1.本发明涉及人类肠道微生物群及其代谢能力。特别是一种证明健康人类肠道微生物群发酵能力方面的功能异质性的方法。更具体地,本发明提供了一种用于基于对受试者肠道微生物群的发酵或代谢能力的分析和测量来预测对不同膳食纤维的反应的计算机模拟(in silico)或体外(in vitro)方法,以及用于预测受试者对不同膳食纤维的反应的计算机软件产品和装置。
背景技术:
2.宿主和肠道菌群之间的共生关系与宿主饮食密切相关。例如,反刍动物摄入的大部分热量来自微生物对其饮食中原本难以消化的多糖的发酵。在人类中,对膳食纤维和其他微生物群可利用碳水化合物(mac)的发酵仅占总热量摄入的一小部分,但由此产生的代谢物还发挥着其他重要的生理作用(davie,2003;kuo,2013)。特别是短链脂肪酸(scfa)乙酸、丙酸和丁酸(肠道中微生物发酵膳食纤维的主要副产品)对宿主的生理机能产生了多种影响。虽然人们早就知道丁酸是结肠细胞的主要能量来源(roediger,1980),因此对于维持健康的肠道至关重要,但最近的研究表明,微生物产生和周转乙酸的增加已被证明可通过肠-脑轴介导的过程激活葡萄糖刺激的胰岛素分泌,这可能导致胰岛素抵抗和随后的肥胖,此外还会增加患者罹患2型糖尿病的风险(perry et al.,2016)。与此相反,丙酸和丁酸会激活糖异生,这对宿主代谢和血浆葡萄糖的调节具有有益作用(de vadder et al.,2014;zhao et al.,2018)。事实上,增加的丁酸的产生与口服葡萄糖耐量试验后改善的胰岛素反应有因果关系,而异常的丙酸的产生与增加的2型糖尿病风险有关(sanna et al.,2019)。
3.虽然这些作用中的许多作用是由肠上皮细胞中的短链脂肪酸受体介导的,但结肠scfa的一个关键特性是它们作为组蛋白去乙酰化酶(hdac)抑制剂的活性(davie,2003)。这种在宿主细胞中调节基因表达的能力使微生物衍生的scfa与越来越多的临床适应症相关联。特别是丁酸,已提出其通过多种机制对宿主的免疫系统施加抗炎压力,所述多种机制包括调节性t细胞和产生il-10的t细胞的分化、il-6的产生的下调、促炎性t细胞的凋亡和对结肠上皮中ifn-γ介导的炎症的抑制(zimmerman et al.,2012)。这些数据补充了在产生丁酸的生物体例如普拉梭菌(faecalibacteriumprausnitzii)和炎症性肠病(ibd)中发现的关联(machiels et al.,2013),表明scfa可能在疾病病因学中发挥重要作用。此外,结肠微生物群产生的scfa可以被吸收到血流中的这一事实表明,它们对基因表达的影响可能超越肠道,并以目前知之甚少的方式影响远端组织。因此,提高我们对肠道中scfa产生的定量理解具有重要的临床意义,以便可以针对特定的临床结果对其进行调节。
4.scfa是多糖通过在肠道宏基因组中的多种生化途径的发酵的化学最终结果。首先是这些复杂的膳食多糖被编码合适的多糖利用基因座(pul)和糖苷水解酶(gh)的微生物群成员水解成它们的组成单糖,所述多糖利用基因座(pul)和糖苷水解酶(gh)在不同的细菌菌种之间、甚至在同一菌种内的不同菌株之间存在很大差异(sonnenburg et al.,2010)。然后这些单糖可以通过多种发酵途径进行发酵,最终产生乙酸、丙酸或丁酸。重要的是,在
发酵途径之间存在交互作用(cross-talk):例如,人类肠道中最普遍的丁酸生成途径涉及酶丁酰辅酶a:乙酸辅酶a转移酶(duncan et al.,2002),它将丁酸部分交换为乙酸部分并释放游离的丁酸。因此,可用的乙酸池会影响丙酸和丁酸的产生。细菌发酵的其他产物也可以作为scfa产生中的中间体:特别是乳酸,其可以作为进一步发酵成乙酸、丙酸和丁酸的底物。此外,产生scfa的生物本身可能无法直接发酵具体的多糖;相反,他们依赖于其他编码这些碳水化合物活性酶的细菌将这些膳食纤维初步降解为己糖和戊糖。这产生了许多一起发挥作用的协作微生物网络,以产生存在于个体结肠中的整体scfa谱。因此,膳食输入的组合(膳食纤维的种类、数量)和个体的微生物群组成共同决定了他们肠道中产生的scfa的比例和绝对数量,这对生理机能具有潜在的重要影响。由于scfa参与多种不同的生理和疾病相关机制,因此它是独特且有发展前途的靶点。
5.存在着许多本质上极其广泛的公开,涉及通过粪便微生物群测序诊断具体疾病适应症的特征,或涉及用于治疗具体病症的益生菌制剂或细菌联合体。例如,已经公开了用于治疗疾病的具体聚糖聚合物。在wo2018106845 a1(kaleido biosciences inc[us])中,公开了聚糖聚合物的组合物及其制备和制造方法。还提供了使用聚糖聚合物制剂治疗疾病或障碍的方法。
[0006]
用于对微生物群进行测序的现有的粪便采样试剂盒不提供对与人类健康相关的实验可测量且经验证的数量的功能预测,但wo 2015/166489 a2(yeda res&dev[il])除外,它提供了一种预测受试者对食物的反应的方法,特别是通过血糖反应来预测受试者对食物的反应的方法。所述方法包括:选择受试者对其反应未知的食物;访问具有描述所述受试者但没有描述所述受试者对所选食物的反应的数据的第一数据库;访问具有与其他受试者对食物的反应有关的数据的第二数据库,所述其他受试者的反应包括至少一个其他受试者对所选食物或与所述所选食物类似的食物的反应;基于所选食物分析数据库,以估计所述受试者对所选食物的反应。然而,该文献只专注于餐后葡萄糖反应(ppgr),并使用了机器学习以从宏基因组dna测序预测对不同食物的ppgr。此外,这项技术需要血液样本,这无法寄出。
[0007]
wo 2019/046372 a1(univ michigan regents[us])公开了用于增加受试者中丁酸的产生的组合物和方法。具体地,该文献提供了促进受试者中丁酸的产生的组合物、益生菌组合物及其组合。还公开了一种增加受试者肠道中丁酸水平的方法,包括:施用碳水化合物源以及至少一种选自由以下细菌组成的组的第一细菌,所述细菌所属分类单元被鉴定为双歧杆菌属(bifidobacterium spp.),梭菌属(clostridium)seq176、序列100,以及布氏瘤胃球菌(ruminococcus bromii)。该文献描述了促进受试者中丁酸产生的制剂,而不考虑其微生物群组成如何。特别地,它公开了将碳水化合物源和细菌一起施用以达到期望的效果。
[0008]
tingting chen et al:"fiber-utilizing capacity varies in prevotella-versus bacteroides-dominated gut microbiota",scientific reports,vol.7,no.1,1june 2017(2017-06-01)公开了个体的肠道微生物群主要为不同的纤维利用细菌,这些细菌将膳食纤维发酵成已知对人类健康很重要的短链脂肪酸(scfa)。结果表明,粪便接种物中主要为普氏菌属(prevotella)与拟杆菌属(bacteroides),其被鉴定为两种不同肠型,对来自于不同化学结构的纤维的scfa的体外发酵谱有不同的影响。在普氏菌属肠型微生物组中,低聚果糖以及高粱阿拉伯木聚糖和玉米阿拉伯木聚糖显著促进了一种单一的普氏菌属otu,并且具有同样高的以丙酸为主要产物的总scfa产生。与此相反,在以拟杆菌属为主的
微生物群中,这三种纤维富集了不同的otu,导致不同水平和比例的scfa。该文献显示了在两种肠型中的各个差异如何导致对膳食纤维的明显不同的反应。由不同的纤维利用细菌主导的微生物群可能通过从相同的碳水化合物底物产生不同数量和谱的scfa来影响宿主健康。该文献重点介绍了在该领域常用的二分法,即普氏菌属与拟杆菌属主导的微生物群组成,并报告了根据这些所谓的“肠型”进行的分析,其中从每种类型中选择一个供体进行发酵实验。虽然作者的数据表明两个供体的发酵谱存在差异,但这些数据本身并不能证明微生物群组成可以从具体纤维预测scfa产生。相反,他们比较了两种供体微生物群在被相同的纤维挑战时的应答otu和scfa产生,并将这些差异归因于以普氏菌属为主的一种微生物群和以拟杆菌属为主的另一种微生物群。人们不能从这些数据中得出如下结论:即对微生物群组成的了解可以预测对具体纤维的代谢反应,因为在这两个类别中的每一个中都只有n=1个实例,而且并没有分析、统计或其他方面可以表明,采用新样本的微生物群组成作为输入的模型可以返回有关这些样本中的scfa产生的预测,如果它们受到不同纤维的挑战。
[0009]
us 2018/357375 a1(cutcliffe colleen[us]et al)提供了在拟进行定制化微生物疗法的个体中确定微生物组代谢途径的代谢图谱、鉴定微生物组代谢途径的存在和评估其丰度的方法。在一个方面,该公开提供了一种从包括多种不同生物体的群体的样本中确定代谢途径丰度的方法。
[0010]
该方法可用于评估微生物群中包括scfa产生途径在内的给定代谢途径的价值。这些数据可用于预测微生物群的scfa产生能力的潜力,因为该途径的丰度越高,通过该途径的最大代谢通量(metabolic flux)就越高。然而,没有提及从给定的输入评估给定代谢物的产生速率,也没有考虑到这一事实,即具有使用上述方法计算出的丰度的具体途径可以响应于不同输入(膳食或其他)而产生随上游过程不同而不同的代谢通量,在scfa产生的情况下,该具体途径取决于能够将所关注的具体纤维降解成作为具体scfa产生途径的输入的化学实体的上游糖苷水解酶或多糖利用基因座。
[0011]
riley l hughes et al:"the role of the gut microbiome in predicting response to diet 20and the development of precision nutrition models",advances in nutrition,vol.10,no.6,21june 2019(2019-06-21),pages 953-978报道了医疗保健越来越关注个人层面的健康。在快速发展的精准营养领域,研究人员的目标在于鉴定遗传学、表观遗传学和微生物组是如何相互作用以塑造个体对饮食的反应。有了这种了解,就可以预测个性化的反应,并可以为个人量身定制饮食建议。通过对这些复杂数据源的整合,精准营养研究的一个重要方面是用于研究响应于饮食的个体间变异性的方法学。这篇文章研究了肠道微生物群对响应于饮食的个体间变异性的贡献。并对研究人员用于设计和开展此类研究的方法以及用于分析结果的统计和生物信息学方法进行了评述。本文还评述了这些研究的结果,讨论了当前认知的差距,并总结了用于未来的研究的方向。总之,此综述总结了当前的认知状况,并为未来研究肠道微生物组在精准营养中的作用奠定了基础。该文献引用了几篇文章,这些文章描述了与对纤维干预的反应相关的普氏菌属与拟杆菌属(p:b)的比率,以及具体细菌进化枝(例如双歧杆菌(bifidobacteria))的丰度。然而,这种“对纤维的反应”是指下述的对纤维的反应,即作为响应于膳食纤维消耗的总scfa输出(即响应于纤维的scfa的总产生量),而不是指对各纤维的反应与各代谢物/scfa之间的具体关联。响应于具体纤维结构的具体scfa/代谢物的产生速率与源自基因和代谢途径组合
的具体宏基因组序列的相对丰度之间没有联系。
[0012]
仍然没有用于测量或预测响应不同膳食化合物的短链脂肪酸(scfa)产生(以及诸如寡糖、单糖、乳酸、甲酸和琥珀酸的中间发酵代谢物)的诊断可用解决方案。
[0013]
本发明的一个目的是为了更好地了解健康人群的微生物群中的不同发酵能力的程度,并提出用于测量响应于以具体膳食纤维挑战的个体粪便微生物群的scfa产生的活体外(ex vivo)参照标准。进行这些活体外实验允许申请人能够准确地量化scfa的积累,并避免与体内scfa产生的测量(侵入性)或对来自通过时生粪(raw fecal)的scfa的测量(肠道上皮对scfa的吸收程度未知)相关的技术困难。
技术实现要素:
[0014]
人类肠道微生物群以其在不同个体之间的高度异质性组成而闻名。然而,关于其在将多糖发酵成短链脂肪酸(scfa)的能力方面的功能性差异的了解相对较少。通过对健康人类供体的活体外测量,申请人证明个体的微生物代谢表型(mmp)存在显著差异,反映了他们的微生物群组成的差异,并导致了从相同的输入产生不同数量和比例的scfa。申请人还证明,可以使用16s rrna测序从组成预测这些mmp的方方面面。从在体内使用相同膳食纤维进行的实验中,申请人证明了摄入的纤维团在通过时几乎完全被微生物群消耗。申请人利用这些活体外数据构建了体内scfa产生和吸收的模型,并认为scfa的吸收量的个体间差异与产生差异直接相关。总之,这些数据表明,基于个体mmp的个性化膳食纤维补充是治疗与scfa产生相关的疾病的一种有吸引力且具有成本效益的治疗策略。
[0015]
待解决的技术问题有几个层次。第一层是从存在于粪便样本中的核酸或代谢物预测个体微生物群代谢能力的能力,这为封套式诊断试剂盒提供了可能性,该封套式诊断试剂盒可以保持样本“稳定”长到足以使其可以从世界上的任何地方运送到测序或治疗设施的时间。例如,这是通过在从活粪便微生物群获得的实验测量数据库上训练机器学习算法来实现的。第二层是根据这些预测制定个性化的饮食建议,所述预测例如应该消耗哪些具体的膳食纤维以最大限度地产生给定的短链脂肪酸。第三层是将这些个性化的饮食建议应用于具体的临床适应症,例如在诸如炎症性肠病或阿尔茨海默病的患者中最大化丁酸产生。
[0016]
本发明的一个目的是提供一种用于从核酸序列预测对不同膳食纤维的反应的计算机模拟(in silico)方法,所述方法基于对通过响应于所述不同膳食纤维的短链脂肪酸(scfa)的产生速率来评估的受试者肠道微生物群的发酵或代谢能力的分析,所述计算机模拟方法应用于从先前收集的所述受试者的稳定化生物样本中获得的具体微生物群衍生的核酸的丰度数据,所述计算机模拟方法包括以下步骤:
[0017]-从所述稳定化生物样本中分离和提取所述具体微生物群衍生的核酸以及微生物群衍生的代谢物,其中所述具体微生物群衍生的核酸的具体序列从包含来自健康受试者或患有特定疾病的受试者的宏基因组测序数据的训练数据库中鉴定,并将其与体外测量的数据配对,所述体外测量的数据与来自不同膳食纤维的scfa和其他发酵代谢物的产生速率相关,所述scfa和其他发酵代谢物选自包括丙酸、丁酸、乙酸、乳酸、甲酸、琥珀酸、异丁酸、戊酸和异戊酸的列表;
[0018]-确定和量化来自所述稳定化生物样本的所述具体微生物群衍生的核酸的相对丰
度以生成第一输入,其中所述具体微生物群衍生的核酸由以下基因序列组成:特异于能够水解或切割所述不同膳食纤维的复合多糖的糖苷水解酶和/或多糖水解酶的基因序列,以及特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶的基因序列;
[0019]-应用机器学习算法以从这些具体微生物群衍生的核酸序列预测响应于以不同膳食纤维挑战的scfa和其他发酵代谢物的产生速率,从而获得对所述不同膳食纤维的反应。
[0020]
本发明的另一个目的是提供一种计算机软件产品,其包括存储有程序指令的计算机可读介质,在被数据处理器读取时,所述程序指令使所述数据处理器接收从受试者样本中收集的数据,并执行根据本发明的计算机模拟方法。
[0021]
本发明的另一个目的是提供一种用于预测受试者对不同膳食纤维的反应的装置,所述装置包括:用户界面,其配置为接收受试者样本;模块,用于从所述受试者样本中提取微生物群衍生的核酸以及用于对所述核酸进行量化和/或测序;以及数据处理器,其具有存储本发明的计算机软件产品的计算机可读介质。
[0022]
对于本领域技术人员来说,通过参考以下说明性附图和所附权利要求进行的对随后的详细描述的仔细研究,本发明的其他目的和优点将而变得显而易见。
附图说明
[0023]
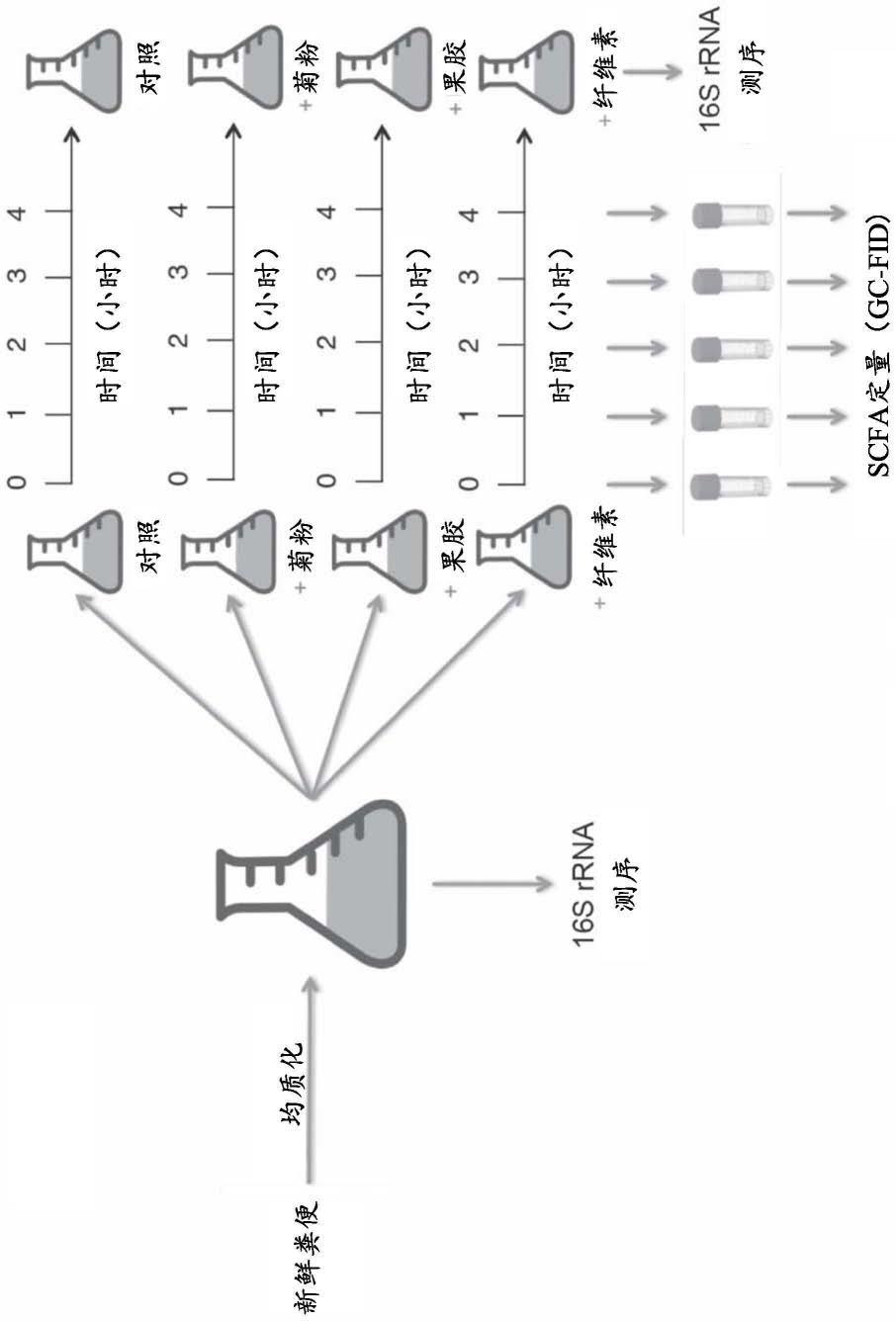
图1:分析设置和采样频率示意图。
[0024]
图2:两个不同参与者中响应于菊粉(i)、果胶(p)、纤维素(ce)和对照(ct)的随时间变化的丁酸浓度的24小时示踪。参与者c仅从菊粉产生丁酸,而参与者g还仅从果胶产生丁酸。在用于计算产生速率的0-4小时窗口中,两个参与者都具有线性产生规律。
[0025]
图3:不同参与者中的自菊粉(inul)和果胶(pect)的醋酸、丙酸和丁酸的产生速率,以在所有参与者中计算得出的z分数表示。每行代表来自单个样本的测量值。在活体外测量每个参与者响应于每个条件的scfa的产生速率(以mm/h为单位)。对每个条件计算在2h和4h的时间点之间的产生速率,并减去来自对照条件(没有添加物)的产生速率。纤维素时间点与对照是无法区分的,因此未呈现。
[0026]
图4:不同rfc的auc值,其经过训练可用于预测高或低的粪便基线scfa含量、或响应于具体膳食纤维的高或低的活体外scfa的产生速率。根据研究中所有参与者的z分数来定义高产生量和低产生量。
[0027]
图5:(a)丙酸和丁酸产生速率与未指定的毛螺菌科(lachnospiraceae)的otu的相对丰度之间的关系,显示了其相对丰度与特别响应于菊粉的丁酸产生之间的关系。(inul:菊粉;pect:果胶)。(b)类似的但针对普氏菌(prevotella copri)otu的相对丰度与响应于菊粉的丙酸产生的关系。
[0028]
图6:显示了个体mmp随时间推移是稳定的一般特征。(a)在相隔至少六个月的两个时间点内,响应于菊粉和果胶的每种scfa的连续产生速率,表示为相对于数据集中群体的z分数。(b)相同的数据,但被折叠(collapse)到响应于给定纤维的给定scfa的高或低的产生者(暗=高,亮=低)。
[0029]
图7:模型参数示意图。
[0030]
图8:来自先前的研究中的参与者粪便中的菊粉浓度,在该研究中在持续的(缺乏纤维的)饮食背景下,在第4天、第5天和第6天向参与者喂食10g菊粉。
[0031]
图9:参与者h中的预测丁酸吸收量与粪便中的预测丁酸排泄量作为结肠上皮吸收速率常数的函数,假设输送时间为12小时。已明确显示出了文中讨论的通过透析袋和caco单层方法测量的速率常数值。
[0032]
图10:使用每个受试者12小时的输送时间和透析袋速率常数参数预测的每种scfa的吸收量(以mmol为单位)。
[0033]
图11:在t=0、2h、4h、6h和24h时测量的六个不同的供体(供体id:k、s、z、ee、1和2)中的菊粉浓度,使用菊粉特异性elisa分析确定。
[0034]
图12:每个条件和每个时间点(0h、4h、24h)下16s rrna群落的shannon多样性指数。
[0035]
图13:显示了在研究中的所有参与者中测量的随时间推移的浆液的ph(ctrl:对照,inul:菊粉,pect:果胶,cell:纤维素)。
[0036]
图14:显示了样本的bristol评分(bss:bristol粪便量表)、产生的总scfa和qpcr扩增循环(ct值)之间的成对线性回归。
[0037]
图15:说明了微生物scfa产生是通过微生物群不同成员的合作进行的。(a)说明了膳食纤维降解所涉及的一般步骤的示意图。具有相对丰度xi的细菌otu i将多糖(膳食纤维)f水解成寡糖o。然后这些寡糖被具有相对丰度xj的otuj发酵成还原的中间体p。最后,p可以通过具有相对丰度xk的otu k进一步发酵成scfa。此外,otu进行给定反应的能力本身可以被另外的otu抑制(例如,xi被xo抑制)。(b)给定scfa的总产生速率的整体测量,φscfa(x),其本身是粪便微生物群组成x的函数,并且对应于使用申请人的活体外实验测量的量。
具体实施方式
[0038]
虽然在本发明的实施或测试中可以使用与本文描述的方法和材料相似或等同的方法和材料,但本文描述了合适的方法和材料。本文提及的所有公布、专利申请、专利和其他参考文献均通过引用整体并入本文。提供本文讨论的公布和申请仅仅是因为其公开早于本技术的申请日。本文中的任何内容均不应被解释为承认本发明无权因在先发明而早于此类公布。此外,材料、方法和示例仅是说明性的而不是限制性的。
[0039]
在矛盾的情况下,以本说明书(包括定义)为准。除非另有定义,否则本文使用的所有技术和科学术语具有与本文主题所属领域的技术人员通常理解的相同含义。如本文所用,提供以下定义以便于理解本发明。
[0040]
术语“包含”通常以包括的含义使用,即允许存在一个或多个特征或组件/分。
[0041]
如在说明书和权利要求书中所使用的,单数形式“a”、“an”和“所述”包括复数指代对象,除非上下文另有明确规定。
[0042]
如本文所用,术语“受试者”或“患者”在本领域中是公认的,并且在本文中可互换使用以指代哺乳动物,并且最优选地,指代人类。在一些实施方案中,所述受试者可以是正常受试者。该术语不表示具体的年龄或性别。因此,旨在涵盖成年和新生受试者,无论是男性/雄性还是女性/雌性。
[0043]
术语“膳食纤维”是本领域公认的,可与“复合多糖”或“复合碳水化合物”互换使用以指代碳水化合物。复合多糖或其他膳食纤维成分包括菊粉、低聚果糖、抗性淀粉、木质素、丹宁酸、纤维素、半纤维素(包括木葡聚糖、葡糖醛酸木聚糖、阿拉伯葡糖醛酸木聚糖、阿拉
伯木聚糖、heteroxylan、甘露聚糖和β-葡聚糖)、洋车前子、聚葡萄糖、几丁质、壳聚糖、果胶(包括同型半乳糖醛酸聚糖(homogalacturonan)/聚半乳糖醛酸聚糖(polygalacturonan)和鼠李糖半乳糖醛酸聚糖i和鼠李糖半乳糖醛酸聚糖ii)、阿拉伯聚糖和蒟蒻/魔芋(葡甘露聚糖)。
[0044]
术语“短链脂肪酸”在本领域中是公认的,并且在本文中可互换使用以指代甲酸、乙酸、丙酸、丁酸、戊酸、异丁酸和异戊酸。
[0045]
术语“中间发酵代谢物”是指将复合多糖和氨基酸发酵成短链脂肪酸的过程中的中间生化实体。这些包括但不限于寡糖、单糖(例如果糖、葡萄糖、半乳糖醛酸)、乳酸、甲酸和琥珀酸。
[0046]
术语“宏基因组测序”既指对从复杂生态系统(例如粪便)中分离的dna进行的鸟枪法测序,也指对来自生态系统的具体标志物基因进行的dna测序(例如16s rrna测序)。
[0047]
术语“宏转录组测序”是指对来自复杂生态系统的rna转录物进行的测序。
[0048]
术语“代谢组学”是指使用分析化学技术(例如质谱,其可以与色谱或纯化技术相结合,也可以不与色谱或纯化技术相结合)对来自样本的一种或多种分析物进行的测量。
[0049]
一般而言,术语“预防”、“控制”和“治疗”包括预防疾病或症状的发展,包括但不限于可能易感疾病或症状但尚未被诊断出患有该疾病或症状的受试者;抑制疾病的症状,即抑制或延缓其进展;以及缓解疾病症状,即使疾病或症状消退,或使症状进展逆转。
[0050]
根据本发明的一些实施方案,可以控制或治疗所有类型的肥胖症,包括但不限于体质型肥胖症、外因型肥胖症、高胰岛素性肥胖症、增生性肥大性肥胖症、肥大性肥胖症、甲状腺功能减退性肥胖症和病态肥胖症。例如,本发明实施方案可用于减缓、停止或逆转体重增加,特别是体脂肪增加,从而导致体重的维持或减少。体重或体脂的减少可通过降低血压、总胆固醇、低密度脂蛋白胆固醇和甘油三酯来预防心血管疾病,并可缓解与诸如高血压、冠心病、2型糖尿病、高脂血症、骨关节炎、睡眠呼吸暂停症和退行性关节病的慢性病症相关的症状。
[0051]
代谢综合征,或称x综合征,是一种复杂的多因素疾病,伴有各种异常,包括高血压、高甘油三酯血症、高血糖症、低hdl-c水平和腹部肥胖。具有这些特征的个体通常表现出血栓前期和促炎状态。现有数据表明,代谢综合征确实是一种综合征(一组危险因素)。
[0052]
每一种与代谢综合征相关的障碍本身都是危险因素,可以促进动脉粥样硬化、心血管疾病、中风、全身性微血管和大血管并发症以及其他不良健康后果。然而,当这些因素同时出现时,它们预示着增加的心血管疾病、中风和全身性微血管和大血管并发症的风险。
[0053]
在本发明的背景下,“涉及代谢和/或免疫和/或炎症的障碍和/或病理状况”是指人体能量代谢和/或免疫系统的正确功能和/或炎症状态的改变(局部的、在肠道水平、和/或全身性),这导致了临床表现。所述临床表现优选为:肥胖症、2型糖尿病、代谢综合征、非酒精性脂肪肝变性、胰岛素抵抗、高胆固醇血症、糖代谢的调控解除(deregulation)、心血管疾病、高血压、克罗恩病、溃疡性结肠炎、类风湿性关节炎、憩室病、肠易激综合征、变态反应、食物不耐受、腹泻、便秘、结肠炎和肠炎。
[0054]
如本文所用,术语“微生物组”是指在确定环境中的微生物(细菌、真菌、原生生物和病毒)、它们的遗传元件(基因组)的总体。换句话说,“微生物组”是指持续地或短暂地生活在受试者(例如,人类受试者)体内和体表上的微生物群落(“微生物群”)的遗传内容,包
括真核生物、古生菌、细菌和病毒(包括细菌病毒(例如噬菌体)),其中“遗传内容”包括基因组dna、诸如核糖体rna和信使rna的rna、表观基因组、质粒和所有其他类型的遗传信息。在一些实施方案中,微生物组具体指生态位中微生物群落的遗传内容。
[0055]
如本文所用,“微生物群”是指(持续地或短暂地)出现在受试者(例如人类受试者)体内或体表的微生物群落,包括真核生物、古生菌、细菌和病毒(包括细菌病毒,例如噬菌体)。在一些实施方案中,微生物群具体指生态位中的微生物群落。
[0056]
本发明的实施方案包括对可以依赖微生物特征(microbial signature)的作为微生物组组成和/或活性的代表的认识。微生物特征包括作为微生物组组成和/或活性指标的数据点。因此,根据本发明,可以通过检测微生物特征中的一种或多种特征来检测和/或分析微生物组的变化。
[0057]
如本文所用,术语“代谢组”是指个体中存在的代谢物的集合。人类代谢组包括天然小分子(天然可生物合成的非聚合化合物),它们是一般代谢反应的参与者,并且是细胞维持、生长和正常功能所必需的。通常,代谢物是小分子化合物,例如代谢途径的酶的底物、此类途径的中间体或通过代谢途径获得的产物。
[0058]
如本文所用,术语“微生物群衍生的代谢物”是指衍生自微生物群或存在于微生物群的生物体内的并因此在存在的情况下可以从受试者的样本中检测到的任何代谢物。作为一个复杂的生态系统,微生物群包含大量不同的生物体,每一种生物体都产生代谢物作为其细胞内代谢过程的一部分。
[0059]
根据本发明的一些实施方案的数据库的分析包括执行机器学习或统计建模过程。
[0060]
如本文所用,术语“机器学习”指的是体现为计算机程序的过程,所述计算机程序配置为从先前收集的数据中归纳出模式、规律或规则,以开发出对未来数据的适当反应,或以某种有意义的方式描述数据。
[0061]
当数据库包含多维条目时,机器学习的使用是特别有利的,但也不是完全有利的。
[0062]
组和受试者数据库可以用作训练集,机器学习过程可以从该训练集提取出最能描述该数据集的参数。一旦这些参数被提取出来,它们可用于预测对所选食物的反应。
[0063]
在机器学习中,可以通过监督学习或无监督学习来获取信息。在本发明的一些实施方案中,机器学习过程包括监督学习过程或者是监督学习过程。在监督学习中,使用全局或局部目标函数来优化学习系统的结构。换句话说,在监督学习中有一个期望的反应,系统使用该期望的反应来指导学习。
[0064]
在本发明的一些实施方案中,机器学习过程包括无监督学习过程或者是无监督学习过程。在无监督学习中,通常没有目标函数。特别是,该学习系统没有配置一套规则。根据本发明的一些实施方案的无监督学习的一种形式是无监督聚类,其中数据对象是没有先验分类标记的。
[0065]
适用于本发明实施方案的“机器学习”过程的代表性示例包括但不限于聚类、关联规则算法、特征评估算法、子集选择算法、支持向量机、分类规则、成本敏感分类器、投票算法、堆栈算法、贝叶斯网络、决策树、神经网络、基于实例的算法、线性建模算法、k-毗邻分析、集成学习算法、概率模型、图形模型、回归方法、梯度上升法、奇异值分解法和主成分分析。在神经网络模型中,自组织映射和自适应共振理论是常用的无监督学习算法。自适应共振理论模型允许集群的数量随着问题的大小而变化,并允许用户通过被称为警戒参数的用
户定义常数来控制相同集群的成员之间的相似程度。
[0066]
计算机模拟方法是意为关于生物实验的“在计算机上或通过计算机模拟执行”的表达。
[0067]
细菌群落中具体细菌的“相对丰度”,或等同地,宏基因组序列集合/群体中具体核酸序列的“相对丰度”,被定义为该具体细菌或核酸序列占总群体的比例。因此,它不是绝对丰度的度量,而是与群体中其他细菌/核酸序列相比的相对丰度。因此,它的取值介于0至100%,或介于以小数形式表示的0至1。
[0068]
本发明的一个目的是提供一种用于从核酸序列预测对不同膳食纤维的反应的计算机模拟或体外方法,所述方法基于对通过响应于所述不同膳食纤维的短链脂肪酸(scfa)的产生速率来评估的受试者肠道微生物群的发酵或代谢能力的分析,所述计算机模拟方法应用于从先前收集的所述受试者的稳定化生物样本获得的具体微生物群衍生的核酸的丰度数据,所述计算机模拟方法包括以下步骤:
[0069]-从所述稳定化生物样本中分离和提取所述具体微生物群衍生的核酸以及微生物群衍生的代谢物,其中所述具体微生物群衍生的核酸的具体序列从包含来自健康受试者或患有特定疾病的受试者的宏基因组测序数据的训练数据库中鉴定,并与体外测量的数据配对,所述体外测量的数据与来自不同膳食纤维的scfa和其他发酵代谢物的产生速率相关,所述scfa和其他发酵代谢物包括丙酸、丁酸、乙酸、乳酸、甲酸、琥珀酸、异丁酸、戊酸和异戊酸的列表;
[0070]-确定和量化来自所述稳定化生物样本的所述具体微生物群衍生的核酸的相对丰度以生成第一输入,其中所述具体微生物群衍生的核酸由以下基因序列组成:特异于能够水解或切割所述不同膳食纤维的复合多糖的糖苷水解酶和/或多糖水解酶的基因序列,以及特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶和/或负责降解所述不同膳食纤维的酶的基因序列;
[0071]-应用机器学习算法以从这些具体微生物群衍生的核酸序列预测响应于以不同膳食纤维挑战的scfa和其他发酵代谢物的产生速率,从而获得对所述不同膳食纤维的反应。
[0072]
与现有技术相反,在本发明中所述稳定化生物样本不打算使用膳食纤维进行处理。相反,本发明试图仅从核酸来预测个体的发酵能力(即他们从某些膳食纤维输入中产生某些代谢物的能力)。这因为在包含对粪便样本的分析的数据库上训练的机器学习算法而成为可能,所述粪便样本受到了膳食纤维的挑战,并且对于所述粪便样本,除了对它们的核酸进行测序外,还通过实验测量了其代谢物。对机器学习算法进行训练以从核酸序列预测这些实验性的纤维挑战结果,使得它们可以简单地应用于只需测序的并且因此可以在室温下在稳定缓冲液中保存更长时间(例如,以允许邮寄/运输)的其他新样本。
[0073]
因此,本发明涉及一种使用在包括与具体纤维的孵育在内的数据上训练过的算法从新样本中的核酸进行预测(并因此只能预测对这些具体纤维的反应)的计算机模拟方法,所述数据包括对上述代谢物的分析。
[0074]
顺便说一下,本发明与wo 2019/046372 a1(univ michigan regents[us])是显著不同的,因为使用膳食纤维促进丁酸产生需要对微生物群组成的了解,原因在于并非所有纤维都会被给定个体的微生物群很好地发酵成scfa,而其他的则可以。
[0075]
优选地,训练数据库包括根据预定集的分类组分类的数据,其中所述分析包括根
据具体类型或表型对受试者进行分类。
[0076]
根据本发明的一个实施方案,该软件还包括用于将与受试者的微生物组相关的数据与某种医疗状况、身体状况或对某种治疗的可能反应联系起来的算法集。
[0077]
特别地,本发明的计算机模拟或体外方法(即软件)还包括将受试者分层到不同治疗计划的步骤,所述不同治疗计划包括个性化饮食建议或旨在提高受试者微生物群发酵能力的其他治疗策略,从而建立受试者的微生物组谱。
[0078]
根据本发明的一个实施方案,所述个性化饮食建议是通过鉴定导致由受试者的微生物群最高地和最低地产生所关注代谢物的膳食纤维输入来制定的,从而为受试者建立个人护理产品。
[0079]
根据本发明的另一个实施方案,旨在改善受试者微生物群发酵能力的治疗策略包括一种或多种益生元、一种或多种益生菌、一种或多种抗生素或任何其他用于治疗性治疗涉及代谢性和/或免疫和/或炎症的疾病或障碍的药物。
[0080]
在本发明的背景下,不同的治疗计划包括对涉及代谢性和/或免疫和/或炎症的疾病或障碍的易感性,所述疾病或障碍选自包括以下的组:肥胖症,代谢综合征或疾病,诸如2型糖尿病的糖尿病,胰岛素缺乏相关障碍,胰岛素抵抗相关障碍,肠道糖异生障碍,炎症障碍,炎症性肠病,在神经退行性疾病、抑郁症或焦虑障碍背景下的全身性或局部炎症,食物不耐受,腹泻,便秘,结肠炎和肠炎,变态反应,类风湿性关节炎和癌症免疫疗法应用。
[0081]
优选地,所述受试者的生物样本选自包括直肠拭子、粪便样本、活检组织或粘膜层样本的组。更优选地,所述受试者的生物样本是粪便样本。所述样本也可以安全运输。
[0082]
有利的是,本发明的体外方法允许确定和量化不同细菌核酸的相对丰度,其通过对包括16s rrna在内的具体标志物基因的dna测序、鸟枪法宏基因组学dna测序、对具体基因的pcr或qpcr进行,或通过包括毛细管电泳在内的核酸定量方法进行。
[0083]
根据本发明的一个实施方案,所关注的具体代谢物包括由短链脂肪酸丙酸和丁酸组成的短链脂肪酸评估。然而,所关注的其他具体代谢物包括乙酸、乳酸、甲酸、琥珀酸、异丁酸、戊酸和异戊酸。
[0084]
根据另一个实施方案,负责降解所述不同膳食纤维的其他酶选自包括源自所述受试者肠道微生物群的细菌、古细菌或真菌生物体的组。根据个体的不同,它们的微生物群降解具体纤维的集体能力可能来自几种生物体的相互作用;例如,来自于微生物群中共生真菌的某种酶可以将纤维降解为其组成性寡糖或单糖(例如将果胶降解为半乳糖醛酸),该组成性寡糖或单糖随后被细菌生物体发酵为短链脂肪酸。
[0085]
优选地,所述机器学习算法包括监督学习过程。更优选地,所述机器学习算法包括选自由以下组成的组的至少一个过程:聚类、支持向量机、线性建模、k-毗邻分析、决策树学习、集成学习过程、神经网络、概率模型、图形模型、贝叶斯网络和关联规则学习。
[0086]
允许用户鉴定个体发酵能力的机器学习算法由几个层组成。第一层由各个分类器或回归器组成,所述分类器或回归器使用具体核酸序列的相对丰度作为输入来预测响应于具体膳食纤维的具体代谢物(例如scfa)的产生速率。这些各个分类器或回归器将会在上述数据库中进行训练。仅这一层的分类器或回归器就可以描述个体的发酵能力。然后可以将来自该层的结果输入到第二层,所述第二层将这些具体代谢物的结果结合起来以鉴定出可以产生所需的代谢物产生谱(例如,最大限度地提高丁酸的产生,同时最大限度地减少丙酸
的产生)的膳食纤维的最佳混合或组合。
[0087]
在本发明的背景下,不同的膳食纤维是选自包括以下的组的复合多糖或其他膳食成分:菊粉、低聚果糖、抗性淀粉、木质素、丹宁酸、纤维素、半纤维素(包括木葡聚糖、葡糖醛酸木聚糖、阿拉伯葡糖醛酸木聚糖、阿拉伯木聚糖、异木聚糖(heteroxylan)、甘露聚糖和β-葡聚糖)、洋车前子、聚葡萄糖、几丁质、壳聚糖、果胶(包括同型半乳糖醛酸聚糖/聚半乳糖醛酸聚糖和鼠李糖半乳糖醛酸聚糖i和鼠李糖半乳糖醛酸聚糖ii)、阿拉伯聚糖和蒟蒻/魔芋(葡甘露聚糖)。
[0088]
根据本发明的优选实施方案,使用擦拭物并用核酸稳定溶液(例如乙醇、rnalater等)处理擦拭物,将用于预测对不同膳食纤维的反应的计算机模拟或体外方法应用于受试者的样本粪便。一旦稳定下来,粪便中的核酸在室温下就是稳定的,并可以进行邮寄。样本也可以安全运输。
[0089]
黏膜层样本可在内窥镜检查过程中通过活检收集,或在门诊处作为新鲜粪便样本收集。
[0090]
从样本中提取微生物群衍生的核酸。
[0091]
然后,所述方法通过对具体标志物基因(例如16s rrna)的dna测序、鸟枪法宏基因组dna测序、对具体基因的pcr或qpcr(定量pcr)、或另一种核酸定量方法(例如毛细管电泳)中的任一种来确定和/或量化来自粪便或粘膜样本的不同核酸序列的相对丰度。
[0092]
dna测序(最初是16s rrna扩增子测序)必然包括形成针对关键特征(即特异于能够水解或切割所述不同膳食纤维的复合多糖的糖苷水解酶和/或多糖水解酶的基因序列、以及特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶的基因序列)的一组pcr引物。然而,也可以使用利用毛细管电泳对具体序列的相对丰度进行定量的方法以及技术人员已知的具有足够解析度的核酸定量的任何其他方法。
[0093]
下一步是将这些核酸定量数据或核酸测序数据输入到由预测微生物群发酵能力的统计模型或机器学习算法组成的软件中。更具体地说,所述软件预测微生物群从不同的饮食输入中产生具体代谢物的能力。关键代谢物是短链脂肪酸丙酸和丁酸。所述模型是从数据库训练/开发的。所关注的其他代谢物包括乙酸、乳酸、甲酸、琥珀酸、异丁酸、戊酸和异戊酸。
[0094]
特别的是,所述软件或算法将具体核酸序列的相对丰度作为输入;通过预测响应于一组具体膳食输入的每种相关代谢物的产生速率或产生能力的相对量度(例如,与抽样群体相比,高vs低,与抽样群体相比的z分数),对微生物群产生具体代谢物的能力进行评分。具体膳食输入包括各个类型的膳食纤维和复合多糖。这是使用作用于输入特征的统计或机器学习模型来实现的。此外,该模型可能会替代性返回单个变量,即具体的发酵“类型”(其中在人群中观察到的不同发酵能力的连续体被离散为具体类型/表型,例如1型、2型、3型等)。此外,该模型可以使用输入特征的加权的线性或非线性组合以及一个或多个统计分类器或回归器的分级组合根据相关特征来计算得分。
[0095]
上述收集的信息用于将用户/患者分层到不同治疗计划的方法中,所述治疗计划可能包括个性化饮食建议或其他旨在提高受试者微生物群发酵能力的治疗策略(例如使用益生元、益生菌、合生元(即益生元 益生菌的治疗)或粪便微生物群移植)。
[0096]
有利的是,所述方法允许对导致微生物群产生最高和最低的关注代谢物的饮食输
入进行鉴定,并允许使用这些结果来制定个性化饮食/营养建议。这可以以包含相关膳食输入的具体补充剂的形式,或作为旨在产生具体代谢物的不同膳食成分(如具体的水果、蔬菜、谷物等)的一般建议。后述的一般建议可以从包含存在于不同成分的每种具体膳食输入的量的查找表中计算出来。
[0097]
训练数据库的建设:
[0098]
收集粪便样本、粪便活检组织或黏膜活检组织。
[0099]
在厌氧条件下处理样本,包括将样本均质成浆液并将其稀释至保持微生物群代谢活性但允许进行所有必要的下游发酵实验的浓度。
[0100]
取样本的基线等分试样进行核酸定量。
[0101]
取基线等分试样用于代谢物的量化(如上所述)。
[0102]
于厌氧条件和37摄氏度(人体温度)中孵育样本的各等分试样。在存在具体膳食成分或添加物的情况下,于厌氧条件和37摄氏度(人体温度)中孵育等分试样。为了清楚起见,从这一时间点开始,每个不同的添加物(包括一个或多个没有添加物的对照)都被称为具体的“添加物条件”。以不同的时间间隔(例如每小时)对每个添加物条件进行采样。
[0103]
从每个关注的时间点量化关注的代谢物的存在/不存在或浓度。
[0104]
将这些数据组合成具体的产生速率或产生能力(例如,诸如能或不能产生代谢物的二元能力)。
[0105]
将初始样本的核酸定量数据(其构成关于微生物群组成的信息)以及样本微生物群响应于不同的添加物而产生不同代谢物的能力存储在数据库中。
[0106]
根据一个具体实施方案,本发明提供了一种用于预测对不同膳食纤维的反应的体外方法,所述方法基于对通过响应于所述不同膳食纤维的短链脂肪酸(scfa)的产生速率来评估的受试者肠道微生物群的发酵或代谢能力的分析和测量,所述方法应用于先前收集的所述受试者的稳定化生物样本中,所述方法包括以下步骤:
[0107]-从所述稳定化生物样本中分离和提取微生物群衍生的核酸以及微生物群衍生的代谢物;
[0108]-确定和量化来自所述稳定化生物样本的所述微生物群衍生的代谢物的相对丰度和所述不同的微生物群衍生的核酸的相对丰度以产生第一输入,其中所述不同的微生物群衍生的核酸由以下组成:特异于能够水解或切割所述不同膳食纤维的复合多糖的糖苷水解酶和/或多糖水解酶的基因序列,以及特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶的基因序列;
[0109]-将所述第一输入应用到具有与对来自其他健康受试者或患有具体疾病的受试者的微生物群的发酵能力的反应有关的数据的训练数据库中,并在由统计模型和/或机器学习算法组成的软件中分析所述生成的数据库,其中所述统计模型和/或机器学习算法配置为预测所述受试者的肠道微生物群从所述不同膳食纤维产生关注的具体代谢物的能力,其中所述关注的具体代谢物包括短链脂肪酸评估,并且其中所述机器学习算法从所述生成的数据库中提取参数以预测所述受试者对所述不同膳食纤维的反应。
[0110]
所提及的技术特征如上所述。
[0111]
根据又一个实施方案,本发明提供了一种用于预测对不同膳食纤维的反应的体外方法,所述方法基于对通过响应于所述不同膳食纤维的短链脂肪酸(scfa)的产生速率来评
估的受试者肠道微生物群的发酵或代谢能力的分析和测量,所述方法应用于先前收集的所述受试者的稳定化生物样本中,所述方法包括以下步骤:
[0112]-在存在所述不同膳食纤维的情况下培养所述稳定化生物样本,以检测所述受试者肠道微生物群的发酵或代谢能力;
[0113]-从所述稳定化生物样本中分离和提取微生物群衍生的核酸以及微生物群衍生的代谢物;
[0114]-确定和量化来自所述稳定化生物样本的所述微生物群衍生的代谢物的相对丰度和所述不同的微生物群衍生的核酸的相对丰度以产生第一输入,其中所述不同的微生物群衍生的核酸由以下组成:特异于能够水解或切割所述不同膳食纤维的复合多糖的糖苷水解酶和/或多糖水解酶的基因序列,以及特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶的基因序列;
[0115]-将所述第一输入应用到具有与对来自其他健康受试者或患有具体疾病的受试者的微生物群的发酵能力的反应有关的数据的训练数据库中,并在由统计模型和/或机器学习算法组成的软件中分析所述生成的数据库,其中所述统计模型和/或机器学习算法配置为预测所述受试者的肠道微生物群从所述不同膳食纤维中产生关注的具体代谢物的能力,其中所述关注的具体代谢物包括对选自包括丙酸、丁酸、乙酸、乳酸、甲酸、琥珀酸、异丁酸、戊酸和异戊酸的列表的短链脂肪酸的评估,并且其中所述机器学习算法从所述生成的数据库中提取参数以预测所述受试者对所述不同膳食纤维的反应。
[0116]
所提及的技术特征如上所述。
[0117]
本发明的另一个目的是提供一种计算机软件产品,其包括存储有程序指令的计算机可读介质,在被数据处理器读取时,所述程序指令使所述数据处理器接收从受试者样本中收集的数据,并执行根据本发明的体外方法。
[0118]
执行本实施方案的方法的计算机软件或程序通常可以在诸如但不限于cd-rom或闪存介质的分发介质上分发给用户。计算机程序可以从分发介质复制到硬盘或类似的中间存储介质。在本发明的一些实施方案中,执行本实施方案的方法的计算机程序可以通过允许用户经由通信网络例如因特网从远程位置下载程序以分发给用户。计算机程序的运行可以通过将计算机指令从它们的分发介质或它们的中间存储介质加载到计算机的执行存储器中,配置计算机使其根据本发明的方法进行操作。所有这些操作对于计算机系统领域的技术人员来说是众所周知的。
[0119]
本发明的另一个目的是提供一种用于预测受试者对不同膳食纤维的反应的装置,所述装置包括:用户界面,其配置为接收受试者样本;模块,用于从所述受试者样本中提取微生物群衍生的核酸以及用于对所述核酸进行量化和/或测序;以及数据处理器,其具有存储本发明的计算机软件产品的计算机可读介质。
[0120]
总之,所述装置:
[0121]
1)在所述用户界面接收受试者的样本,
[0122]
2)提取微生物群衍生的核酸并对其进行量化/测序;
[0123]
3)使用所述数据处理器运行所述软件。
[0124]
本发明的装置适用于多维分析过程,所述多维分析过程可选地并且优选地包括机器学习过程,例如上述过程中的一个或多个。所述分析的目的是为了确定数据中允许在数
据库中不同条目之间找到相似性的模式。例如,所述分析可以包括对数据库条目可以被分类到其中的分类组进行定义,以及根据分类组对数据库条目进行分类。随后,条目可以根据它们的分类进行标记。多维分析可以附加地或替代地包括决策树的构建。随后,可以更新数据库以包括构建的决策树。多维分析可以附加地或替代地包括在数据库中的特征之间的提取关联模式和/或关联规则。随后,可以更新数据库以包括关联模式和/或关联规则。多维分析可以附加地或替代地包括对数据库中的特征进行排序,例如,使用特征评估算法。随后,可以更新数据库以包括排序。多维分析可以附加地或替代地包括从数据库中的数据的至少一部分构建贝叶斯网络。随后,可以更新数据库以包括构建的贝叶斯网络。其他类型的分析也在考虑之中。
[0125]
术语“决策树”是指任何类型的基于树的学习算法,包括但不限于模型树、分类树和回归树。
[0126]
根据一个优选实施方案,本发明的装置还包括pcr试剂盒,用于对从训练数据库鉴定的所述具体微生物群衍生的核酸的相对丰度进行测序和量化,所述pcr试剂盒包括针对从训练数据库中鉴定的具有最佳纤维反应预测性的核酸序列的引物,并且所述pcr试剂盒还任选地包括针对特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶以及负责降解所述不同的膳食纤维的其他酶的基因和代谢途径的序列的引物。
[0127]
然后可以从公众可得的参考基因组中提炼出这些具体的核酸测序引物,以确保它们在不同个体之间保持特异性,所述个体可能含有由于菌株之间的微小突变而在这些具体序列中略有不同的细菌菌株。所述试剂盒允许专门对这些标志物进行测序,从而有效量化个体肠道微生物群中这些标志物的相对丰度,并提供机器学习算法所需的准确输入以做出准确预测。
[0128]
根据一个优选实施方案,所述pcr试剂盒还包括针对特异于来自细菌乙酸、丙酸和丁酸产生途径的关键酶以及负责降解具体纤维的酶的基因和代谢途径的序列的引物。负责降解具体纤维的酶可以是细菌的、古细菌的或真菌的酶,因此可以从肠道微生物群的任何部分提取。
[0129]
如上所述,某些真菌可以包含可以降解膳食纤维的酶,因此它们也有助于个体的发酵能力。例如,果胶酶有时不仅存在于细菌中,还存在于真菌中,其可特异性地将果胶降解为寡糖和单糖,这些寡糖和单糖可以进一步被细菌微生物群发酵。与之相反,降解果聚糖(如菊粉)的果聚糖酶通常存在于细菌中。
[0130]
如上所述,本发明旨在通过诸如粪便核酸测序(宏基因组dna或宏转录组rna)以及粪便代谢组学/代谢物谱的分析来确定个体肠道微生物群的发酵或代谢能力。它允许用户或患者或受试者量化其肠道微生物群发酵不同多糖或膳食化合物以产生具有生理或临床意义的具体代谢物的能力。此外,本发明允许用户或患者确定哪种(些)多糖或膳食化合物可以作为补充剂食用,以增加或减少他们的肠道微生物群产生具体代谢物。
[0131]
因此,本发明构成了诊断/预后和用于针对具体临床结果制定个性化饮食策略的参照标准。所述参照标准包括将诊断与具体的膳食纤维或化合物(以粉末形式,或整合到功能性食品中)配对。生理应用包括最大限度地减少炎症、增加肠道糖异生或最大限度地提高通过发酵过程产生的肠道微生物群衍生的代谢物(包括但不限于短链脂肪酸(scfa))可以赋予的任何其他健康益处。临床应用包括但不限于提高癌症免疫治疗的疗效,在炎症性肠
病或类似疾病的背景下减少肠道炎症,在神经变性、类风湿性关节炎、抑郁症或焦虑症的背景下减少全身性或局部炎症,在代谢疾病或肥胖症的背景下改善代谢健康,以及预防2型糖尿病。此外,可以形成一种装置或设备,其包括测序/核酸检测/分析化学能力、以及用于预测微生物群代谢能力和制定所得个性化饮食建议的软件。
[0132]
制定这些预测的软件由在由志愿者粪便微生物群的实验测量值组成的数据库上训练的统计和/或机器学习模型组成。更具体地说,对于每个志愿者的微生物群,所述数据库包含生粪的宏基因组dna测序数据和/或宏转录组rna测序数据和/或代谢组学数据,所述数据与从样本中的活微生物群厌氧获得的实验测量值相配对。这些实验测量值包括但不一定限于对响应于具体化学添加物随时间产生的scfa和中间发酵代谢物的时间解析测量值。
[0133]
这些化学添加物是指具体的膳食纤维、复合多糖或其他膳食成分,但也可以包括与上述化学物质组合的药物/药品。特别地,这些添加物包括一组膳食纤维,其涵盖包括(但不限于)以下的膳食纤维的主要类别:
[0134]-菊粉
[0135]-低聚果糖
[0136]-抗性淀粉
[0137]-木质素
[0138]-丹宁酸
[0139]-纤维素
[0140]-半纤维素(包括木葡聚糖、葡糖醛酸木聚糖、阿拉伯葡糖醛酸木聚糖、阿拉伯木聚糖、异木聚糖(heteroxylan)、甘露聚糖和β-葡聚糖)
[0141]-洋车前子
[0142]-聚葡萄糖
[0143]-几丁质
[0144]-壳聚糖
[0145]-果胶(包括同型半乳糖醛酸聚糖/聚半乳糖醛酸聚糖和鼠李糖半乳糖醛酸聚糖i和鼠李糖半乳糖醛酸聚糖ii)
[0146]-阿拉伯聚糖
[0147]-蒟蒻/魔芋(葡甘露聚糖)。
[0148]
该数据库还可以采用具体细菌菌株(和伴随的标志物基因序列)及其实验测量的代谢能力的形式。
[0149]
预测算法使用的关键特征包括:
[0150]-针对水解或切割具体复合多糖或膳食纤维的糖苷水解酶和多糖利用基因座
[0151]-丁酸产生途径的标志物基因,包括丁酰辅酶a(coa):乙酸coa转移酶、丁酸激酶和磷酸转丁酰酶(phosphotransbutyrylase)
[0152]-丙酸产生途径的标志物基因,包括甲基丙二酰辅酶a脱羧酶亚基α(mmda)(琥珀酸途径的标志物)、乳酰辅酶a脱水酶亚基α(lcda)(丙烯酸途径的标志物)、醛脱氢酶(pdup)。
[0153]-乙酸产生途径的标志物基因,包括乙酰辅酶a合酶和一氧化碳脱氢酶(wood-ljungdahl途径的标志物)。
[0154]
它们还可以包括已知含有上述酶的具体细菌菌株的具体核酸标志物。
[0155]
本发明的一个优点是仅使用具体的宏基因组或宏转录组测序特征或来自粪便的代谢组/代谢物特征来预测个体从不同膳食成分产生scfa的能力;具体而言,令人惊讶的是,本发明允许预测微生物群对食物的代谢反应及其对具体代谢物的产生,而不是预测个体或患者的反应(例如餐后葡萄糖反应)。
[0156]
有利的是,本发明允许使用来自粪便收集封套式试剂盒的核酸测序进行预测,其中粪便中的核酸是稳定化的。值得注意的是,用于量化个人的微生物群代谢能力的现有技术实验方案涉及活粪便微生物群的厌氧培养,这需要新鲜的粪便样本(在通过的数小时内进行厌氧处理),因此费力、成本高且在物流运输方面高度限制(特别是对于临床应用)。
[0157]
有趣的是,本发明还允许从膳食补充剂列表中选择具体补充剂,所述补充剂将根据个体微生物群最大化给定的scfa或代谢物的产生。
[0158]
此外,本发明还可应用于疾病治疗背景下的诊断工具或方法,无论是最大化患有炎症性肠病、神经变性、类风湿性关节炎、抑郁症或焦虑症的患者中的丁酸产生,最大化肥胖、糖尿病前期/糖尿病(2型)或代谢综合征患者中的丁酸和丙酸产生,或最大化接受癌症免疫疗法的患者中的丙酸产生。
[0159]
如本发明的实施例中所示,申请人已经使用活体外设置来测量响应于不同膳食纤维输入的肠道微生物的scfa产生的个体间差异。申请人已经表明,不同个体的mmp存在显著差异,即他们的微生物群将给定纤维底物发酵成给定scfa的能力存在显著差异。此外,申请人表明,mmp在一定程度上可以从粪便微生物群落组成中预测,这比从生粪scfa含量中预测的程度更大。
[0160]
此外,申请人表明,随着时间的推移,个体mmp的主要特征相对稳定,这与个体mmp与其微生物群落组成有关的事实是一致的。尽管申请人没有研究此类情况,但使用广谱抗生素或对微生物群进行其他极端扰乱的治疗可能会对个体的mmp产生重大影响,同时对其生理学产生影响。
[0161]
为了探索申请人的活体外结果与对体内scfa产生和吸收的影响之间的关系,申请人试图开发出该过程的现象学模型。申请人发现,scfa的吸收量反映了在所考虑的不同参数方案中产生的量,尽管绝对量随其模型中使用的上皮吸收速率常数的改变而发生了显著变化。这突出了该变量在了解粪便scfa的量与临床应用的体内产生和吸收之间的关系方面是至关重要的。因此,除了定量描述这些速率常数的行为和在不同浓度的scfa和肠道不同区域内的潜在吸收动力学之外,获得对这些速率常数的准确测量对于该领域也非常重要。
[0162]
申请人的模型还表明,在增加结肠转运时间会导致scfa的吸收量显著增加的同时这些scfa在肠道中仍保持在可观的浓度,或者仍有底物要发酵。在申请人的活体外实验以及图8中使用的体内菊粉补充实验中,给予微生物群以高度可利用的粉状菊粉作为底物。尽管在活体外和在体内,大部分菊粉分别在24小时内或在通过时被微生物群发酵,申请人的活体外测量表明,在所考虑的输入浓度(10g/l)下,该过程在约12小时-24小时时在大多数参与者中发生(图11)。因此,不太可利用的纤维输入形式(例如,对于菊粉,以生菊苣根的形式)可能需要更长的时间来发酵,在这种情况下结肠转运时间是决定scfa产生量的重要变量。此外,旨在提高scfa产生量的膳食纤维补充治疗策略将受益于这些纤维的高度可利用形式(纯化粉末),而不是在数量上高的这些谷物、水果和蔬菜,微生物群可能需要更长的时间去发酵这些谷物、水果和蔬菜,因此可能会产生大量被排泄的未发酵底物。
[0163]
申请人的模型表明,不同个体间的mmp差异与scfa吸收量之间的关联具有高度的稳健性,尽管吸收速率常数的改变对scfa的吸收量和排泄量都有显著影响。重要的是,根据吸收速率常数的实际大小,粪便scfa浓度与体内的产生和吸收之间的关系可能完全不能提供有用信息。粪便scfa浓度与产生和吸收之间的差异、结肠转运时间以及是否消耗了全部可发酵底物有关。例如,在产生速率显著超过吸收速率的情况下(如本研究中所考虑的两组吸收速率常数参数),粪便scfa随转运时间(τ)和可发酵底物耗尽时间(t1)之间的差异而异。如果τ》t1,则粪便scfa浓度将主要随其间粪便被转运但没有进一步产生scfa(并且管腔scfa仅被吸收)的时间而异。这是一个重要的考虑因素,表明在不了解这些其他变量的情况下,粪便scfa的量可能不是相关的关注量。换句话说,粪便scfa浓度与临床结果变量之间缺乏关联并不一定表明微生物scfa不参与疾病过程。
[0164]
总之,申请人的数据表明,对患者mmp的量化了解可以为个性化膳食补充剂策略提供信息,所述策略旨在增加患者肠道中具体scfa的产生和吸收。更广泛的是,他们提出了通过两种独立但互补的方式调节患者中scfa产生的参照标准:根据其现有mmp对饮食输入进行修改,以及根据给定的关注mmp对其微生物群的潜在群落组成进行修改。
[0165]
本领域的技术人员将理解,本文描述的发明是易于相对于具体描述的那些进行变化和修改的。应当理解,本发明包括所有不背离其本质或基本特征的这些变化和修改。本发明还包括在本说明书中单独或共同提及或指示的所有步骤、特征、组合物和化合物,以及所述步骤或特征的任何和所有组合或任何两个或更多个。因此,本公开在所有方面都被认为是说明性的而不是限制性的,本发明的范围由所附权利要求表明,并且等同的含义和范围内的所有变化都旨在包含在其中。
[0166]
在本说明书中引用了多篇参考文献,每一篇都通过引用整体并入本文。
[0167]
参考以下实施例将更充分地理解前述描述。然而,这样的实施例只是实施本发明的方法的示例,并不旨在限制本发明的范围。
[0168]
实施例
[0169]
材料和方法:
[0170]
人类参与者
[0171]
健康的人类参与者被允许参加这项研究。
[0172]
粪便样本处理和活体外设置
[0173]
收集新鲜粪便样本并称重,然后转移到厌氧条件下,在该条件中将它们在含有0.1%l-半胱氨酸的还原pbs中稀释,稀释比例为1g/5ml。在将样本等分到96孔板中之前,将其均质化成浆液。将未改变的浆液样本作为基线样本,然后从储备溶液中添加菊粉、果胶和纤维素,使它们的最终浆液浓度分别为10g/l、5g/l和20g/l。菊粉和果胶的浓度是根据我们能够获得的膳食纤维完全溶解的最大储备浓度来确定的。测量了四种条件:菊粉、果胶、纤维素和对照(无添加物)。样本在37℃厌氧培养,并在每个时间点收集两个生物学重复(96孔板中的两个不同的孔)。在2小时和4小时后收集所有参与者的样本,并将样本邮寄出去以在gc-fid上对其进行scfa定量。线性产生速率的测量是在2小时和4小时之间进行的,因为这允许申请人在测量时间间隔中的准确度最大化,而不会由于设置期间引入的条件之间的延迟而引入假象。从振动器上的大烧瓶中的浆液获得的结果与在96孔板格式中获得的数据具有良好的一致性。
[0174]
对活体外设置中产生的气体的测量
[0175]
从活体外设置适应:
[0176]
粪便样本的制备、均质化以及不同的条件的制备与传统的活体外实验相同。一旦制备好条件预混料(mastermix)(对照无纤维、含10g/l菊粉、5g/l果胶或20g/l纤维素),就将它们转移到充满100%n2而没有可检测量的co2和h2的乙烯基厌氧室中。对于每个参与者,将每种条件下的2ml的最终粪便浆液一式三份添加到60ml玻璃血清瓶(supelco,bellefonte pa)中。使用带有ptfe/硅胶隔垫(supelco,bellefonte pa)的磁性卷边密封件将每个参与者的共12个瓶子密封在同一腔室中,并于37℃孵育24小时。
[0177]
气体测量:
[0178]
使用气相色谱法测定顶部空间气体的浓度。申请人使用了shimadzu gc-2014气相色谱仪(gc),其配置有保持在140℃的填充柱(carboxen-1000,5'x1/8”(supelco,bellefonte pa))、氩气载体fas、热导率和甲烷化器(methanizer)-火焰离子化检测器。约23℃的实验室温度下的来自每个血清瓶的顶部空间(0.20cm3)的子样本通过气密注射器取出并感染到柱子上。通过将样本和已知浓度的标准品的分压进行比较来确定气体浓度。
[0179]
根据标准品评估的分析的准确度为
±
5%。
[0180]
用于其他目的的样本收集:
[0181]
在本实验结束时,从每个血清瓶中取出1ml的粪便浆液并进行gc测量以用于scfa分析。
[0182]
短链脂肪酸测量
[0183]
气相色谱分析:
[0184]
使用带有火焰离子化检测器(fid)(agilenttechnologies,santa clara,ca)的agilent 7890b系统进行色谱分析。将涂覆有0.25μm涂层厚度的30m x0.25mm的高分辨率气相色谱毛细管柱(db-ffap)用于挥发性酸(agilent technologies),并将涂覆有0.50μm涂层厚度的30m x0.25mm的高分辨率气相色谱毛细管柱(db-ffap)用于非挥发性酸。将氮气用作载气。烘箱温度为145℃,fid和进样口设置为225℃。进样量为1μl,并且每次分析的运行时间为12分钟。使用openlab chemstation软件(agilent technologies)进行色谱图和数据整合。
[0185]
标准品溶液:
[0186]
使用含有10mm的乙酸、丙酸、异丁酸、丁酸、异戊酸、戊酸、异己酸、己酸和庚酸的挥发性酸混合物(supelco crm46975,bellefonte,pa)。制备含有1%的2-甲基戊酸(sigma-aldrich st.louis,mo)的标准品储备溶液作为用于挥发性酸萃取的内部标准品对照。使用含有10mm的丙酮酸和乳酸以及5mm的草酰乙酸、草酸、甲基丙二酸、丙二酸、反丁烯二酸和琥珀酸的非挥发性酸混合物(supelco,46985-u,bellefonte,pa)。制备含有50mm的苯甲酸(sigma-aldrich st.louis,mo)的标准品储备溶液作为用于非挥发性酸萃取的内部标准品对照。
[0187]
样本制备:
[0188]
样本在-80℃冷冻保存直至进行分析。将样本从冰箱中取出并使其解冻。将生粪样本转移至2ml的试管中,确定粪便重量并向每个样本中加入1.5ml的hplc水。将样本涡旋5分钟,直至材料均质化。用50%的硫酸将生粪悬浮液和解冻的粪便浆液样本的ph调节至2-3。
将酸化的样本保持在室温并涡旋10分钟。将样本以5000g离心10分钟。将1000ul的澄清的上清液转移到带有ptfe面橡胶内衬螺旋盖的玻璃管中,用于进一步的处理。向挥发性样本中加入50ul的内部标准品(1%的2-甲基戊酸溶液)和1ml的无水乙醚。颠倒混合试管10分钟,然后以2000g离心1分钟。将上层乙醚转移到agilent取样瓶中进行分析。对于非挥发性萃取,将50ul的内部标准品(50mm的苯甲酸溶液)和1ml的三氟化硼-甲醇溶液(sigma-aldrich st.louis,mo)加入到含有1000ul的澄清上清液的每支试管中。这些试管在室温下孵育过夜。向每个试管中加入2ml的水和1ml的氯仿。颠倒混合试管10分钟,然后以2000g离心1分钟。将下层氯仿转移到agilent取样瓶中进行分析。
[0189]
酸的定量:
[0190]
使用1ml的每种标准品混合物,并按照所述用于样本的处理方式对其进行处理。将标准品混合物中酸的保留时间和峰高用作未知样本的参考。通过这些酸的具体保留时间以及确定并表示为每克生粪材料样本的mm浓度和每毫升粪便浆液的mm浓度的浓度来对它们进行鉴定。
[0191]
16s rrna测序
[0192]
提取细节:
[0193]
对于dna提取,将mobiopowersoil 96试剂盒(现为qiagen cat no./id:12955-4)稍作修改后使用。将所有样本在冰上解冻,并将来自每个样本的250ul的5倍稀释的粪便浆液(来自活体外分析)转移到mobio high throughput powersoil bead plate(12955-4bp)中用于样本装载步骤。然后申请人在同一天按照制造商的方案来实施方案。
[0194]
文库准备细节,包括引物:
[0195]
使用靶向16s rrna基因v4区域的两步pcr法构建双端illumina测序文库,之前已由preheim等人进行了描述(preheim et al,2013)。
[0196]
测序细节:
[0197]
所有双端文库被多重化于流通槽中(每个流通槽最多汇集200个单独的样本),并在illumina miseq平台上进行双端测序,在每个末端上有150个碱基。
[0198]
菊粉特异性elisa
[0199]
申请人使用biopal的菊粉免疫分析试剂盒(biopal worcester ma)来测量来自粪便样本的滤液中的菊粉浓度,并遵循制造商的方案。如果粪便样本来自活体外分析,那它们是已经5倍稀释的粪便浆液,需要对其进行离心(10'000xg,2分钟),然后使上清液通过0.2μm的注射器式过滤器(pall corporation,portwashingtonny)。如果粪便样本来自体内饮食研究,则将它们在冰上解冻后均质成5倍稀释的粪便浆液(在pbs缓冲液中),然后进行与活体外样本类似的离心和过滤。粪便滤液保存在-80℃中,当从冰箱中取出时应解冻并始终保持在冰上,并稀释至适当的稀释度以在菊粉elisa试剂盒的检测范围内。作为参考,活体外菊粉条件需要稀释至少1,000倍,而活体外对照或体内样本只需稀释至少10倍。
[0200]
肠道单层scfa吸收的测量
[0201]
肠道单层模型:
[0202]
申请人使用了不含免疫组分的肠道单层系统,肠道单层的制备如先前wlk chen等人所述(chen et al.,2017b)。简言之,caco-2细胞或c2bbe1细胞与ht29-mtx-e21细胞(sigma)以9:1的比例(1x105个细胞/cm2)在500μl的接种培养基中接种到涂覆有鼠尾胶原蛋
白i(corning 354236)的transwell小室(insert)(corning 3460)上。在transwell板的顶部和底部隔室加入500ul和1.5ml的接种培养基。每2天至3天更换一次培养基。在肠道小室成熟20天后,细胞单层就可以使用了。
[0203]
细胞单层完整性评价:
[0204]
使用evom2和enfohm-12(world precision instruments)于37℃通过teer(跨上皮电阻)量化屏障完整性。申请人在时长24小时的实验的开始和结束时测量了细胞单层的完整性。在中间的时间点,申请人在显微镜下观察了细胞单层的(不)连续性。
[0205]
scfa来源:
[0206]
为了将scfa添加到哺乳动物细胞培养基中而不显著改变其ph,申请人使用了关注的scfa的盐形式:丁酸钠(sigma,b5887)、丙酸钠(alfaaesar,a17440)和乙酸钠(sigma,s2889)。
[0207]
scfa吸收实验:
[0208]
scfa被添加到顶侧的肠道细胞培养基中,而基底侧不变,并且实验进行24小时。在每个时间点(0小时、2小时、4小时、8小时和24小时),申请人从每个肠道细胞transwell小室的顶侧收集100μl,从基底侧收集200μl(wlk chen等人之前所做的实验表明所取体积不会影响细胞培养)。从顶侧和基底侧收集的培养基保存在-80℃直至scfa分析。除对照外,每个条件以一式三份进行。申请人进行了5种不同浓度的scfa组合。在不同条件下,丁酸:丙酸:乙酸的比率保持不变,分别为1:1:5。不同的丁酸浓度分别为40mm、20mm、10mm、5mm和2mm以及相应量的丙酸和乙酸。对照是单一的scfa(5mm的丁酸、5mm的丙酸或25mm的乙酸)或不添加scfa。
[0209]
在24小时,洗涤、裂解所有细胞单层并冷冻保存在-80℃,以便在需要时进行进一步的分析。
[0210]
16s rrna测序分析
[0211]
对原始的16s rrnaillumina双端测序读段进行合并、去多重化,并使用usearch8以q=25的截止值进行质量修整,然后修整为226个碱基的共同长度。然后使用uparse(edgar 2013)将经去除冗余处理(dereplicated)的读段聚类到otu中以达到97%的一致性。使用0.5的不确定性截止值(cole 2005)的rdp分类器将otu质心分配到一个分类。
[0212]
机器学习
[0213]
随机森林分类器(rfc)是使用scikit-learn python软件包构建的。使用5倍交叉验证构建平均接收器操作员特征(roc),从中计算auc。
[0214]
实施例1:
[0215]
scfa产生的活体外测量
[0216]
为了测量响应于不同膳食多糖的活体外scfa产生,将40个健康人类参与者的粪便在厌氧条件下均质化成浆液,并添加菊粉、果胶或纤维素(参见方法部分)。然后允许浆液随时间推移而演化,并在规则时间间隔获取样本,以量化每个时间点的scfa含量(图1)。为了确定合适的取样频率,申请人进行了试点实验,其中申请人分析了每个scfa浓度在24小时里的时间内的轨迹。申请人发现,在24小时的时间点之前,只有一小部分参与者似乎收敛至最终scfa浓度,但所有参与者在0至4小时的时间窗口内都表现出了线性的产生速率(图2)。这些数据与使用菊粉特异性elisa分析从粪便中测量的菊粉随时间推移的浓度非常一致:4
小时后,大部分菊粉底物仍然存在,但在测试的六名参与者中,有五名参与者的菊粉底物到24小时时几乎完全被消耗掉了(图11)。由于申请人试图在最能模拟结肠条件的环境中进行实验,申请人分析了16s rrna数据,并确定了群落结构或多样性在时间点0小时至4小时之间没有显著改变,而在4小时至24小时之间观察到了微小的变化(图12)。此外,由于酸性scfa的积累而导致的变化仅限于在前四个小时期间从大约中性下降到5.5(图13)。综合这些数据,申请人决定使用在0至4小时之间观察到的线性产生状态作为测量活体外scfa产生(作为体内scfa产生的代表)的最合适的时期。因此,申请人选择测量在时间点0小时、2小时和4小时的两个生物学重复,并计算在时间点2小时和4小时之间的产生速率。样本的bristol粪便量表与产生的总scfa浓度之间的线性回归,以及样本的bristol粪便量表与粪便中16s rrna的qpcr扩增的ct值之间的线性回归并未表明粪便稠度或密度会引起任何明显的假象,认为粪便稠度或微生物负荷的校正因子是不需要的,并且各种稠度的样本可以相互比较(图14)。
[0217]
申请人发现参与者的scfa的产生谱差异很大(图3)。在相同的膳食纤维输入下,不同的微生物群产生显著不同数量和比例的scfa。申请人将响应于不同膳食纤维所得的scfa产生速率定义为个体的微生物代谢表型(mmp),其中所述scfa产生速率被量化为与数据集中的其他参与者相比的标准化分数。mmp的层次聚类表明了可识别的群体:例如,具有i型mmp的个体是从菊粉生产丙酸的有力生产者;相比之下,具有ii型mmp的参与者是从果胶生产丙酸的有力生产者(图3)。因此,提高个体的具体scfa的产生并不一定依赖于相同的多糖来达到相同的效果;换句话说,相同的多糖在不同的个体身上会产生不同的效果,这取决于他们的mmp。这些数据表明,当涉及到肠道中纤维的功能性降解和产生的scfa时,健康人群中存在显著的异质性,但mmp聚类成可识别的类型,可用于指导未来的饮食干预。
[0218]
实施例2:
[0219]
从群落组成预测微生物代谢表型
[0220]
然后申请人提出了以下问题,即申请人是否可以仅从群落组成来预测参与者的mmp,mmp在此定义为在与不同纤维孵育之前从粪便的16s rrna测序获得的97%de novo otu的相对丰度。申请人对随机森林分类器(rfc)进行了训练,以预测给定微生物群响应于给定纤维的给定scfa的生产速率是高还是低,生产速率的高低分别由大于等于0或小于0的生产速率z分数来定义。性能是因scfa而异的,在预测响应于菊粉(auc=0.87)和果胶(auc=0.79)的丁酸产生中获得了最高准确度(图4)。
[0221]
申请人还测试了是否可以通过16s rrna测序预测原始(straight)粪便scfa含量,并发现乙酸和丁酸的预测能力适中(分别为auc=0.76和auc=0.73)。尽管这些数据表明群落组成在一定程度上可以预测所得粪便中scfa的含量,但人工检验具体otu特征(这些特征在菊粉特异性rfc和果胶特异性rfc的重要性方面排名很高)发现这些特征往往只与响应于该具体纤维的scfa产生有关。例如,未指定的毛螺菌科(lachnospiraceae)otu的相对丰度仅与响应于菊粉的丁酸产生相关(图5a)。相比之下,具体的普氏菌(prevotella copri)otu似乎仅与从菊粉产生丙酸有关(图5b)。这些数据与普氏菌属的成员是已知的丙酸生产者(chen et al.,2017a)且多糖降解能力不同(filippis et al.,2019)的事实是一致的。因此,i型mmp(图3)与后述的普氏菌otu的高相对丰度有关。类似地,已知的丁酸生产科(例如毛螺菌科和瘤胃菌科(ruminococcaceae))在当在科水平折叠(collapsed)的16s rrna数据
上对响应于菊粉的高/低scfa产生的rfc进行训练时的重要性方面排名很高。这些结果表明,各otu可以预测从具体多糖中产生scfa的能力,这可能是由于它们具体的多糖降解机制和内部发酵途径。
[0222]
实施例3:
[0223]
个体mmp随时间推移的稳定性
[0224]
虽然已知个体的肠道微生物群在没有大扰动的情况下可以是长期相对稳定的,但尚不清楚的是,个体的mmp是否也会随着时间的推移而同样稳定。因此,申请人在间隔至少6个月的时间点对八名参与者重复进行了实验(图6a)。尽管观察到时间点之间的一些可变性,但每个个体的mmp的极值通常是保守的(图6b)。在来源于对所有个体在两个时间点的每个scfa:纤维对进行两两比较获得的列联表上进行的fisher检验表明这种稳定性具有统计学意义(p=0.003;双侧fisher检验)。这些结果与mmp与微生物群具体成员的相对丰度相关的事实是一致的:与一般人群相比,具有较高相对丰度的上述普氏菌otu的个体可能在时间点之间的这六个月期间保持该otu的高相对丰度,从而保持了他们随时间推移从菊粉产生高水平丙酸的能力。
[0225]
实施例4:
[0226]
scfa产生模型
[0227]
为了更好地了解体内微生物scfa产生,申请人试图开发该过程的定量模型(图7)。在给定的参与者中,当消耗一定量的膳食纤维[f]时,它会发酵成乙酸、丙酸和丁酸,其速率随其具体微生物群而变化。正如定义的那样,一个人的mmp是个体的粪便微生物群对以具体膳食纤维进行的挑战的合计总反应。从纤维输入产生scfa的过程需要几个步骤(图15b)。第一步涉及将纤维/多糖(f)分解成更小的寡糖和单糖(o),这通常是通过细胞外酶编码的水解反应实现的(sonnenburg et al.,2010);第二步由将o发酵成还原的生化物质p(例如乳酸、乙酸、丙酮酸)组成,所述生化物质p可以作为第三步(产生最终产物或相关scfa的最终发酵反应)的底物。因此,一个人的mmp是所有这些各个反应的总和(图15a)。
[0228]
原则上,部分或全部膳食纤维可以在粪便中排出,而不在结肠中经历任何发酵。因此,申请人试图确定是否可以预期消耗量的膳食纤维在典型的食物团通过肠道所花费的时间内被完全发酵。申请人从之前的体内研究测量了粪便中的菊粉浓度,在该之前的体内研究中给予参与者申请人在活体外实验中使用的来自同一供应商的10g菊粉作为针对恒定饮食背景的每日补充剂(gurry et al.,2018),申请人发现在菊粉消耗后的几天内,粪便中的菊粉浓度范围为从不可检出到25mg/l(图8),而输入浓度约为10g/l(前提是我们估计肠道中的粪便体积约为1l;参见方法部分)。这些数据表明,菊粉团在粪便通过时几乎完全被微生物群所降解,这与申请人的菊粉在活体外在24小时里被耗竭的测量结果是一致的(图11)。因此,申请人推测这种膳食纤维的排泄率为零并且它几乎完全在肠道内被消耗(即[f]排泄=0)。
[0229]
微生物衍生的scfa的肠道管腔浓度是管腔内净产生和宿主上皮吸收之间的平衡。不幸的是,关于肠道上皮中的细胞对给定scfa的吸收速率的定量了解是有限的。以前使用透析袋技术收集的数据表明,scfa的吸收速率与scfa的典型生理浓度处的浓度呈线性关系(mcneil et al.,1978)。此外,研究表明,scfa的吸收表现出出乎意料适度的ph依赖性(bugaut,1987)。因此,申请人将吸收速率建模为与管腔内scfa浓度成比例。申请人试图估
计该速率常数的数量级,以更好地了解结肠中微生物群产生的scfa的命运(即吸收量与排泄量之间的平衡)。为此,申请人使用在trans-well中生长的caco细胞单层,并在24小时的持续时间里测量从细胞单层的顶侧和基底侧取样的培养基的scfa浓度(详见方法和支持信息部分)。假设活体外测量的scfa产生速率代表具有相同输入的体内产生速率,并明确包括吸收速率参数,申请人获得以下描述结肠腔中每种scfa浓度的时间演化的现象学方程系统:
[0230][0231][0232][0233]
其中以及分别是响应于给定输入浓度的纤维[f]而产生的乙酸、丙酸和丁酸的主要产生速率,定义了个体mmp。
[0234]
为了更好地理解吸收速率常数对肠道上皮的scfa吸收量的影响,申请人使用该模型计算给定参与者中随该速率常数而变的丁酸吸收量:
[0235][0236]
其中b
吸收
是丁酸吸收量(以mmol为单位),v
结肠
和s
结肠
分别是结肠的体积和表面积,[b]是结肠中丁酸的浓度,γb是丁酸的吸收速率,单位为mmoll-1h-1cm-2。申请人发现,假设转运时间为12小时,申请人在本研究中使用的速率常数(透析袋和caco单层衍生的速率常数)对所得动力学具有显著不同的影响:在透析袋常数(0.0019mmoll-1h-1cm-2)的情况下,产生的大部分丁酸被排出体外,而在caco单层常数(0.091mmoll-1h-1cm-2)的情况下,大量的丁酸被吸收(约40%)而其余的被排出(图9)。这表明,根据吸收速率常数的值,排泄的粪便中的scfa的量与肠道中产生和吸收的scfa的量之间的关系不一定相关。
[0237]
转运时间是在考虑的吸收速率数量级上控制肠道scfa吸收量的重要变量。这种关系在具有来自相同的输入的不同的丁酸产生速率的不同的参与者之间与比例因子相关。然而,申请人的模型不能解释纤维底物的耗竭,对粪便中菊粉浓度的测量(图8和图11)表明,这可能在粪便排出时发生。当然,只要仍有scfa被吸收和纤维底物被发酵,那么减缓结肠转运时间只是增加吸收的scfa的有效方法。尽管如此,从申请人的模型中可以清楚地看出,尽管假设参与者之间的吸收速率常数相同且转运时间为12小时,但不同的个体从给定的纤维中吸收的scfa的量仍然是不同的(图10),这反映了他们的产生差异和总mmp产生总是高于吸收,并且吸收与产生的浓度呈线性关系。改变吸收速率常数参数只会影响吸收量的规模,但个体之间的相对比率并没有改变。
[0238]
实施例5:
[0239]
微生物的scfa产生模型
[0240]
图15a说明了涉及微生物群的将膳食纤维降解为scfa的步骤。在此参照标准中,对于每种类型的单体m、中间体p和scfa,存在描述每种化学物质浓度的时间演化的不同方程。
实际上,该系统是不需要的(sparse),因为在绝大多数otu中,速率常数πi1、πj2和πk3为零或接近于零,原因在于只有一小部分otu具有必要的生化能力。对于给定浓度的膳食纤维[f],所有对具体scfa的给定生产速率的有所贡献方程的组合可以通过单个参数来概括,该参数是微生物群组成的函数(图15b)。因此,在活体外条件下,
[0241][0242]
实施例6:
[0243]
对结肠上皮吸收的scfa的评估
[0244]
申请人将结肠近似为直径3cm的圆柱体。这对应于约9.42cm的周长。因此,结肠的每个1cm厚的横截面部分具有约9.42cm2的表面积,申请人将其四舍五入为10cm2。估计结肠的长度为150cm,申请人获得的总表面积约为1,500cm2。假设结肠充满粪便,申请人获得的内部粪便总体积达到1,060cm3,申请人将其近似为1l。
[0245]
使用caco细胞单层实验估计的给定scfa的吸收速率常数υscfa以mol cm-2h-1l-1为单位。因此,整个结肠的吸收速率为每个scfa的1,500υscfamol h-1l-1。根据申请人将全部粪便近似为1l,这相当于每种scfa的吸收速率约为1,500γscfamol h-1。
[0246]
实施例7:
[0247]
活体外和体内之间的比较
[0248]
接下来申请人试图确定在活体外条件下观察到的scfa或群落结构的变化是否与理论上可以在体内测量的一致。为此,申请人求助于来自于一项先前研究的数据集,在该研究中,给参与者安排完全由缺乏纤维的液体营养膳食补充剂组成的固定饮食,持续六天(gurry et al.,2018)。在后三天,参与者被随机分配一种添加物,在恒定的流质饮食背景下,所述添加物每天以规定的剂量食用。这些添加物包括菊粉、果胶和纤维素,并使用了这三种纤维的完全相同的来源,为我们提供了理想的比较数据集。
[0249]
在活体外条件下,申请人观察到在某些参与者中菊粉团被完全降解(图6a)。申请人试图确定是否可以在消耗相似浓度的菊粉团的参与者的体内观察到相似程度的降解。因此,使用菊粉特异性elisa分析对粪便中的残留菊粉进行定量。申请人发现,正如预期的那样,在第1天、第2天和第3天,参与者的粪便中没有发现可检测的菊粉,但在某些粪便样品中在摄入菊粉之后的几天里可以检测到菊粉(图8)。然而,这些天检测到的菊粉只占总菊粉消耗量(10g/天)的一小部分,假设每日粪便总量为1l,则总菊粉消耗量相当于约10g/l,该浓度是在活体外实验中使用的浓度。这表明大部分可利用的菊粉也在摄入后的典型粪便通过时间内在体内被消耗。此外,由于10g的菊粉剂量是显著大于通常在典型饮食中消耗的剂量,因此申请人可以得出结论,在普通饮食中以与这些实验中使用的菊粉粉末类似可利用的大部分菊粉在体内被发酵。当然,来自天然来源和典型膳食纤维来源的菊粉可能不含像这些实验中使用的纯化形式的可利用形式的纤维。
[0250]
参考文献
[0251]
bugaut,m.(1987).occurrence,absorption and metabolism of short chain fatty acids in the digestive tract of mammals.comp.biochem.physiol.part b comp.biochem.86,439-472.
mediates a microbiomc-brain-β-cell axis to promote metabolic syndrome.nature 534,213-217.
[0265]
preheim,s.p.,perrotta,a.r.,martin-platero,a.m.,gupta,a.,and alm,e.j.(2013).
[0266]
distribution-based clustering:using ecology to refine the operational taxonomic unit.appl env.microbiol 79,6593-6603.
[0267]
roediger,w.e.(1980).role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man.gut 21,793-798.
[0268]
sanna,s.,zuydam,n.r.van,mahajan,a.,kurilshikov,a.,vila,a.v.,u.,mujagic,z.,masclee,a.a.m.,jonkers,d.m.a.e.,oosting,m.,et al.(2019).causal relationships among the gut microbiome,short-chain fatty acids and metabolic diseases.nat.genet.l.
[0269]
sonnenburg,e.d.,zheng,h.,joglekar,p.,higginbottom,s.k.,firbank,s.j.,bolam,d.n.,and sonnenburg,j.l.(2010).specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations.cell 141,1241-1252.
[0270]
zhao,l.,zhang,f.,ding,x.,wu,g.,lam,y.y.,wang,x.,fu,h.,xue,x.,lu,c.,ma,j.,et al.(2018).gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes.science359,1151-1156.
[0271]
zimmerman,m.a.,singh,n.,martin,p.m.,thangaraju,m.,ganapathy,v.,waller,j.l.,
[0272]
shi,h.,robertson,k.d.,munn,d.h.,and liu,k.(2012).butyrate suppresses colonic inflammation through hdac1-dependent fas upregulation and fas-mediated apoptosis of t cells.am.j.physiol.-gastrointest.liver physiol.302,g1405-g1415.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
