时间优选为3~7小时,更优选为4~6小时,最优选为5小时;所述3,3
’‑
二甲基联苯胺溶液中的溶剂优选为乙醚;所述第三反应过程中优选将正丁基锂溶液缓慢滴加至3,3
’‑
二甲基联苯胺溶液中;所述第三反应优选包括:在低温下反应0.5小时后,自然升温至室温再进行反应;所述低温的温度优选为-15~-25℃,更优选为-20℃。
[0039]
在本发明中,所述第四反应之前优选还包括:
[0040]
将第一反应产物冷却。
[0041]
在本发明中,所述冷却的温度优选为-75~-85℃,更优选为-78~-82℃。
[0042]
在本发明中,所述第四反应过程中优选向反应体系中通入co2;所述第四反应优选包括:
[0043]
低温反应0.5小时后,缓慢升至室温进行,去除多余的co2进行反应。
[0044]
在本发明中,所述低温反应的温度优选为-15~-25℃,更优选为-18~-22℃,最优选为-20℃;所述室温反应的时间优选为过夜。
[0045]
在本发明中,所述溶剂优选为四氢呋喃;所述叔丁基锂溶液中的溶剂优选为戊烷;所述叔丁基锂溶液的浓度优选为1~2mol/l,更优选为1.2~1.8mol/l,最优选为1.4~1.6mol/l。
[0046]
在本发明中,所述第五反应的温度优选为-15~-25℃,更优选为-18~-22℃,最优选为-20℃;所述第五反应的时间优选为1~3小时,更优选为1.5~2.5小时,最优选为2小时;所述第五反应过程中优选缓慢加入叔丁基锂溶液;所述第五反应优选包括:
[0047]
先低温反应0.5小时再进行室温反应。
[0048]
在本发明中,根据预获得的配体结构选择相应结构的茚酮类化合物。
[0049]
在本发明中,所述茚酮类化合物溶液中的溶剂优选为四氢呋喃。
[0050]
在本发明中,所述第六反应过程中优选将茚酮类化合物溶液缓慢滴加至第三反应产物中;所述第六反应优选包括:
[0051]
低温反应后自然升温至室温反应。
[0052]
在本发明中,所述低温反应的温度优选为-15~-25℃,更优选为-18~-22℃,最优选为-20℃;所述低温反应的时间优选为0.5小时;所述室温反应的时间优选为过夜。
[0053]
在本发明中,所述第六反应完成后优选还包括:
[0054]
将得到的反应产物进行提纯后处理,得到配体。
[0055]
在本发明中,所述提纯后处理优选包括:加水、加酸进行处理。
[0056]
在本发明中,所述提纯后处理的方法优选包括:
[0057]
向所述反应产物中加水和盐酸进行反应后萃取,中和,收集有机相,干燥除水,过滤,得到配体。
[0058]
在本发明中,所述反应的时间优选为0.3~0.7小时,更优选为0.4~0.6小时,最优选为0.5小时;所述萃取的试剂优选为二氯甲烷;所述中和的试剂优选包括:三乙醇胺和/或碳酸氢钠水溶液;所述干燥除水优选采用无水硫酸钠进行干燥;所述过滤后优选还包括:
[0059]
将得到的滤液去除溶剂后过柱子,得到配体。
[0060]
在本发明中,所述去除溶剂的方法优选为旋蒸;所述过柱子过程中优选采用石油醚和乙酸乙酯;所述石油醚和乙酸乙酯的体积比优选为(45~55):1,更优选为(48~52):1,
最优选为50:1。
[0061]
在本发明中,所述正丁基锂溶液中的溶剂优选为己烷;所述正丁基锂溶液的浓度优选为2~3mol/l,更优选为2.5mol.l。
[0062]
在本发明中,所述第一反应优选在低温氮气保护下进行,所述低温氮气的温度优选为-15~-25℃,更优选为-18~-22℃,最优选为-20℃;所述第一反应的优选包括:
[0063]
低温反应后再缓慢升温至室温反应。
[0064]
在本发明中,所述低温反应的时间优选为0.5小时;所述室温反应的时间优选为室温反应过夜。
[0065]
在本发明中,所述mx4中的m选择元素与上述技术方案所述式i中的一致;x选自卤素。
[0066]
在本发明中,所述mx4的溶液中的溶剂优选为己烷,更优选为正己烷。
[0067]
在本发明中,所述第二反应优选在低温氮气的保护下进行,所述低温氮气的温度优选为-15~-25℃,更优选为-18~-22℃,最优选为-20℃;所述第二反应优选包括:
[0068]
低温反应后再缓慢升温至室温反应。
[0069]
在本发明中,所述低温反应的时间优选为0.5小时;所述室温反应的时间优选为室温反应过夜。
[0070]
在本发明中,所述第二反应完成后优选还包括:
[0071]
将得到的反应产物进行过滤、浓缩、重结晶,得到x为卤素的式i结构化合物。
[0072]
在本发明中,所述过滤优选采用滤针进行;所述浓缩优选为真空浓缩;所述重结晶优选采用己烷,更优选采用超干己烷;所述重结晶优选为低温下缓慢重结晶;所得x为卤素的式i结构化合物为暗红色块状晶体。
[0073]
在本发明中,得到x为卤素的式i结构化合物后优选还包括:
[0074]
将所述x为卤素的式i结构化合物进行烷基化反应,得到x为烷基的式i结构化合物。
[0075]
在本发明中,所述烷基化反应的方法包括:
[0076]
将x为卤素的式i结构化合物溶液和格氏试剂进行烷基化反应。
[0077]
在本发明中,所述x为卤素的式i结构化合物溶液中的溶剂优选为己烷,更优选为正己烷。
[0078]
在本发明中,可根据预获得的式i结构化合物选择相应的格氏试剂。
[0079]
在本发明中,所述烷基化反应完成后优选还包括:
[0080]
将得到的反应产物进行纯化,得到x为烷基的式i结构化合物。
[0081]
在本发明中,所述纯化的方法与上述技术方案所述第二反应完成后进行的过滤、浓缩、重结晶方法一致,在此不再赘述。
[0082]
在本发明中,所述式i结构化合物的制备方法优选包括:
[0083]
在-20℃的氮气保护下,将适量的正丁基锂己烷溶液缓慢滴加至3,3
’‑
二甲基联苯胺的乙醚溶液中,低温反应半小时后,自然升至室温,然后室温下反应一段时间;之后将反应液在-78℃下冷却数分钟,接着向反应体系通入co2(g),低温反应半小时后,缓慢升至室温,除去多余的co2(g)并反应一段时间;反应结束后,继续降至-20℃,并缓慢加入适量叔丁基锂的戊烷溶液、四氢呋喃;低温反应一段时间后,接着将相应茚酮类化合物的四氢呋喃溶
液缓慢滴加至反应产物中,低温反应后自然升至室温反应一段时间;待反应结束后,经过加水、加酸等一系列提纯后处理,即可得到相应结构的配体。
[0084]
在低温氮气保护下,将适量的正丁基锂己烷溶液缓慢滴加至上述制备的配体中,低温反应0.5小时然后室温下反应一段时间,得到配体的锂盐;之后,在低温氮气保护下,将配体的锂盐缓慢滴加至冷却的mx4己烷溶液中,室温反应一段时间;最后,将反应得到的粗产物进行过滤、浓缩、重结晶,得到x为卤素的式i结构茂金属化合物。
[0085]
在低温氮气保护下,向x为卤素的式i结构茂金属化合物正己烷溶液中,缓慢加入相应格氏试剂(grignard reagent)进行烷基化反应,经纯化处理,得到x为烷基的式i结构茂金属化合物。
[0086]
本发明提供了一种烯烃聚合的方法,包括:
[0087]
所述烯烃聚合过程中采用的催化剂包括:上述技术方案所述的非对称芳基桥连茂金属化合物。
[0088]
在本发明中,所述烯烃聚合优选为乙烯和α-烯烃共聚,更优选为更优选为乙烯和1-辛烯共聚。
[0089]
在本发明中,所述非对称芳基桥连茂金属化合物优选作为主催化剂;所述烯烃聚合过程中采用的催化剂优选还包括:助催化剂。
[0090]
在本发明中,所述助催化剂优选包括:烷基铝类化合物和/或有机硼化合物。
[0091]
在本发明中,所述烷基铝类化合物优选选自烷基铝氧烷、改性烷基铝氧烷和烷基铝中的一种或几种;所述烷基优选为甲基;所述烷基铝化合物优选选自甲基铝氧烷和改性甲基铝氧烷中的一种或几种。
[0092]
在本发明中,所述助催化剂和主催化剂的摩尔比优选为(100~5000):1,更优选为(500~2000):1,最优选为(700~1000):1。
[0093]
在本发明中,所述烯烃聚合物过程中的压力(乙烯压力)优选为0.1~10mpa,更优选为0.1~5mpa,最优选为4mpa;所述烯烃聚合物过程中的温度优选为20~200℃,更优选为100~180℃,最优选为140℃。
[0094]
在本发明中,所述烯烃聚合方法优选包括:
[0095]
在乙烯氛围下,将聚合溶剂、共聚单体助催化剂溶液注入聚合装置中,设定聚合压力、聚合温度,通入氮气,通入主催化剂溶液进行聚合反应;聚合完成后加入终止剂终止聚合反应。
[0096]
在本发明中,所述乙烯氛围优选为常压乙烯氛围;所述注入优选采用导针进行注入;所述聚合溶剂优选为正己烷;所述聚合装置优选为高压釜;所述聚合压力优选通过乙烯压力进行控制,可通过乙烯流量计控制乙烯压力;所述聚合物温度可通过聚合装置进行设定并稳定温度。
[0097]
在本发明中,所述主催化剂溶液中的溶剂优选为甲苯;所述主催化剂溶液优选在手套箱中进行保存;所述通入氮气优选通过过渡仓持续通入氮气,优选将主催化剂溶液注入过渡仓,以高于聚合物压力的氮气将主催化剂溶液压入高压釜内;所述加入主催化剂溶液后优选将乙烯压力调至聚合压力并开始计时进行聚合反应时间;优选将终止剂预先加入到过渡仓中,待到聚合时间后,将终止剂压入高压釜终止聚合反应。
[0098]
在本发明中,所述烯烃聚合之前优选还包括:
[0099]
对所述烯烃单体进行精制。
[0100]
在本发明中,所述精制的方法优选包括:
[0101]
对烯烃单体和聚合溶剂分别依次进行干燥、蒸馏、收集。
[0102]
在本发明中,所述干燥优选在氮气的保护下进行;所述干燥优选采用金属钠进行干燥;所述干燥优选为过夜干燥,用于除水。
[0103]
在本发明中,所述蒸馏优选为常压蒸馏,去除前馏分。
[0104]
在本发明中,所述收集优选在氮气持续通入的条件下将蒸馏后所需的馏分收集至溶剂瓶氮气保存。在本发明中,所述溶剂瓶优选为烘干的溶剂瓶;所述溶剂瓶中优选设置有分子筛。
[0105]
在本发明中,所述烯烃聚合之前优选还包括:
[0106]
对聚合装置进行预处理。
[0107]
在本发明中,所述预处理的方法优选包括:
[0108]
依次进行清洗、干燥置换、稳定温度、去除氮气、持续通入乙烯。
[0109]
在本发明中,所述清洗优选为采用水和乙醇对聚合装置进行彻底清洗,以去除残余的物料和杂质;所述乙醇优选为无水乙醇。
[0110]
在本发明中,所述干燥置换优选为真空干燥并以氮气进行置换,彻底除去高压釜内的水氧;所述真空干燥的温度优选为120~160℃,更优选为130~150℃,最优选为140℃;所述真空干燥的时间优选为4~5小时,更优选为4.5小时。
[0111]
在本发明中,所述稳定温度的温度优选为聚合溶剂沸点以下的预定温度。
[0112]
在本发明中,所述去除氮气优选为真空除去氮气。
[0113]
在本发明中,所述持续通入乙烯优选为持续通入常压乙烯。
[0114]
在本发明中,所述烯烃聚合完成后优选还包括:
[0115]
降温、泄压、对得到的反应产物进行沉降、过滤、干燥。
[0116]
在本发明中,所述降温优选将温度降至聚合溶剂沸点;所述泄压优选为对高压釜泄压,然后打开聚合装置,将得到的共聚物倒出。在本发明中,所述沉降优选采用酸化乙醇沉降;所述酸化乙醇的浓度优选为5~15%,更优选为8~12%,最优选为10%。在本发明中,所述干燥优选在真空烘箱中进行;所述干燥的温度优选为50~70℃,更优选为55~65℃,最优选为60℃;所述干燥的时间优选为8~12小时,更优选为9~11小时,最优选为10小时。
[0117]
在本发明中,乙烯和1-辛烯共聚的方法优选包括:
[0118]
原料的精制:在氮气氛围保护下,利用金属钠分别对聚合溶剂正己烷和共聚单体1-辛烯进行过夜干燥(除水),之后进行常压蒸馏;去除前馏分(去除最佳),用烘干且装有适量分子筛(也可不装)的溶剂瓶进行收集所需馏分(务必保证氮气持续通入),氮气保护存放。
[0119]
聚合装置的预处理:利用水和无水乙醇对装置进行彻底清洗,去除残余的物料和杂质后,在140℃下进行真空干燥4~5小时并以氮气进行置换,彻底除去高压釜中的水氧;然后,将聚合装置设置并稳定到预定温度(正己烷沸点以下),真空除去高压釜中氮气,并持续通入常压乙烯,为共聚反应加料做好准备。
[0120]
共聚反应加料:在常压乙烯氛围下,利用导针依次将定量的聚合溶剂正己烷、共聚单体1-辛烯和助催化剂mao(甲基铝氧烷)溶液注入高压釜内;然后打开乙烯流量计,将乙烯
压力设到略低预定聚合压力,并把聚合装置设置并稳定到预定聚合温度;此时,需要到手套箱称取主催化剂并以甲苯配成溶液待用;待高压釜内温度稳定,保持过渡仓氮气持续通入,将配好的催化剂溶液注入过渡仓,然后以高于聚合压力的氮气将催化剂压入釜内,立刻将乙烯压力调到预定压力并开始计时;聚合结束前,预先将终止剂加到过渡仓,待到预定时间,立刻压入终止聚合反应。
[0121]
聚合后处理:在聚合结束后,将温度降至溶剂沸点左右开始对釜泄压,待到釜内压力泄完后,打开聚合装置,将共聚物倒入烧杯中并以适量的10%酸化乙醇进行沉降,过滤得到的聚合物于真空烘箱60℃下干燥8~12h。
[0122]
本发明以新型非对称芳基桥连茂金属配合物作为主催化剂,辅以烷基铝和有机硼化物或烷基铝氧烷(mao)作为助催化剂,可以高活性、高选择性催化烯烃聚合及α-烯烃共聚。与现有茂金属催化剂相比,本发明提到的新型茂金属配合物以大位阻苯环桥连,使得活性中心空间变大,从而进一步提高了催化剂的活性。此外,可以通过调节与氮原子相连芳基上的取代基的种类,调节金属中心的电荷密度与周围的空间位阻,从而实现对新型配合物催化性能的灵活调控。
[0123]
实验结果表明,本发明提供的茂金属化合物在催化乙烯/1-辛烯共聚时,共聚物中共聚单体1-辛烯插入率最高可达11.2%,相对分子质量最高可达27.3
×
104g/mol。
具体实施方式
[0124]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0125]
本发明以下实施例中所用原料均为市售商品。
[0126]
实施例1
[0127]
配体l1的制备:将3,3
’‑
二甲基联苯胺(0.98g,5mmol)加至10ml的乙醚溶液中溶解后,在低温槽内降温至-20℃下冷却并恒温5分钟;之后,缓慢向混合溶液中滴加(2.2ml,5.5mmol)正丁基锂的己烷溶液(2.5m),低温反应0.5小时后,将反应体系缓慢升至室温并室温下反应5小时;反应结束后,将反应温度降至-78℃后,向反应体系通入co2(g);低温反应0.5小时后,缓慢升至室温并除去剩余的co2(g),室温下反应过夜;
[0128]
然后,将反应产物降温至-20℃后,依次缓慢向反应液中加入(0.53ml,6.5mmol)四氢呋喃和(4.06ml,6.5mmol)叔丁基锂的戊烷溶液(1.6m)之后,低温反应0.5小时后继续室温下反应2小时;之后,降温至-20℃后,将1-茚酮(0.36g,2.75mmol)的四氢呋喃(5ml)溶液缓慢滴加至反应体系中,低温反应0.5小时后,缓慢升至室温并反应过夜;
[0129]
反应结束后,依次向反应液中加入水(2ml)、盐酸(6n,20ml),反应0.5小时后用二氯甲烷萃取,并分别以三乙醇胺、碳酸氢钠水溶液进行中和,收集有机相并以无水硫酸钠干燥除水,过滤并收集滤液;最后,滤液经过旋蒸除去溶剂后,利用过柱子(石油醚:乙酸乙酯=50:1)得到式l1结构配体(产量0.67g,产率=43%)。
[0130]
对本发明实施例1制备的产物进行核磁共振检测,检测结果为:1h nmr(400mhz,298k,cdcl3):δ10.48(s,lh),7.53~7.47(m,2h),7.39~7.34(m,3h),7.24~7.08(m,4h),
6.95(m,lh),6.82(m,lh),6.58(d,lh),6.39(d,lh),4.74(m,lh),2.33(s,3h),2.29(s,3h).anal.calcd.for c
23h21
n:c,88.71;h,6.80;n,4.50;found:c,88.32;h,6.34;n,4.35;本发明实施例1制备的配体具有式l1结构。
[0131]
实施例2
[0132]
配体l3的制备:将3,3
’‑
二甲基联苯胺(0.98g,5mmol)加至10ml的乙醚溶液中溶解后,在低温槽内降温至-20℃下冷却并恒温5分钟;之后,缓慢向混合溶液中滴加(2.2ml,5.5mmol)正丁基锂的己烷溶液(2.5m),低温反应0.5小时后,将反应体系缓慢升至室温并室温下反应5小时;反应结束后,将反应温度降至-78℃后,向反应体系通入co2(g);低温反应0.5小时后,缓慢升至室温并除去剩余的co2(g),室温下反应过夜;
[0133]
然后,将得到的反应产物降温至-20℃后,依次缓慢向反应液中加入(0.53ml,6.5mmol)四氢呋喃和(4.06ml,6.5mmol)叔丁基锂的戊烷溶液(1.6m)之后,低温反应0.5小时后继续室温下反应2小时;之后,降温至-20℃后,将5,6-二甲基-2,3-二氢-1h-茚-1-酮(0.44g,2.75mmol)的四氢呋喃(5ml)溶液缓慢滴加至反应体系中,低温反应0.5小时后,缓慢升至室温并反应过夜;
[0134]
反应结束后,依次向反应液中加入水(2ml)、盐酸(6n,20ml),反应0.5小时后用二氯甲烷萃取,并分别以三乙醇胺、碳酸氢钠水溶液进行中和,收集有机相并以无水硫酸钠干燥除水,过滤并收集滤液;最后,滤液经过旋蒸除去溶剂后,利用过柱子(石油醚:乙酸乙酯=50:1)得到式l3的配体(产量0.70g,产率=41%)。
[0135]
对本发明实施例2制备的产物进行核磁共振检测,检测结果为:1h nmr(400mhz,298k,cdcl3):δ10.48(s,lh),7.53~7.47(m,2h),7.25(m,2h),7.15~7.08(m,2h),6.95(m,lh),6.82(m,lh),6.58(d,lh),6.39(d,lh),4.74(m,lh),2.33(s,6h),2.29(s,6h).anal.calcd.for c
25h25
n:c,88.45;h,7.42;n,4.13;found:c,88.29;h,7.89;n,4.57;本发明实施例2制备的配体具有式l3结构。
[0136]
实施例3
[0137]
配体l5的制备:将3,3
’‑
二甲基联苯胺(0.98g,5mmol)加至10ml的乙醚溶液中溶解后,在低温槽内降温至-20℃下冷却并恒温5分钟;之后,缓慢向混合溶液中滴加(2.2ml,5.5mmol)正丁基锂的己烷溶液(2.5m),低温反应0.5小时后,将反应体系缓慢升至室温并室温下反应5小时;反应结束后,将反应温度降至-78℃后,向反应体系通入co2(g);低温反应0.5小时后,缓慢升至室温并除去剩余的co2(g),室温下反应过夜;
[0138]
然后,将上述得到的反应产物降温至-20℃后,依次缓慢向反应液中加入(0.53ml,6.5mmol)四氢呋喃和(4.06ml,6.5mmol)叔丁基锂的戊烷溶液(1.6m)之后,低温反应0.5小时后继续室温下反应2小时;之后,降温至-20℃后,将3,5,6,7-四氢-s-吲哚乙酸-1(2h)-酮(0.47g,2.75mmol)的四氢呋喃(5ml)溶液缓慢滴加至反应体系中,低温反应0.5小时后,缓慢升至室温并反应过夜;
[0139]
反应结束后,依次向反应液中加入水(2ml)、盐酸(6n,20ml),反应0.5小时后用二氯甲烷萃取,并分别以三乙醇胺、碳酸氢钠水溶液进行中和,收集有机相并以无水硫酸钠干燥除水,过滤并收集滤液;最后,滤液经过旋蒸除去溶剂后,利用过柱子(石油醚:乙酸乙酯=50:1)得到式l5结构的配体(产量0.63g,产率=36%)。
[0140]
对本发明实施例3制备的产物进行核磁共振检测,检测结果为:1h nmr(400mhz,
298k,cdcl3):δ10.48(s,lh),7.53~7.47(m,2h),7.32(s,1h),7.15~7.08(m,2h),6.95(m,lh),6.82(m,lh),6.58(d,lh),6.39(d,lh),4.74(m,lh),2.85(s,4h),2.33(s,6h),2.29(s,6h),2.07(m,2h).anal.calcd.for c
26h25
n:c,88.85;h,7.17;n,3.99;found:c,88.26;h,7.45;n,3.85;本发明实施例3制备的配体具有式l5结构。
[0141]
按照与实施例1~实施例3相类似的方法,采用不同的原料获得式l1~式l10结构的配体。
[0142]
实施例4
[0143]
茂金属配合物c1的制备:在-20℃下,将正丁基锂(0.16ml,0.41mmol)的己烷溶液(2.5m)缓慢滴加到配体l1(62mg,0.2mmol)中低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;
[0144]
之后,将反应体系降至-25℃后,缓慢向其加入四氯化钛(19mg,0.1mmol)的己烷溶液,低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;
[0145]
反应结束后,利用滤针进行过滤,收集滤液并真空下浓缩溶液至5ml,沿壁缓慢加入20ml超干己烷,低温下缓慢重结晶,最后得到暗红色块状晶体的式c1结构的过渡金属化合物(获得量=69mg,产率=81%)。
[0146]
对本发明实施例4制备的产物进行核磁共振检测,检测结果如下:1h nmr(400mhz,298k,c6d6):δ7.39~7.34(m,3h),7.25(m,1h),7.18~7.08(m,5h),6.95(m,lh),6.79(m,lh),6.58(d,lh),6.39(d,1h),2.29(s,3h),2.27(s,3h).anal.calcd.for c
23h19
cl2nti:c,64.52;h,4.47;n,3.27;found:c,64.12;h,4.72;n,3.52;本发明实施例4制备的产物具有式c1结构。
[0147]
实施例5
[0148]
茂金属配合物c3的制备:在-20℃下,将正丁基锂(0.16ml,0.41mmol)的己烷溶液(2.5m)缓慢滴加到配体l3(68mg,0.2mmol)中低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;
[0149]
之后,将反应体系降至-25℃后,缓慢向其加入四氯化钛(18.9mg,0.1mmol)的己烷溶液,低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;
[0150]
反应结束后,利用滤针进行过滤,收集滤液并真空下浓缩溶液至5ml,沿壁缓慢加入20ml超干己烷,低温下缓慢重结晶,最后得到暗红色块状晶体的式c3结构过渡金属化合物(获得量=71mg,产率=78%)。
[0151]
对实施例5制备的产物进行核磁共振检测,检测结果为:1hnmr(400mhz,298k,c6d6):δ7.25(m,2h),7.15~7.08(m,5h),6.95(m,lh),6.79(m,lh),6.58(d,lh),6.39(d,1h),2.31(s,3h),2.29(s,6h),2.27(s,3h).anal.calcd.for c
25h23
cl2nti:c,65.82;h,5.08;n,3.07;found:c,65.37;h,5.87;n,3.12;本发明实施例5制备的产物具有式c3结构。
[0152]
实施例6
[0153]
茂金属配合物c11的制备:在-20℃下,将正丁基锂(0.16ml,0.41mmol)的己烷溶液(2.5m)缓慢滴加到配体l10(70mg,0.2mmol)中低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;
[0154]
之后,将反应体系降至-25℃后,缓慢向其加入四氯化钛(18.9mg,0.1mmol)的己烷溶液,低温反应0.5小时后,将反应液缓慢升至室温并于室温下反应过夜;然后,将反应液再
次降温至-20℃,并向其中缓慢加入memgbr(0.18ml,3.0m,2.05当量);
[0155]
反应结束后,利用滤针进行过滤,收集滤液并真空下浓缩溶液至5ml,沿壁缓慢加入20ml超干己烷,低温下缓慢重结晶,最后得到暗红色块状晶体的式c11结构过渡金属化合物(获得量=91mg,产率=84%)。
[0156]
对本发明实施例6制备的产物进行核磁共振检测,检测结果为:1h nmr(400mhz,298k,c6d6):δ7.36(m,4h),7.25~7.08(m,6h),6.95~6.94(m,2h),6.79(m,lh),6.58(d,lh),6.79(m,1h),2.85(m,8h),2.29(s,3h),2.27(s,3h),2.07(m,4h),1.02(s,6h).anal.calcd.for c
37h37
nti:c,81.76;h,6.86;n,2.58;found:c,81.39;h,6.93;n,2.45;本发明实施例6制备的产物具有式c11结构。
[0157]
按照与实施例4~实施例6相类似的方法,采用不同结构的配体获得式c1~式c11结构化合物。
[0158]
实施例7
[0159]
茂金属配合物c1催化乙烯/1-辛烯共聚:在常压乙烯氛围下,利用双针依次将定量的聚合溶剂正己烷(0.5l)、共聚单体1-辛烯(105ml)和助催化剂甲基铝氧烷(mao)的甲苯溶液(1.0ml,1.5m),注入1.0l的高压釜内;然后打开乙烯流量计,将乙烯压力设到略低预定聚合压力4.0mpa,并把聚合装置设置并稳定到预定聚合温度140℃;
[0160]
之后,在手套箱称取主催化剂式c1结构化合物(1.5μmol)并以甲苯配成溶液待用;待高压釜内温度稳定,保持过渡仓氮气持续通入,将配好的主催化剂溶液注入过渡仓,然后以高于聚合压力的氮气将催化剂压入釜内,并立刻将乙烯压力调到预定压力并开始计时;聚合结束前,预先将终止剂加到过渡仓,待到预定聚合时间10分钟后,立刻终止聚合反应;
[0161]
聚合结束后,将温度降至溶剂沸点左右开始对釜泄压,待到釜内压力泄完后,打开聚合装置,将共聚物倒入烧杯中并以适量的10%酸化乙醇进行沉降,将过滤得到的聚合物于真空烘箱60℃下干燥8h。
[0162]
对本发明实施例7制备的聚合物进行核磁1h-nmr及gpc测定,检测结果为,共聚过程中的催化活性为0.62
×
108g/(mol
·
h),共聚物中1-辛烯插入率达9.2%,相对分子质量达21.3
×
104g/mol。
[0163]
实施例8
[0164]
茂金属配合物c2催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c2结构化合物代替式c1结构化合物。
[0165]
实施例9
[0166]
茂金属配合物c3催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c3结构化合物代替式c1结构化合物。
[0167]
实施例10
[0168]
茂金属配合物c4催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c4结构化合物代替式c1结构化合物。
[0169]
实施例11
[0170]
茂金属配合物c5催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c5结构化合物代替式c1结构化合物。
[0171]
实施例12
[0172]
茂金属配合物c6催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c6结构化合物代替式c1结构化合物。
[0173]
实施例13
[0174]
茂金属配合物c7催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c7结构化合物代替式c1结构化合物。
[0175]
实施例14
[0176]
茂金属配合物c8催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c8结构化合物代替式c1结构化合物。
[0177]
实施例15
[0178]
茂金属配合物c9催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c9结构化合物代替式c1结构化合物。
[0179]
实施例16
[0180]
茂金属配合物c10催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c10结构化合物代替式c1结构化合物。
[0181]
实施例17
[0182]
茂金属配合物c11催化乙烯/1-辛烯共聚:按照实施例7的聚合方法进行共聚,与实施例7的区别在于,采用式c11结构化合物代替式c1结构化合物。
[0183]
按照实施例7的方法对乙烯/1-辛烯共聚的性能检测检测,检测结果如下:
[0184][0185]
本发明以新型非对称芳基桥连茂金属配合物作为主催化剂,辅以烷基铝和有机硼化物或烷基铝氧烷(mao)作为助催化剂,可以高活性、高选择性催化烯烃聚合及α-烯烃共聚。与现有茂金属催化剂相比,本发明提到的新型茂金属配合物以大位阻苯环桥连,使得活性中心空间变大,从而进一步提高了催化剂的活性。此外,可以通过调节与氮原子相连芳基上的取代基的种类,调节金属中心的电荷密度与周围的空间位阻,从而实现对新型配合物催化性能的灵活调控。
[0186]
虽然已参考本发明的特定实施例描述并说明本发明,但是这些描述和说明并不限制本发明。所属领域的技术人员可清晰地理解,在不脱离如由所附权利要求书定义的本发明的真实精神和范围的情况下,可进行各种改变,以使特定情形、材料、物质组成、物质、方法或过程适宜于本技术的目标、精神和范围。所有此类修改都意图在此所附权利要求书的
范围内。虽然已参考按特定次序执行的特定操作描述本文中所公开的方法,但应理解,可在不脱离本发明的教示的情况下组合、细分或重新排序这些操作以形成等效方法。因此,除非本文中特别指示,否则操作的次序和分组并非本技术的限制。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。