1.本发明属于检测方法技术领域,尤其涉及一种石墨炉原子吸收法的基体效应的校正方法。
背景技术:
2.基体效应是指试样中与被分析物共存的一种或多种组分所引起的种种干扰。石墨炉升温程序分为四阶段:干燥、灰化、原子化、清除;干燥能够去除水分;灰化能够去除基体溶剂(基体);原子化能够将试样中目标元素转化为原子;清除能够去除石墨管内残留的目标待测元素。背景吸收干扰包括分子吸收,可能在原子化过程中产生,也可能在原子前产生;原子化过程中基体溶液生成的非特征气体分子、氧化物及盐类分子对特征辐射吸收引起的干扰;分子吸收也可能先于原子吸收信号前产生,主要是样品蒸气在石墨炉内分布的不均匀性,导致背景吸收空间分布的不均匀性。背景吸收干扰还包括光散射,在原子化中产生的固体微粒、气溶胶对光散射,使被散射的光偏离光路不为检测器所检测。综上,基体效应产生背景吸收干扰,分子吸收干扰和光散射干扰,它们可能发生在原子化前的干燥和灰化阶段中,也可能发生在原子化过程中,如果发生在原子化前的干燥和灰化阶段,可能使原子化信号降低;如果发生在原子化过程中,可能使原子化信号升高/降低,如果干扰严重,根本无法完成石墨炉原子吸收检测。
3.基体干扰的检测方法适用于有一定浓度的样品,具体为:取同一试样两份,其中一份稀释5倍(1 4),稀释后试样的测定值(不得低于检出限的10倍)乘以稀释倍数与未稀释的测定值比较,相对偏差在
±
10%范围内视为无干扰、否则,表明有干扰存在,可采取稀释或标准加入法消除;当稀释后试样浓度低于检出限的10倍时,可将标准加入法曲线斜率与标准曲线斜率进行比较,相对偏差在
±
3%范围内视为无干扰;否则,表明有基体干扰存在。
4.校正干扰的方法有基于光谱校正干扰,如氘灯法,氘灯是连续光源,在仪器光谱通带范围内如果有共存元素的强烈吸收,会被误作为“背景”而在总吸光度内扣除,在石墨炉中这种现象更为明显。塞曼效应法校正背景,设备较为复杂,不可能应用于所有的原子吸收光谱仪。空心阴极灯自吸法校正背景,利用大电流时空心阴极灯的发射谱线变宽产生自吸,以此测量背景吸收,但是一些元素谱线很容易自吸,对灵敏度影响较小,而另一些元素空心阴极灯的谱线较难发生自吸,经过背景校正,灵敏度损失较大。基于光谱校正干扰的缺点为:操作复杂且对原子吸收光谱仪提出要求较高,仪器管理方面,需要对背景校正能力测试,合格方可用。
5.校正干扰的方法还有标准加入法校正干扰,具体为:分别量取四份等量的同一待测试样,配制相同四份溶液,第1份不加标准溶液,第2份、第3份、第4份按比例加入不同浓度的标准溶液,分别为:c
x
、c
x
c0、c
x
2c0、c
x
3c0,加入标准溶液的质量浓度约等于0.5倍量的试样质量浓度,即c
x
≈0.5c0。在相同测试条件下,测定四份溶液的吸光度,以加入标准溶液的质量浓度为横坐标,以其对应的吸光度为纵坐标,建立校准曲线,曲线反向延伸与浓度轴的交点即为待测试样的质量浓度;该方法只适用于质量浓度和吸光度呈线性的区域。标准
加入法注意事项:加入标准溶液后所引起的体积误差不应超过0.5%;采用标准加入法只能抵消基体效应带来的影响,不能消除背景吸收的影响;只适用于试样浓度与吸光度呈线性的区域。标准加入法适用性判断:测定待测试样的吸光度为a,从标准曲线上查得浓度为x,再向待测试样加入标准溶液,加标浓度为s,测定其吸光度为b,从标准曲线上查得浓度为y;用标准加入法适用性判断公式判断,当c=x时,即s/(y-x)=1时,才能用标准溶液校正曲线法(简称:标准曲线法);当存在基体效应时,s/(y-x)在0.5~1.5之间,可用标准加入法,s/(y-x)超出范围时,标准加入法不再适用,必须预先分离基体后才能测定。
6.现有原子吸收技术检测中,针对试样存在大量基体和基体含量不明情况下,一般采用标准加入法消除基体干扰。标准加入法适用范围更加严格,校准曲线不仅要严格在线性范围内,而且吸光度值最好在0.100~0.200范围内;且标准加入法工作量大,尤其不适合大批量样品分析测定;标准加入法由于是将不同浓度标准溶液加在试样中,因此,每条校准曲线只能测量一个试样;如果待测试样的浓度很高或者很低不适合标准加入法时,预先应分离基体,但分离基体步骤繁琐且基体成分不明时,难以分离。
技术实现要素:
7.有鉴于此,本发明的目的在于提供一种石墨炉原子吸收法的基体效应的校正方法,本发明提供的方法较为简单,能够准确的校正基体效应。
8.本发明提供了一种石墨炉原子吸收法的基体效应的校正方法,包括:
9.按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号;
10.若所述吸光度-实时信号不理想,则利用待测样品的吸光度-时间实时信号优化升温程序,得到优化后的升温程序;
11.按照优化后的升温程序检测标准曲线上浓度点对应的吸光度,得到标准曲线;
12.对标准曲线进行检验;
13.采用标准曲线法检测待测样品,利用平行样校正并检验是否存在基体效应的干扰。
14.优选的,所述按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号之前还包括:
15.寻峰,所述寻峰的能量为98~102%;
16.校核能量,所述校核能量包括:
17.调整光路至最佳,校核能量为100%,放置石墨管后,能量不低于75%。
18.优选的,所述预设升温程序包括:
19.一步干燥、一步灰化、原子化和清除;
20.所述一步干燥的升温时间为4~6s,干燥温度为90~110℃,保持时间为8~12s;
21.所述一步灰化的升温时间为4~6s,灰化温度为不损失待测元素的最高温度,保持时间为8~12s;
22.所述原子化的温度为待测元素吸光度信号最大值处对应的最低温度,保持时间为2~4s;
23.所述清除的温度为高于原子化温度90~110℃,保持时间为1~2s。
24.优选的,所述按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信
号之前还还包括:
25.按照预设升温程序检查标准曲线零点浓度值的吸光度-时间实时信号;和/或;
26.按照预设升温程序检查空烧石墨管的吸光度-时间实时信号。
27.优选的,所述利用待测样品吸光度-时间实时信号优化升温程序的方法包括:
28.按照所述预设升温程序检测待测样品检查得到的吸光度-时间实时信号,若所述吸光度-实时信号不理想,则查看吸光度-实时信号中异常出峰位置;若异常出峰位置对应的温度<120℃,则优化干燥阶段;若异常出峰位置对应的温度≥120℃且<原子化温度,则优化灰化阶段;
29.所述利用待测样品吸光度-时间实时信号优化升温程序的方法还包括:
30.若待测样品吸光度对应的浓度≥标准曲线次高点浓度,则对待测样品进行稀释后再进行优化升温程序,稀释后的浓度c1满足:3倍测定下限≤c1<标准曲线次高点浓度。
31.优选的,所述优化干燥阶段的方法包括:
32.在预设升温程序的干燥阶段增加一步干燥,进行两步干燥,检查吸光度-时间实时信号,若吸光度-时间实时信号不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度<120℃,则继续增加一步干燥,进行三步干燥,检查吸光度-时间实时信号,若吸光度-实时信号还不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度<120℃,则对应三步干燥中异常出峰的温度时间,在不改变三步干燥温度的情况下,增加相应干燥阶段的升温时间和保持时间;
33.所述优化灰化阶段的方法包括:
34.在预设升温程序的灰化阶段增加一步灰化,进行两步灰化,检查吸光度-时间实时信号,若吸光度-时间实时信号不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度≥120℃且<原子化温度,则继续增加一步灰化,进行三步灰化,检查吸光度-时间实时信号,若吸光度-时间实时信号还不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度≥120℃且<原子化温度,则对应三步灰化中异常出峰的温度时间,在不改变三步灰化温度的情况下,增加相应灰化阶段的升温时间和保持时间。
35.优选的,所述利用待测样品吸光度-时间实时信号优化升温程序的方法还包括:
36.若所述优化干燥阶段和优化灰化阶段的总增加时间>90s,则对待测样品稀释后再进行优化升温程序,所述稀释的倍数≤20倍。
37.优选的,所述异常出峰还包括:
38.原子化峰型异常,所述原子化峰型异常包括:
39.原子化峰不为尖锐单峰,原子化峰有拖尾现象和/或原子化峰峰尾高于空烧值水平。
40.优选的,所述对标准曲线进行检验包括:
41.检测标准曲线的斜率、截距、回归标准偏差、残差。
42.优选的,利用平行样校正并检验是否存在基体效应的干扰的方法包括:
43.按照优化后的升温程序检测待测样品的浓度为c1:
44.若1倍测定下限≤c1<2倍测定下限,对平行样加标处理得到c2溶液,加标倍数不低于1.25倍,不超过3倍,加标倍数为j
加标
;如果c1=(85~115%)(1/j
加标
)
×
c2,则视为原子化期
间没有干扰;
45.若2倍测定下限≤c1<3倍测定下限,对平行样加标或稀释处理得到c2溶液,加标倍数为j
加标
,稀释倍数为n
检测稀释
,加标或稀释的c2溶液同时满足:2倍测定下限≤c2<4倍测定下限;如果c1=(90~110%)(n
检测稀释
或1/j
加标
)
×
c2,则视为原子化期间没有干扰;
46.若3倍测定下限≤c1<标准曲线次高点浓度,对平行样稀释处理得到c2,稀释倍数为n
检测稀释
,2倍测定下限≤c2;如果c1=(95~105%)n
检测稀释
×
c2,则视为原子化期间没有干扰;
47.若标准曲线次高点浓度≤c1,对平行样两次稀释处理得到溶液cc1和cc2,cc1浓度>cc2浓度,稀释倍数分别为:n
检测稀释1
和n
检测稀释2
;同时满足:3倍测定下限≤cc1<标准曲线次高点浓度,3倍测定下限≤cc2<标准曲线次高点浓度;如果n
检测稀释1
cc1=(95~105%)n
检测稀释2
×
cc2,则视为原子化期间没有干扰;
48.若检出限≤c1<测定下限,对平行样加标或稀释处理得到c2溶液,加标倍数不低于1.25倍,不超过3倍,加标倍数为j
加标
;稀释倍数不低于1.25倍,不超过2倍,稀释倍数为n
检测稀释
;稀释得到c2溶液浓度应不小于检出限;如果c1=(75~125%)(1/j
加标
)
×
c2,则视为原子化期间没有干扰;
49.若c1<检出限,且优化升温程序时没有执行稀释,判断原子化是否存在干扰:若测定c1溶液7次,t
单侧检验(7次,0.95)
×s7次c1溶液测定浓度值
≤mdl检出限,则视为无原子化干扰;
50.若c1<检出限,且校正升温程序时执行稀释,1<n
升温稀释
≤10,执行富集程序直至富集后浓度大于检出限,富集倍数记为f
富集
,1<f
富集
≤10,c
试样最终测定结果
=n
升温稀释
×
(1/f
富集
)
×
c1;富集倍数如果超过10倍还未大于检出限,停止富集,试样检测结果为未检出;判断原子化是否存在干扰:若测定未富集前c1溶液7次,t
单侧检验(7次,0.95)
×s7次未富集前c1溶液测定浓度值
≤mdl检出限;同时富集两次,为cc1和cc2,如果cc1=(70~130%)cc2;满足以上两条,则视为无原子化干扰。
51.本发明在石墨炉原子吸收环境样品检测领域应用,校正环境试样中基体对原子吸收测定中带来的干扰,为了实现基体效应精密化校正方法,需要基于最小二乘法的一元线性回归标准曲线的线性范围内检验,保证最小二乘法的一元线性回归标准曲线是一条直线,才能校正原子化过程中的基体效应干扰。基体效应产生的分子吸收干扰和光散射干扰如果发生在原子化信号前的干燥和灰化阶段,在石墨炉原子吸收光谱仪实时信号中可以显示,可以通过单独优化石墨炉升温程序或者结合稀释富集程序的方法消除,并利用稀释富集程序校正原子化过程中的基体效应干扰。
52.本发明提供的基体抗干扰方法直接利用标准曲线法,优化石墨炉升温程序,稀释富集校正试样基体带来的化学干扰,一条标准曲线可以实现大批量试样的检测,当稀释的试样浓度不适合原子吸收检测范围时,采用石墨炉富集方法将试样浓度浓缩至石墨炉适合的测定范围内即可。
附图说明
53.图1为本发明中abs-时间实时信号的理想信号图;
54.图2为本发明中abs-时间实时信号的不理想信号图;
55.图3为本发明实施例中未经稀释的试样吸光度-时间实时信号曲线图;
56.图4为本发明实施例中稀释后的试样吸光度-时间实时信号曲线图;
57.图5为本发明实施例中得到的吸光度-浓度标准曲线;
58.图6为本发明实施例中得到的浓度-吸光度标准曲线。
具体实施方式
59.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
60.本发明提供了一种石墨炉原子吸收法的基体效应的校正方法,包括:
61.按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号;
62.若所述吸光度-实时信号不理想,则利用待测样品的吸光度-时间实时信号优化升温程序,得到优化后的升温程序;
63.按照优化后的升温程序检测标准曲线上浓度点对应的吸光度,得到标准曲线;
64.对标准曲线进行检验;
65.采用标准曲线法检测待测样品,利用平行样校正并检验是否存在基体效应的干扰。
66.在本发明中,所述按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号之前优选还包括:
67.调整石墨炉仪器参数。
68.在本发明中,所述调整石墨炉仪器参数优选包括:
69.进行寻峰。
70.在本发明中,所述进行寻峰的寻峰结果优选不超过特征值的
±
0.15;原则上,寻峰时能量优选为98~102%,更优选不低于99%;若不满足上述条件,优选进行重新寻峰。
71.在本发明中,所述调整石墨炉仪器参数优选还包括:
72.调整石墨炉光路,校核能量。
73.在本发明中,所述校核能量过程中的积分方式优选为峰高法。
74.在本发明中,所述调整石墨炉光路、校核能量优选包括:
75.未放置石墨管前,调整石墨炉光路至最佳,校核能量为100%;放置石墨管后,能量不低于75%,优选不低于80%;
76.若不满足上述条件优选重新调整石墨炉光路,满足上述条件后再次校核能量为98~102%。
77.在本发明中,优选放置石墨管后能量损失一般不超过20%,特殊情况不超过25%。
78.在本发明中,所述预设升温程序优选包括:
79.一步干燥、一步灰化、原子化和清除。
80.在本发明中,所述一步干燥的升温时间优选为4~6s,更优选为5s;干燥温度优选为90~110℃,更优选为100℃;保持时间优选为8~12s,更优选为10s。在本发明中,所述一步灰化的升温时间优选为4~6s,更优选为5s;灰化温度优选为不损失待测元素的最高温度;保持时间优选为8~12s,更优选为10s。在本发明中,所述原子化的温度优选为待测元素吸光度信号最大值处对应的最低温度(吸光度信号最高峰对应的最低温度),保持时间优选为2~4s,更优选为3s。在本发明中,所述清除的温度优选为高于原子化温度90~110℃,更
优选为高于原子化温度100℃;保持时间优选为1~2s,更优选为1s。
81.在本发明中,所述预设升温程序的时间优选为30~35s,更优选为31~34s,最优选为32~33s。
82.在本发明中,所述预设升温程序优选包括:
83.一步干燥(100℃:5s 10s) 一步灰化(不损失待测元素的最高温度:5s 10s) 原子化(原子化信号最大的最低温度:0s 3s) 清除(原子化温度 100℃:0s 1s)。
84.在本发明中,标准曲线最高浓度点的吸光度-时间信号为理想信号的条件优选包括:
85.干燥阶段吸光度逐渐降低;灰化阶段abs(吸光度)基本等同于空烧值(接近零);原子化阶段起原子化峰(最后10s内),峰型目测尖锐狭窄;清除阶段峰尾返回空烧值(接近零)。
86.在本发明中,理想的吸光度-时间实时信号无基体干扰,最后10s内的起峰为原子化峰,在原子化峰之前出现小包小峰均为存在干扰,为干燥、灰化阶段的假吸收峰,如图1所示。
87.在本发明中,标准曲线最高浓度点的吸光度-时间信号为不理想信号的条件优选包括:
88.原子化峰前有干扰峰。
89.在本发明中,不理想的吸光度-时间实时信号有基体干扰,除了原子化阶段起峰,在干燥、灰化阶段也起峰,如图2所示。
90.在本发明中,所述按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号之前还还包括:
91.按照预设升温程序检查空烧石墨管的吸光度-时间实时信号。
92.在本发明中,所述空烧石墨管的次数优选为10~12次,更优选为11次。
93.在本发明中,优选所述空烧石墨管时abs-时间(吸光度-时间)实时信号为平整直线,否则更换石墨管。
94.在本发明中,优选所述空烧石墨管时得到的吸光度值满足下述条件,否则更换石墨管:
95.若1800℃≤t
原子化温度
<2000℃,则a
空烧11次最大值
≤0.005;
96.若2000℃≤t
原子化温度
<2200℃,则a
空烧11次最大值
≤0.010;
97.若2200℃≤t
原子化温度
<2500℃,则a
空烧11次最大值
≤0.015;
98.若2500℃≤t
原子化温度
,则a
空烧11次最大值
≤0.018。
99.在本发明中,空烧石墨管能够了解基线,由于后续检测原子化峰型时需要回到基线,即这个空烧石墨管时吸光度值水平。
100.在本发明中,所述按照预设升温程序检查标准曲线最高浓度值的吸光度-时间实时信号之前还还包括:
101.按照预设升温程序检查标准曲线零点浓度值的吸光度-时间实时信号。
102.在本发明中,所述标准曲线零点为空白溶液;所述空白溶液优选选自:水,硝酸和盐酸混合液。在本发明中,所述水优选超纯水;所述硝酸和盐酸混合液中硝酸和盐酸的体积比优选为1:(98~99),更优选为1:99;所述硝酸和盐酸优选为优级纯。
103.在本发明中,检测吸光度-时间实时信号过程中石墨炉一次进样量优选为5~20μl,更优选为10μl。
104.在本发明中,检测标准曲线零点浓度值的吸光度-时间实时信号的次数优选为7~21次,更优选为10~20次,最优选为15次。
105.在本发明中,优选标准曲线零点浓度值的abs-时间实时信号为吸光度接近于0的平整直线,否则重新石墨炉光路,并且校核能量,重新放置石墨管和/或更换标准曲线空白溶液。在本发明中,所述重新石墨炉光路,并且校核能量,重新放置石墨管的方法与上述技术方案所述调整石墨炉仪器参数的方法一致,在此不再赘述。
106.在本发明中,优选标准曲线零点浓度值的吸光度值满足下述条件,否则更换标准曲线的空白溶液或重新测定标准曲线空白溶液的abs-时间实时信号:
107.若1800℃≤t
原子化温度
<2000℃,则a
标准曲线空白溶液21次最大值
≤0.006;
108.若2000℃≤t
原子化温度
<2200℃,则a
标准曲线空白溶液21次最大值
≤0.012;
109.若2200℃≤t
原子化温度
<2500℃,则a
标准曲线空白溶液21次最大值
≤0.018;
110.若2500℃≤t
原子化温度
,则a
标准曲线空白溶液21次最大值
≤0.030;和/或;
111.a
单次标准曲线空白值
∈a
n次标准曲线空白平均值
±
t
双侧检验(n-1,0.99)
×sn次标准溶液空白值吸光度标准偏差
。
112.在本发明中,检测标准曲线零点浓度值的吸光度可以查看标准曲线基体,因为标准曲线零点溶液为标准曲线基体,可以获得,但待测样品基体完全无法获得。
113.在本发明中,所述利用待测样品吸光度-时间实时信号或结合稀释富集程序优化升温程序的方法包括:
114.按照所述预设升温程序检测待测样品检查得到的吸光度-时间实时信号,若所述吸光度-实时信号不理想,则查看吸光度-实时信号中异常出峰位置;若异常出峰位置对应的温度<120℃,则优化干燥阶段;若异常出峰位置对应的温度≥120℃且<原子化温度,则优化灰化阶段。
115.在本发明中,所述结合稀释富集程序优化升温程序优选包括:
116.若待测样品吸光度对应的浓度≥标准曲线次高点浓度,优选按照检测样品要求进行稀释操作,此时的稀释为检测稀释,检测稀释后浓度c1满足:3倍测定下限≤c1<标准曲线次高点浓度,优选按照检测稀释后浓度优化升温程序。
117.在本发明中,所述结合稀释富集程序优化升温程序优选还包括:
118.若优化干燥阶段和优化灰化阶段升温程序总时间增加大于90s,且待测样品吸光度对应浓度<标准曲线次高点浓度,优选进行优化升温程序时同时结合稀释程序(升温稀释,n
升温稀释
),当升温稀释后的对应浓度<检出限,升温稀释优选执行富集程序,富集倍数≤升温稀释倍数;优选n
升温稀释
≤10倍,但以不稀释为最佳,如果稀释,此阶段稀释优选不大于10倍,特殊情况不得大于20倍。
119.在本发明中,所述理想信号与不理想信号的条件与上述技术方案所述一致,在此不再赘述。
120.在本发明中,所述优化干燥阶段的方法优选包括:
121.在预设升温程序的干燥阶段增加一步干燥,进行两步干燥,检查吸光度-时间实时信号,若吸光度-时间实时信号不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度<120℃,则继续增加一步干燥,进行三步干燥,检查吸
光度-时间实时信号,若吸光度-实时信号还不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度<120℃,则对应三步干燥中异常出峰的温度时间,在不改变三步干燥温度的情况下,增加相应干燥阶段的升温时间和保持时间。
122.在本发明中,所述两步干燥的温度差优选为15~25℃,更优选为20℃,如两步干燥的温度可设定为95℃,110℃,升温时间为5s,保持时间为10s。在本发明中,所述三步干燥的温差优选为15~20℃,如三步干燥的温度可设定为80℃,95℃,110℃,升温时间为5s,保持时间为10s。在本发明中,所述优选干燥阶段的温度优选为75~120℃,更优选为80~110℃,最优选为90~100℃。
123.在本发明中,所述增加相应干燥阶段的升温时间和保持时间过程中,升温时间优选增加4~6s,更优选为5s;保持时间优选增加8~12s,更优选为10s。在本发明中,所述优化干燥阶段升温时间优选最长为15s,由5s开始,每次增加5s;保持时间优选最长为50s,由10s开始,每次增加10~20s。
124.在本发明中,所述两步干燥的升温程序优选包括:
125.干燥1(80℃~95℃:5s 10s) 干燥2(100℃~110℃:5s 10s)。
126.在本发明中,所述三步干燥的升温程序优选包括:
127.干燥1(75~80℃:5s 10s) 干燥2(95℃:5s 10s) 干燥3(110~120℃:5s 10s)。
128.在本发明中,所述三步干燥的升温程序更优选包括:
129.干燥1(75~80℃:5s/10s/15s...... 10s/20s/30s/40s/50s.....) 干燥2(95℃:5s/10s/15s...... 10s/20s/30s/40s/50s.....) 干燥3(110~120℃:5s/10s/15s...... 10s/20s/30s/40s/50s.....);
130.在本发明中,所述优化灰化阶段的方法优选包括:
131.在预设升温程序的灰化阶段增加一步灰化,进行两步灰化,检查吸光度-时间实时信号,若吸光度-时间实时信号不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度≥120℃且<原子化温度,则继续增加一步灰化,进行三步灰化,检查吸光度-时间实时信号,若吸光度-时间实时信号还不理想继续进行优化,检查吸光度-时间实时信号异常出峰的位置,若异常出峰位置对应的温度≥120℃且<原子化温度,则对应三步灰化中异常出峰的温度时间,在不改变三步灰化温度的情况下,增加相应灰化阶段的升温时间和保持时间。
132.在本发明中,优选所述两步灰化的最终灰化温度为第二阶段灰化温度,第一阶段的灰化温度优选为480~520℃,更优选为500℃;升温时间优选为4~6s,更优选为5,保持时间优选为8~12s,更优选为10s。
133.在本发明中,所述两步灰化的升温程序优选包括:
134.灰化1(500℃:5s 10s) 灰化2(灰化最高温度:5s 10s)。
135.在本发明中,所述三步灰化的升温程序优选包括:
136.灰化1(500℃:5s 10s) 灰化2(0.5
×
(500℃ 最高灰化温度)
±
100℃:5s 10s) 灰化3(灰化最高温度:5s 10s)。
137.在本发明中,所述三步灰化的升温程序更优选包括:
138.灰化1(500℃:5s/10s/15s...... 10s/20s/30s.....) 灰化2(0.5
×
(500℃ 最高灰化温度)
±
100℃:5s/10s/15s...... 10s/20s/30s.....) 灰化3(灰化最高温度:5s/
10s/15s...... 10s/20s/30s.....)。
139.在本发明中,所述异常出峰优选还包括:
140.原子化峰型异常;
141.所述原子化峰型异常优选包括:
142.原子化峰不为尖锐单峰,原子化峰有拖尾现象和/或原子化峰峰尾高于空烧值水平。
143.在本发明中,所述原子化峰型优选为尖锐的单峰型,若不为单峰如双峰、m峰优选进行第一优化原子化升温程序。
144.在本发明中,所述第一优化原子化升温程序的方法优选包括:
145.降低原子化温度。
146.在本发明中,降低原子化温度优选降低50~100℃,更优选为60~90℃,最优选为70~80℃。
147.在本发明中,所述第一优化原子化升温程序优选包括:
148.原子化(t
原子化
/t
原子化-50℃/t
原子化-100℃:原子化时间不变)。
149.在本发明中,若原子化峰型有“拖尾”现象,优选进行第二优化原子化升温程序,所述第二优化原子化升温程序优选包括:
150.增加原子化保持时间,提高原子化温度。
151.在本发明中,所述增加原子化保持时间优选为增加保持时间1s,原子化保持时间优选不超过5s。在本发明中,所述提高原子化温度优选为提高温度不超过500℃,优选每次提高100℃。
152.在本发明中,所述第二优化原子化升温程序优选包括:
153.原子化(t
原子化
/t
原子化
100℃/t
原子化
200℃/t
原子化
300℃/t
原子化
400℃/t
原子化
500℃:0s 2s/3s/4s/5s)。
154.在本发明中,若原子化峰尾高于空烧值水平,优选进行优化清除升温程序,所述优化清除升温程序的方法优选包括:
155.提高清除温度,增加清除保持时间。
156.在本发明中,优选针对“记忆”元素,为避免全程空烧带来的检测时间无效延长,进行优化清除升温程序,“记忆”元素一般具有两个特征,第一,原子化温度相对较高,一般大于等于2200℃,例如,al、be、ba;第二,原子化后峰型容易拖尾,需要多空烧几次才能完成清除。
157.在本发明中,所述提高清除温度优选为每次提高温度80~120℃,更优选为100℃;所述增加清除保持时间优选为每次增加保持时间1~2s,更优选为1s;所述清除时间优选不超过5s;所述增加清除温度优选不超过300℃
158.在本发明中,所述优化清除升温程序优选包括:
159.清除(t
清除
/t
清除
100℃/t
清除
200℃/t
清除
300℃:0s 1s/2s/3s/4s/5s)。
160.在本发明中,所述优化清除升温程序后优选原子化峰尾基本返回空烧值水品,更优选包括:
161.优化清除后再次空烧的石墨管的吸光度与原空烧石墨管11次的最大空烧吸光度的差值不大于0.005。
162.在本发明中,若优化升温程序的总时长过长,还没有消除干扰,优选为了缩短优化升温程序的总时间适当稀释待测样品,稀释倍数为n
升温稀释
,优选n
升温稀释
≤20倍,更优选n
升温稀释
≤10倍,最优选以不稀释为最佳。在本发明中,根据优化升温程序的总时间、待测样品吸光度、标准曲线最高浓度点吸光度可以初步判断是否适合执行稀释的优化升温。
163.在本发明中,利用配置好的标准曲线的浓度溶液,按照优化后的升温程序进行检测,保证标准曲线和待测样品升温程序完全一致。
164.在本发明中,所述得到标准曲线的方法优选包括:
165.根据待测样品的浓度和测定线性范围,取6~10个标准曲线浓度点做一元线性回归方程,得到标准曲线。
166.在本发明中,优选所述标准曲线的回归点个数≤7时,石墨炉法回归系数不低于0.997;所述标准曲线的回归点个数8≤回归点个数≤10时,石墨炉法回归系数不低于0.995。
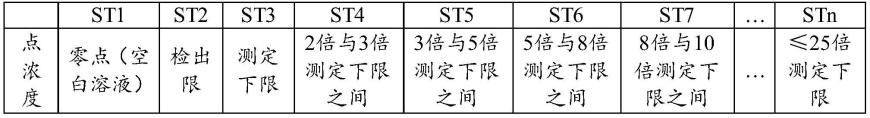
167.在本发明中,优选按照下表进行标准曲线浓度点的设置:
[0168][0169]
在本发明中,所述标准曲线最高点浓度优选不超过测定下限的25倍;优选st2和st3合并为一点,取在二者中间,优选2倍测定下限以上的浓度点间隔均匀。
[0170]
在本发明中,优选设置所述标准曲线的最高浓度点吸光度值<0.6,若标准曲线的最高浓度点的吸光度值大于0.6,可降低最高点浓度或在保证没有光谱干扰的条件下,重新设置目标元素空心阴极灯光源狭缝的宽度,进而降低信噪比。
[0171]
在本发明中,所述标准曲线的进样rsd要求优选包括:
[0172]
标准曲线上的浓度点优选重复测量两次或者两次以上,取平均值,零点和检出限附近点尽量降低rsd;和/或;
[0173]
测定7~21次空白溶液时,得出仪器当时状态下最大漂移量,测定下限点要求测量rsd<10%;和/或;
[0174]
吸光度值<0.050时,两次测量值sd<3倍空白溶液漂移量,rsd<10%;和/或;
[0175]
0.050≤吸光度值<0.100时,两次测量值sd<5倍空白溶液漂移量,rsd<10%;和/或
[0176]
0.100≤吸光度值时,两次测量值sd<10倍空白溶液漂移量,rsd<5%。
[0177]
在本发明中,空白溶液漂移量为测定空白溶液7~21次的测定值,1倍漂移量=空白溶液测定最大值-空白溶液测定最小值。
[0178]
在本发明中,所述对标准曲线进行检验优选包括:
[0179]
检测标准曲线的斜率、截距、回归标准偏差、残差。
[0180]
在本发明中,所述对标准曲线进行检验的方法优选包括:
[0181]
逐一去除标准曲线上各点,如果曲线有n个点,重新对剩余点做最小二乘法一元线性回归曲线,分别得到新曲线斜率b0.....b
n-1
、截距v0......v
n-1
、回归标准偏差s
a/c0
......s
a/cn-1
、被去除点在新回归曲线中残差d0......d
n-1
,如满足下述条件则检验合格:
[0182]
新斜率b0.....b
n-1
∈[b
平均-stdev(b0.....b
n-1
)
×
t
双侧(n-2,0.05)
,b
平均
stdev
(b0.....b
n-1
)
×
t
双侧(n-2,0.05)
];和/或;
[0183]
截距v0......v
n-1
∈[v
平均-stdev(v0.....v
n-1
)
×
t
双侧(n-2,0.05)
,v
平均
stdev(v0.....v
n-1
)
×
t
双侧(n-2,0.05)
];和/或;
[0184]
回归标准偏差s
a/c0
......s
a/cn-1
∈[s
a/c平均-stdev(s
a/c0
......s
a/cn-1
)
×
t
双侧(n-2,
0.05)
,s
a/c平均
stdev(s
a/c0
......s
a/cn-1
)
×
t
双侧(n-2,0.05)
];和/或;
[0185]
被去除点在新回归曲线中残差d0......d
n-1
∈[d
平均-stdev(d0......d
n-1
)
×
t
双侧(n-2,
0.05)
,d
平均
stdev(d0......d
n-1
)
×
t
双侧(n-2,0.05)
]。
[0186]
在本发明中,所述检验优选还包括:
[0187]
计算t0,若t0<t
单侧(n-2,0.05)
,则检测合格。
[0188]
在本发明中,所述t0的计算方法优选包括:
[0189][0190]
其中,
[0191]
在本发明中,去除零浓度点后,重新计算剩余点回归方程,计算因变量回归标准偏差s
a/c
,在新的回归方程中计算c=0时因变量残差d0,计算标准曲线上t0;a0为被去除的零点测定的吸光度值(两次或多次平均值),c0=0,为零点去掉后c0=0在剩余点新回归方程中估计值。
[0192]
在本发明中,所述检验优选还包括:
[0193]
测试待测样品后,回测待测样品两端最近的标准曲线浓度点,若满足下述条件则检验合格:
[0194]
若待测样品的浓度点在1倍和2倍测定下限之间,则回测误差<10%;
[0195]
若待测样品的浓度点在3倍和5倍测定下限之间,则回测误差<7%;
[0196]
若待测样品的浓度点在6倍和10倍测定下限之间,则回测误差<5%;
[0197]
若待测样品的浓度点高于10倍测定下限,则回测误差<3%。
[0198]
在本发明中,检测待测样品,利用不同范围平行样检测,检验并校正待测样品干扰。在本发明中,利用平行样检验是否存在基体效应的干扰并校正的方法优选包括:
[0199]
按照优化后的升温程序检测待测样品的浓度为c1(若优化升温程序时没有稀释待测样品,即为原待测样品,如果优化升温程序时执行稀释,则为升温程序稀释后的待测样品):
[0200]
若测定下限≤c1<标准曲线次高点:
[0201]
若1倍测定下限≤c1<2倍测定下限,对平行样加标处理得到c2溶液,加标倍数不得低于1.25倍,不得超过3倍,加标倍数为j
加标
;如果c1=(85~115%)(1/j
加标
)
×
c2,则视为原子化期间没有干扰;此时,c
试样最终测定结果
=n
升温稀释
×
c1=n
升温稀释
×
(1/j
加标
)
×
c2,其中j
加标
∈[1.25,3],如果优化升温程序时没有稀释,则n
升温稀释
=1;
[0202]
若2倍测定下限≤c1<3倍测定下限,对平行样加标或稀释处理得到c2溶液,加标倍
数为j
加标
,稀释倍数为n
检测稀释
,加标或稀释的c2溶液同时满足:2倍测定下限≤c2<4倍测定下限;如果c1=(90~110%)(n
检测稀释
或1/j
加标
)
×
c2,则视为原子化期间没有干扰;c
试样最终测定结果
=n
升温稀释
×
c1=n
升温稀释
×
(n
检测稀释
或1/j
加标
)
×
c2,如果优化升温程序时没有稀释,则n
升温稀释
=1;
[0203]
若3倍测定下限≤c1<标准曲线次高点浓度,对平行样稀释处理得到c2,稀释倍数为n
检测稀释
,2倍测定下限≤c2;如果c1=(95~105%)n
检测稀释
×
c2,则视为原子化期间没有干扰;c
试样最终测定结果
=n
升温稀释
×
c1=n
升温稀释
×n检测稀释
×
c2,如果优化升温程序时没有稀释,则n
升温稀释
=1;
[0204]
若标准曲线次高点浓度≤c1,对平行样两次稀释处理得到溶液cc1和cc2(cc1浓度>cc2浓度),稀释倍数分别为:n
检测稀释1
和n
检测稀释2
;同时满足:3倍测定下限≤cc1<标准曲线次高点浓度,3倍测定下限≤cc2<标准曲线次高点浓度;如果n
检测稀释1
cc1=(95~105%)n
检测稀释2
×
cc2,则视为原子化期间没有干扰;c
试样最终测定结果
=n
升温稀释
×n检测稀释1
×
cc1=n
升温稀释
×n检测稀释2
×
cc2,如果优化升温程序时没有稀释,则n
升温稀释
=1;
[0205]
若检出限≤c1<测定下限,对平行样加标或稀释处理得到c2溶液,加标倍数不得低于1.25倍,不得超过3倍,加标倍数为j
加标
;稀释倍数不得低于1.25倍,不得超过2倍,稀释倍数为n
检测稀释
;稀释得到c2溶液浓度应不小于检出限;如果c1=(75~125%)(1/j
加标
)
×
c2,则视为原子化期间没有干扰;此时,c
试样最终测定结果
=n
升温稀释
×
c1=n
升温稀释
×
(1/j
加标
)
×
c2,其中j
加标
∈[1.25,3],如果优化升温程序时没有稀释,则n
升温稀释
=1;
[0206]
若c1<检出限,且优化升温程序时没有执行稀释,即n
升温稀释
=1,此时,不必须执行富集程序,判断原子化是否存在干扰:若测定c1溶液7次,t
单侧检验(7次
,
0.95)
×s7次c1溶液测定浓度值
≤mdl检出限,即为无干扰;测定结果为未检出;
[0207]
若c1<检出限,且校正升温程序时执行稀释,即1<n
升温稀释
≤10,此时,必须执行富集程序直至富集后浓度大于检出限,富集倍数记为f
富集
,1<f
富集
≤10,c
试样最终测定结果
=n
升温稀释
×
(1/f
富集
)
×
c1;富集倍数如果超过10倍还未大于检出限,停止富集,试样检测结果为未检出;判断原子化是否存在干扰:若测定未富集前c1溶液7次,t
单侧检验(7次,0.95)
×s7次未富集前c1溶液测定浓度值
≤mdl检出限;同时富集两次,即为cc1和cc2,如果cc1=(70~130%)cc2;满足以上两条,则视为无原子化干扰。
[0208]
在本发明中,如果优化的干燥、灰化升温程序时间过长,而待测样品吸光度相对较高(对应的浓度也高),适合进行稀释或者待测样品吸光度已经高出了标准曲线最高浓度范围,则检测过程中按照检测要求必须稀释待测样品,此时可以按照稀释后的待测样品优化升温程序,适当降低升温程序时间。如果是待测样品浓度高,按照正常检测要求进行稀释,为检测稀释,用n
检测稀释
表示,检测稀释不需要富集程序;但如果待测样品浓度没有超过标准曲线最高浓度范围,只是单纯为了降低优化升温程序时间而进行的稀释,为升温稀释,用n
升温稀释
表示,n
升温稀释
≤10倍,但以不稀释为最佳,如果稀释,稀释应不大于10倍,特殊情况不得大于20倍;那么后续绘制完标准曲线,正式检测待测样品时候,在待测样品浓度小于检出限时候考虑富集程序;按照优化后的升温程序测定待测样品,记为c1(如果优化升温程序时没有稀释待测样品,即为原待测样品,如果优化升温程序时执行稀释,则为升温程序稀释后的待测样品)。
[0209]
在本发明中,石墨炉富集程序为富集次数=f,总进样次数=富集次数 1=f 1,富集倍数f=f 1,操作程序为,分别在每次进样后执行石墨炉升温程序中的干燥、灰化,然后继续进样,全部进样后,一起执行原子化和清除,理论上相当于富集f 1倍。
[0210]
在本发明中,原子吸收检测法指的是检测大多数金属和少数非金属元素,分为火焰法和石墨炉法,首先通过已知浓度的标准溶液建立,浓度与吸光度的一元线性方程,然后通过测量未知试样的吸光度,间接在标准曲线上求得未知试样的金属浓度值。
[0211]
本发明在石墨炉原子吸收环境样品检测领域应用,校正环境试样中基体对原子吸收测定中带来的干扰,为了实现基体效应精密化校正方法,需要基于最小二乘法的一元线性回归标准曲线的线性范围内检验,保证最小二乘法的一元线性回归标准曲线是一条直线,才能校正原子化过程中的基体效应干扰。基体效应产生的分子吸收干扰和光散射干扰如果发生在原子化信号前的干燥和灰化,在石墨炉原子吸收光谱仪实时信号中可以显示,可以通过单独优化石墨炉升温程序或者结合优化石墨炉升温程序 稀释富集程序的方法消除;基体效应产生的分子吸收干扰和光散射干扰如果发生在原子化过程中,可以利用基体干扰检查方法通过稀释富集程序校正原子化过程中的基体效应干扰。
[0212]
实施例
[0213]
石墨炉(北京普析通用仪器有限责任公司提供的tas-990原子吸收分光光度计)检测铬元素,排除基体效应干扰:
[0214]
本发明为了验证实施效果,自制基体效应干扰溶液,同时也是加标溶液:
[0215]
未加铬基体试样制备:200ml超纯水 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体);
[0216]
加铬基体试样制备500μg/l:200ml超纯水 0.1ml铬标液(1000mg/l) 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体);
[0217]
加铬基体试样制备250μg/l:200ml超纯水 0.05ml铬标液(1000mg/l) 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体)。
[0218]
调整石墨炉仪器参数:
[0219]
铬元素寻峰,特征峰为357.87nm,寻峰结果为357.88nm,满足
±
0.15nm要求;校核能量为100.9%,放置石墨管,调整至能量为80%,再次校核能量为100.5%。
[0220]
铬元素预设升温程序:干燥100℃(5s 10s);灰化1300℃(5s 10s);原子化2000℃(0s 2s);清除2100℃(0s 1s)。
[0221]
空烧石墨管11次:11次空烧石墨管的吸光度值为0.000~0.008,实时信号理想,满足“2000℃≤t
原子化温度
<2200℃,a
空烧11次最大值
≤0.010”的要求。
[0222]
测定标准曲线空白溶液7次:标准曲线空白溶液为超纯水,7次标准曲线空白溶液测定吸光度值为0.001~0.009,实时信号理想,满足“2000℃≤t
原子化温度
<2200℃,a
标准曲线空白溶液21次最大值
≤0.012”和“a
单次标准曲线空白值
∈a
n次标准曲线空白平均值
±
t
双侧检验(n-1,0.99)
×sn次标准溶液空白值吸光度标准偏差”的要求:
[0223][0224]
优化升温程序消除原子化前干扰(干燥/灰化):
[0225]
优化升温程序:三步干燥 三步灰化 原子化 清除,升温程序中总时长为140s,其中0s~45s为干燥,46s~135s为灰化,136s~138s为原子化(起峰),139s~140s为清除。
[0226]
为了缩短升温时间,优化升温程序将有干扰的未加铬试样稀释10倍~20倍,未经稀释的试样吸光度-时间实时信号曲线如图3所示,干扰严重;稀释后的试样的吸光度-时间实时信号如图4所示。(本发明目的是校正基体干扰,自己配置的未添加铬有干扰的试剂就相当于分离的基体溶液,可以看出基体是有严重干扰的,进而证明本发明的方法可行,而且不理想的abs-t实时信号都是来自这个自制的干扰溶液,有干扰的试剂都是分析纯,没有添加铬不代表试剂中就不含有铬,石墨炉检出限是极低的,所以未添加铬元素的无铬试样中检出了铬)
[0227]
优化升温程序:原子化峰型基体干扰消除,为了缓解原子化峰拖尾现象,清除温度定为原子化基础上加200℃,优化的升温程序及参数如下:
[0228]
序号升温程序功能执行温度(℃)升温时间(s)保持时间(s)氩气1干燥180510开2干燥2100510开3干燥3120510开4灰化15005010开5灰化21000510开6灰化31300510开7原子化200003关8清除220002开
[0229]
配制标准曲线溶液,测定标准曲线溶液并检验标准曲线:
[0230]
02.510203040506070800.0020.0260.1010.2010.2740.3590.4660.5550.6720.735
[0231]
按照上表中的数据进行标准曲线绘制如图5和图6所示,所得标准曲线为:
[0232]
c(ng/ml)=107.97a-0.361,a=0.0092c(ng/ml) 0.0039,r=0.99919
[0233]
计算检出限和测定下限:
[0234][0235]
检出限确定方法:
[0236]
按照样品分析的全部步骤,重复n(n≥7)次空白试验,将各测定结果换算为样品中的浓度或含量,计算n次平行测定的标准偏差,检出限=t
(n-1,0.99)
×sn次标准溶液空白值吸光度对应浓度标准偏差
;t值表为:
[0237]
平行测定次数(n)自由度(n-1)t
(n-1,0.99)
763.143872.998
982.8961092.82111102.76416152.60221202.528
[0238]
检验标准曲线:
[0239][0240][0241]
进一步检验c=0时,即标准曲线截距v:
[0242]
t0=0.259<t
单侧(7,0.95)
=1.895,合格。
[0243]
检测待测样品及原子化干扰判定:
[0244]
未加铬基体试样制备:200ml超纯水 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体);
[0245]
加铬基体试样制备500μg/l:200ml超纯水 0.1ml铬标液(1000mg/l) 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体);
[0246]
加铬基体试样制备250μg/l:200ml超纯水 0.05ml铬标液(1000mg/l) 1.5g碳酸钠(固体) 1.0g氢氧化钠(固体) 0.4g氯化镁(固体)。
[0247]
假设并不清楚以上试样的真实浓度,用以下结果验证本发明方法(每个试样都稀释10倍,为了配合优化升温程序而设定的稀释倍数):
[0248][0249][0250]
因为本实施例中基体试样均为自行设计,故用计算的没有基体效应干扰试样最终检测结果计算回收率,进一步验证准确性:
[0251][0252]
因为未加铬基体试样中虽然没特意加铬但已经检出了铬元素,为了进一步说明500μg/l加铬基体试样和250μg/l加铬基体试样检测结果准确性,可从加铬基体试样中去除对应倍数的基体试样。
[0253][0254][0255]
本发明在石墨炉原子吸收检测领域应用,校正环境试样中基体对原子吸收测定中带来的干扰,为了实现基体效应精密化校正方法,首先需要提出检测中基于最小二乘法的一元线性回归标准曲线的线性范围检验,必须保证最小二乘法的一元线性回归标准曲线是一条直线,才能校正原子化过程中基体效应干扰;基体效应产生的分子吸收干扰和光散射干扰如果发生原子化信号前的干燥和灰化,在石墨炉原子吸收光谱仪实时信号中可以显示,可以通过单独优化石墨炉升温程序或者结合优化石墨炉升温程序 稀释富集程序的方法消除;基体效应产生的分子吸收干扰和光散射干扰如果发生原子化过程中,可以利用基体干扰检查方法通过稀释富集程序校正原子化过程中基体效应干扰。
[0256]
虽然已参考本发明的特定实施例描述并说明本发明,但是这些描述和说明并不限制本发明。所属领域的技术人员可清晰地理解,在不脱离如由所附权利要求书定义的本发明的真实精神和范围的情况下,可进行各种改变,以使特定情形、材料、物质组成、物质、方法或过程适宜于本技术的目标、精神和范围。所有此类修改都意图在此所附权利要求书的范围内。虽然已参考按特定次序执行的特定操作描述本文中所公开的方法,但应理解,可在不脱离本发明的教示的情况下组合、细分或重新排序这些操作以形成等效方法。因此,除非本文中特别指示,否则操作的次序和分组并非本技术的限制。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
