检测继发性肝癌的生物标志物
发明领域
1.本发明涉及诊断领域,更具体地说,涉及用于检测继发性肝癌的肽生物标志物领域。更具体地说,本发明涉及羟基化的胶原天然发生肽(nop),其实现对象中继发性肝癌的检测。本发明还涉及此类生物标志物的用途。本发明还涉及治疗,更具体地说,涉及对根据本文所述的方法分型的患有继发性肝癌的对象的治疗。
背景技术:
2.结直肠癌是荷兰第三常见的癌症。2010年至2017年期间,每年有10000-16000名新患者确诊,5000名患者死亡(荷兰综合癌症中心,2018)。在西方世界,原发性肿瘤治愈性手术后患者发生肝转移(结直肠癌肝转移;crlm)的概率为20-40%(riihimaki等人,sci rep.6:29765(2016);figueredo等人,bmc cancer;3:26(2003);al-asfoor等人.,cochrane database syst rev.cd006039(2018);gregoire等人,eur j surg oncol.36:568-74(2010);grossmann等人,colorectal dis.9:787-92(2007))。
3.治愈性手术后,患者将接受为期5年的强化随访计划,包括定期的计算机断层(ct)扫描、超声研究和癌胚抗原(cea)血清测量,以筛查crlm(grossmann等人,colorectal dis.9:787-92(2007);locker等人,j clin oncol.,24:5313-27(2006);pita-fernandez等人,ann oncol.,26:644-56(2015))。
4.此前有报道称,血清cea和尿液中特定胶原天然发生肽(nop)的组合可用于检测crlm(灵敏度85%,特异性84%)(lalmahomed等人,am j cancer res.,,6:321-30(2016))。即使这种组合的灵敏度和特异性高于目前使用技术的灵敏度和特异性,它们仍然可以改进,这在考虑在临床环境中的应用时是有益的。
5.本领域需要更多可用于检测继发性肝癌(如crlm)的生物标志物。
6.本发明的目的是提供此类生物标志物,尤其是羟基化胶原天然发生肽的生物标志物。另外,本发明的目的是改进继发性肝癌检测中的cea血清测量。在癌症患者的原发肿瘤手术切除后的随访期内,可靠检测的实现可大大减少手术次数。
7.发明概述
8.因此,本发明在一个方面提供了一种对对象是否存在继发性肝癌进行分型的方法,包括以下步骤:-在包含来自对象的肽的样品中测量以下肽水平:(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽,或(ii)包含与seq id no:1、seq id no:2或seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽;和-根据测量的肽水平,对所述对象是否存在继发性肝癌进行分型。另外,本发明在一个方面提供了一种对对象是否存在继发性肝癌进行分型的方法,包括以下步骤:-在包含来自对象的肽的样品中测量(i)包含seq id no:1的氨基酸序列的肽或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽;和/或(ii)包含seq id no:4氨基酸序列的肽或包含与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的肽水平;和-根据测量的肽水平,对所述对象是否存在继发性肝癌进行分型。
9.发明人意外地发现了三种新的羟基化胶原天然发生肽(nop)(羟基化胶原nop肽“gnd”(seq id no:1),羟基化胶原nop肽“gpp”(seq id no:2)和羟基化胶原nop肽ger(seq id no:4)),可有利地用于检测患有或曾经患有癌症(如结直肠癌)的对象中的继发性肝癌(表3和表5)。
10.此外,发明人确定,组合使用seq id no:1肽和癌胚抗原(cea)可以更好地检测患有或曾经患有癌症的对象中的继发性肝癌(表4和图3)。与之前基于羟基化胶原nop肽“agp”(seq id no:3)和cea的模型相比,这种组合被证明具有显著更高的预测能力。在作为独立收集样品组的验证组中,灵敏度从80%增加到92%,而特异性从80%增加到90%。这种新组合的灵敏度比目前使用的技术(从57%到70%不等)至少高15-20%。特异性与这些技术相当,从90%到96%不等。总的来说,这种新组合的性能优于目前使用的技术。这种特性在临床上是有益的,因为可以预见crlm等继发性肝癌的早期发现,并降低医疗成本。当将nop肽ger与cea组合使用时,观察到类似的有益效果(实施例2,表5)。
11.在所述分型方法的一个优选实施方式中,将对象分型的方法还包括以下步骤:-将所述测量的肽水平与参考肽水平进行比较:(i)包含seq id no:1、seq id no:2、或seq id no:4的氨基酸序列的所述肽或(ii)包含与seq id no:1、seq id no:2、或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的所述肽;和-根据测量的肽水平和参考肽水平的比较,对所述对象是否存在继发性肝癌进行分型。
12.在所述分型方法的优选实施方式中,所述分型方法还包括以下步骤:将所述测量的肽水平与参考肽水平进行比较:(i)包含seq id no:1氨基酸序列的所述肽,或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的所述肽;和/或(ii)包含seq id no:4氨基酸序列的所述肽或包含与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽;和-根据测量的肽水平和参考肽水平的比较,对所述对象是否存在继发性肝癌进行分型。
13.在所述分型方法的另一优选实施方式中,其中在本发明的分型方法中使用(联用)包含seq id no:1的肽和包含seq id no:4的肽时,所述分型方法还包括以下步骤:将所述测量的肽水平与参考肽水平进行比较:(i)包含seq id no:1氨基酸序列的所述肽,或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的所述肽;和/或(ii)包含seq id no:4氨基酸序列的所述肽或包含与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的所述肽;和-根据测量的肽水平和参考肽水平的比较,将所述对象分型成是否存在继发性肝癌。
14.在所述分型方法的另一优选实施方式中,对象是患有或曾患有原发性癌症,优选原发性结直肠癌的对象。
15.在所述分型方法的另一优选实施方式中,所述对象是患有原发性癌症,优选原发性结直肠癌的对象,并且其中原发性癌症被手术切除。
16.在所述分型方法的另一优选实施方式中,包含来自对象的肽的所述样品是来自所述对象的体液样品,优选尿液样品。
17.在所述分型方法的另一优选实施方式中,所述包含肽的样品是包含胶原天然发生肽(nop)的样品。
18.在所述分型方法的另一优选实施方式中,在包含来自未患癌症或未曾患有癌症的
参考对象的肽的样品中测量所述参考肽水平。
19.在所述分型方法的另一优选实施方式中,当所述肽水平与所述参考肽水平相比增加时,所述对象或所述样品被分型为患有继发性肝癌。
20.在所述分型方法的另一优选实施方式中,所述方法还包括以下步骤:在包含来自所述对象的蛋白质的样品中测量癌胚抗原(cea)蛋白质水平;
[0021]-根据所测得的肽水平和所测得的cea蛋白水平,对所述对象是否存在继发性肝癌进行分型。
[0022]
在包括测量cea蛋白质水平的所述分型方法的另一优选实施方式中,来自所述对象的所述蛋白质是来自所述对象血液样品的蛋白质。
[0023]
在包括测量cea蛋白水平的所述分型方法的另一优选实施方式中,通过使用如下所示的公式,对所述对象或所述样品进行是否存在继发性肝癌的分型:
[0024][0025]
其中“gnd”是seq id no:1的肽或含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的所测肽水平,并且其中“gnd”以质谱所测峰(曲线)下面积表示;而“cea”是所测cea蛋白质水平,以ng cea/ml血清表示。
[0026]
在又一个优选实施方式中,所述分型方法包括测量cea蛋白水平,通过采用下式对所述对象或所述样品进行是否存在继发性肝癌的分型:
[0027][0028]
其中,“ger”是seq id no:4的肽或包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的测量肽水平,其中“ger”表示成质谱测量的峰(曲线)下面积;“cea”是测得的cea蛋白水平,以ng cea/ml血清计。
[0029]
在包括测量cea蛋白质水平的所述分型方法的另一优选实施方式中,所述方法还包括以下步骤:—将所述测量的蛋白质水平与参考cea蛋白质水平进行比较;和-根据(i)测量的肽水平和参考肽水平的比较,以及(ii)测量的cea蛋白水平和参考cea蛋白水平的比较,对所述对象是否存在继发性肝癌进行分型。
[0030]
在包括测量cea蛋白水平的所述分型方法的另一优选实施方式中,在包含来自未患癌症或未患过癌症的参考对象的蛋白质的样品中测量所述参考cea蛋白水平。
[0031]
在包括测量cea蛋白水平的所述分型方法的另一优选实施方式中,当(i)所述肽水平与所述参考肽水平相比增加,且(ii)所述cea蛋白水平与所述参考cea蛋白水平相比增加时,所述对象或所述样样品分型为患有继发性肝癌。
[0032]
在另一方面,本发明提供了(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的用途,用于对对象是否存在继发性肝癌进行分型。本发明还提供了(i)包含seq id no:1氨基酸序列的肽或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽或(ii)包含seq id no:4氨基酸序列的肽或与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的用途,用于对对象是否患有继发性肝癌进行分型。
[0033]
在一个实施方式中,所述用途涉及(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽与癌胚抗原(cea)的组合,用于对对象是否患有继发性肝癌进行分型。在此类用途的实施方式中,包含seq id no:1氨基酸序列的肽或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽和包含seq id no:4氨基酸序列的肽或与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的组合,与癌胚抗原(cea)组合用于对对象是否存在继发性肝癌进行分型。
[0034]
在所述分型方法或所述用途的优选实施方式中,所述继发性肝癌是结直肠癌肝转移(crlm)。
[0035]
在另一方面中,本发明提供包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽,或包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽。在本文所述的方法中,尤其优选包含或由seq id no:1或seq id no:4的氨基酸序列组成的肽,或包含或由与seq id no:1或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列组成的肽。因此,本发明还提供了包含或由seq id no:1的氨基酸序列组成的肽,或包含或由与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列组成的肽,或包含或由seq id no:4氨基酸序列组成的肽,或包含或由与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列组成的肽。
[0036]
在所述肽的优选实施方式中,所述肽包含或由seq id no:1或seq id no:4的氨基酸序列组成。
[0037]
在另一方面,本发明提供了一种针对继发性肝癌的标准护理治疗剂,用于治疗根据本发明分型方法分型为患有继发性肝癌,优选crlm的对象。
[0038]
在另一方面,本发明提供了一种针对继发性肝癌的标准护理治疗剂的用途,用于制造治疗患有继发性肝癌的对象的药物,其中根据本发明的分型方法,所述对象被分型为患有继发性肝癌。
[0039]
在另一方面,本发明提供了一种治疗患有继发性肝癌的对象的方法,包括以下步骤—进行本发明的分型方法;-当所述对象被分型为患有继发性肝癌时,针对继发性肝癌施用治疗有效量的标准护理治疗剂。
[0040]
在另一方面,本发明提供一种测量肽水平的方法,包括以下步骤:-可任选地,提供包含来自对象的肽的样品;-在包含来自对象的肽的样品中测量包含seq id no:1、seq id no:2和/或seq id no:4的氨基酸序列的肽,或包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的肽水平。在该方法的一个实施方式中,可测量以下组合的肽水平:(i)包含seq id no:1氨基酸序列的肽,或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽,和(ii)包含seq id no:2氨基酸序列的肽,或包含与seq id no:2氨基酸序列具有至少90%序列相同性的氨基酸序列的肽。另外,可测量以下组合的肽水平:(i)包含seq id no:1氨基酸序列的肽,或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽,和(ii)包含seq id no:4氨基酸序列的肽,或包含与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽。
[0041]
本发明还提供了一种测量肽水平的方法,包括以下步骤:-任选地,提供包含来自
对象的肽的样品;-在包含来自对象的肽的样品中测量以下肽水平:(i)包含seq id no:1氨基酸序列的肽或包含与seq id no:1氨基酸序列具有至少90%序列相同性的氨基酸序列的肽,和/或(ii)包含seq id no:4氨基酸序列的肽或包含与seq id no:4氨基酸序列具有至少90%序列相同性的氨基酸序列的肽。
[0042]
在上述用于测量肽水平的方法中,该方法可进一步包括以下步骤:-可任选地,提供包含来自所述对象的蛋白质的样品;-在包含来自所述对象的蛋白质的样品中测量癌胚抗原(cea)蛋白质水平。在用于测量肽水平的所述方法的优选实施方式中,所述包含肽的样品是尿液样品,所述包含蛋白质的样品是血液样品,优选血清或血浆样品。
具体实施方式
[0043]
本文使用的术语“分型”是指根据是否存在继发性肝癌(如crlm)对对象进行区分或分级。该术语还包括对继发性肝癌(如crlm)的诊断或检测。优选地,在本发明的分型方法中,分型基于(i)所测量的肽水平和(ii)所述肽的参考肽水平的比较,所述肽包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列,或所述肽包括与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%的序列相同性的氨基酸序列。
[0044]
本文使用的术语“对象”指哺乳动物,更优选灵长类,最优选人类。该术语包括患有或患过恶性肿瘤的患者,优选其中恶性肿瘤是原发性(恶性)肿瘤,例如选自乳腺癌;结直肠癌;肾癌;食管癌;肺癌;皮肤癌;卵巢癌;子宫癌,包括子宫内膜癌和子宫肉瘤;脑癌;胰腺癌和胃癌。众所周知,这些癌症类型的原发性(恶性)肿瘤可以扩散到肝脏。
[0045]
优选地,在本发明的分型方法中,对象是患有或患过原发性(恶性)肿瘤的患者。更优选地,对象是通过手术切除原发性(恶性)肿瘤的患者。更优选地,对象是接受(或经历)原发性(恶性)肿瘤治愈性手术切除的患者。优选对象在手术切除原发性(恶性)肿瘤后尚未发生继发性癌症,如转移。或者,本文所述的对象是患有或患过原发性(恶性)肿瘤且有发展为继发性活癌的风险的患者。原则上,所有接受原发性(恶性)肿瘤手术切除的癌症患者都有发生继发性肝癌的风险。
[0046]
优选地,在本发明的分型方法中,对象是患有或患过原发性(恶性)肿瘤的患者,所述原发性(恶性)肿瘤是结直肠癌。更优选地,对象是通过手术切除原发(恶性)肿瘤的患者,所述原发性(恶性)肿瘤是结直肠癌。甚至更优选地,对象是接受(或经历)原发性(恶性)肿瘤的治愈性手术切除的患者,所述原发性(恶性)肿瘤是结直肠癌。优选对象在手术切除原发性(恶性)肿瘤(结直肠癌)后,尚未发生继发性癌症,包括转移。或者,本文所述的对象是患有或患过原发性(恶性)肿瘤且有发展为继发性肝癌的风险的患者,所述原发性(恶性)肿瘤是结直肠癌。原则上,所有接受原发性(恶性)肿瘤手术切除的癌症患者都有发展为继发性肝癌的风险,所述原发性(恶性)肿瘤是结直肠癌。
[0047]
本文使用的术语“继发性肝癌”包括指存在于肝脏但起源于身体其他部位的癌症。例如,癌症可能起源于结直肠癌(原发性恶性肿瘤),结直肠癌细胞可能扩散或转移到肝脏,形成结直肠起源的肝癌(继发性肝癌)。继发性肝癌可能起源于癌症,包括但不限于:乳腺癌;肺癌;结直肠癌;脑癌;肾癌;食管癌;皮肤癌;卵巢癌;子宫癌,包括子宫内膜癌和子宫肉瘤;胰腺癌和胃癌。最优选继发性肝癌是结直肠癌肝转移(crlm)。
[0048]
术语“原发性肿瘤”和“原发性癌症”在本文中互换使用。
[0049]
本文使用的术语“结直肠癌肝转移”或“crlm”指结直肠癌患者的肝脏中形成转移的公认临床指征。肝脏是结直肠癌患者最常见的转移部位。
[0050]
本文使用的术语“样品”指包含来自对象的肽和/或蛋白质的样品。样品优选是体液样品。此类样品包括但不限于痰、血液、血清、血浆、尿液、腹膜液和胸膜液。最优选地,当要测量本文所述肽的肽水平时,所述样品是尿液样品,且当要测量cea蛋白水平时,所述样品是血清样品。获取此类样品完全在本领域技术人员的常识范畴内。
[0051]
优选地,样品是经处理或制备的样品,例如经处理或制备用于肽或蛋白质水平测量步骤的尿液样品。这种处理或制备是常规的,例如可以包括步骤,其中(胶原)天然发生肽(nop)与样品的其他成分,包括小分子、盐和蛋白质分离。例如,可以使用蛋白质回收柱(如mrp c-18 hi recovery蛋白质柱(4.6
×
50mm)(安捷伦,荷兰阿姆斯特丹)与液相色谱相结合来实现这一点。随后,可收集、干燥、重构(例如,使用水等水性液体,包括0.1%三氟乙酸(tfa)的水溶液)和/或使用肽或蛋白质水平测量技术(包括质谱)分析肽组分,优选nop组分。
[0052]
本文使用的术语“蛋白质”和“肽”指氨基酸残基(氨基酸序列)的聚合物。这些术语还包括修饰的肽或蛋白质,例如稳定同位素标记的(sil)肽。优选地,当本文中提到肽时,其指如本文所述的本发明的羟基化胶原nop肽。优选地,当提到蛋白质时,指的是cea。
[0053]
如本文所使用的术语“天然发生肽”或“nop”包括对象中天然发生的肽。优选地,这种nop是胶原nop,即胶原衍生的nop,更优选羟基化的胶原nop,甚至更优选羟基化胶原nop,其包含seq id no:1、seq id no:2或seq id no:4中任一项的氨基酸序列,以及最优选包含seq id no:1或seq id no:4的氨基酸序列的羟基化的胶原nop。seq id no:4的nop ger肽的优点是不含羟基化的赖氨酸残基。获得具有羟基化的赖氨酸残基的参考用sil nop肽是昂贵的。术语“nop”包括羟基化nop,例如羟基化胶原nop。
[0054]
在根据本发明的分型方法中使用的肽是羟基化的。
[0055]
羟基化是一种将羟基引入氨基酸的过程,并由称为羟化酶的酶实现。肽中要羟基化的主要残基是脯氨酸,但其他氨基酸残基如赖氨酸也可以羟基化。羟基化主要发生在γ-c原子上,形成羟脯氨酸(hyp)。在某些情况下,脯氨酸可能在换而其β-c原子上羟基化。赖氨酸也可以在其δ-c原子上羟基化,形成羟基赖氨酸(hyl)。这些反应可分别由多亚基酶脯氨酰4-羟化酶、脯氨酰3-羟化酶和赖氨酰5-羟化酶催化。此外,半胱氨酸、苯丙氨酸、酪氨酸也是可能羟基化的氨基酸的例子。
[0056]
在根据本发明的分型方法中,测定(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的肽水平。优选地,序列相同性为至少91%、92%、93%、94%、95%、97%、98%或至少99%。优选地,包含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:1中定义的至少1、2、3、4、5或6个羟基化氨基酸残基。更优选地,包含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:1中定义的所有6个羟基化氨基酸残基,因此具有与seq id no:1中定义的相同羟基化模式。优选地,包含与seq id no:2的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:2中定义的至少1、2、3或4个羟基化氨基酸残基。更优选地,包含与seq id no:2的氨基酸序列具有至少90%序列相同性的氨基
酸序列的肽具有seq id no:2中定义的所有4个羟基化氨基酸残基,因此具有与seq id no:2中定义的相同羟基化模式。优选地,包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:4中定义的至少1、2、3、4、5、6或7个羟基化氨基酸残基。更优选地,包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:4中定义的所有7个羟基化氨基酸残基,因此具有与seq id no:4中定义的相同羟基化模式。
[0057]
seq id no:1的肽在六个位置进行羟基化,即位置15(脯氨酸)、位置17(脯氨酸)、位置18(脯氨酸)、位置24(脯氨酸)、位置27(脯氨酸)和位置30(赖氨酸)。优选地,在位置15处,脯氨酸为4-羟脯氨酸。优选地,在位置17处,脯氨酸为3-羟脯氨酸。优选地,在位置18处,脯氨酸为4-羟脯氨酸。优选地,在位置24处,脯氨酸为4-羟脯氨酸。优选地,在位置27处,脯氨酸为4-羟脯氨酸。优选地,在位置30处,赖氨酸为5-羟赖氨酸。最优选地,seq id no:1的肽在15位具有4-羟脯氨酸;17位为3-羟脯氨酸;18位为4-羟脯氨酸;24位为4-羟脯氨酸;27位为4-羟脯氨酸;在30位为5-羟赖氨酸。
[0058]
seq id no:2的肽在四个位置羟基化,即位置8(赖氨酸)、位置9(脯氨酸)、位置15(脯氨酸)和位置21(脯氨酸)。优选地,在位置8处,赖氨酸为5-羟赖氨酸。优选地,在位置9处,脯氨酸为4-羟脯氨酸。优选地,在位置15处,脯氨酸为4-羟脯氨酸。优选地,在位置21处,脯氨酸为4-羟脯氨酸。最优选地,seq id no:2的肽在第8位为5-羟基赖氨酸;第9位为4-羟脯氨酸;第15位为4-羟脯氨酸;第21位为4-羟脯氨酸。
[0059]
seq id no:4的肽在七个位置进行羟基化,即位置6(脯氨酸)、位置9(脯氨酸)、位置15(脯氨酸)、位置21(脯氨酸)、位置24(脯氨酸)、位置33(脯氨酸)和位置35(脯氨酸)。优选地,位置6、9、15、21、24、33和35处的一个或多个羟基化脯氨酸为4-羟脯氨酸(4hyp)。更优选地,位置6、9、15、21、24、33和35处的所有羟脯氨酸均为4-羟脯氨酸(4hyp)。
[0060]
术语“%序列相同性”在本文中定义为在对齐序列并在必要时可选地引入缺口以实现最大序列相同性百分数后,氨基酸序列中与所关注的氨基酸序列中的氨基酸相同的氨基酸百分数。用于比对的方法和计算机程序在本领域中是众所周知的。序列相同性是基于感兴趣的氨基酸序列的几乎整个长度上计算的,优选整个(完整)长度。本领域技术人员理解,将一个氨基酸序列中的连续氨基酸残基与另一个氨基酸序列中的连续氨基酸残基进行比较。此处使用的术语“%序列相同性”,要求仅当目标氨基酸序列中的目标氨基酸残基也在某位置(参考序列(即,seq id no:1、seq id no:2或seq id no:4)中在该位置处具有羟基化的氨基酸残基)被羟基化时,才认为该目标氨基酸残基与参考序列中的羟基化的氨基酸残基相同。
[0061]
本领域技术人员拥有大量已知的方法和手段,用于测量样品中的肽或蛋白质水平,包括测量相对或绝对肽或蛋白质浓度,和/或纵向(同一患者随时间多次取样)或横向(每个患者一个时间点测量)测量。
[0062]
肽或蛋白质分析的示范方法包括但不限于高效液相色谱法(hplc);质谱(ms),优选设置为ms/ms模式;基于lc-ms的肽谱分析,优选hplc-ms,优选设置为ms/ms模式(鸟枪模式/数据相关采集(dda)、数据独立采集(dia)、靶向模式(选择反应监测(srm)、平行反应监测(prm)和多重反应监测(mrm))等。优选使用prm。在本发明中,这些方法用于定量检测所分析样品中是否存在肽或蛋白质,即评价或评估所分析样品中肽或蛋白质的实际量或相对丰
度。在这些实施方式中,定量检测可以是绝对或相对的。因此,术语“水平”或“定量”在用于定量或测量样品中的肽或蛋白质水平时,可以指绝对定量或相对定量。绝对定量可通过纳入一种或多种已知浓度的对照分析物并且将靶标肽或蛋白质的测得水平与已知对照分析物相比照(例如通过产生标准曲线)来实现。或者,相对定量可通过将两种或更多不同肽或蛋白质的测得水平或测得量彼此比较从而得出两种或更多不同肽或蛋白质各自的相对量,例如相对于彼此的相对定量。此外,可使用来自一个或多个对照或参考样品的对照或参考值(或概况)确定相对定量。
[0063]
通过例如ms-ms对羟基化肽的片段化识别羟基的位置,即羟基化模式。确定羟基化模式的其他合适方法是测量任何羟基化肽相互作用的任何方法,例如免疫分析、多重分析、竞争分析、珠、载体芯片、阵列、棒、柱。一种合适的方法可以是免疫分析、多重分析、竞争分析和选择反应监测(srm)。可通过化学发光和/或荧光等任何可用的适当手段指示检测。
[0064]
当ms被用作肽测量工具时,这些肽序列的优点是可以容易地鉴定对应这些肽生成的ms图谱中的峰,并归因于本文所述的羟基化nop肽生物标志物。这种肽的ms峰是其肽水平的量度。然而应该理解的是,肽水平可以通过许多其他方法测量。
[0065]
在本文所述的分型方法中,根据所测得的肽水平对所述对象进行是否存在所述继发性肝癌的分型属于本领域技术人员的常规能力范围。例如,从本技术可以看出,与健康个体相比,患有继发性肝癌的对象样品中本文所述的肽的肽水平增加。该知识允许技术人员设置其认为合适的阈值水平。
[0066]
本发明的一种分型方法还可包括步骤:将测量的肽水平与(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽,或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的参考肽水平进行比较。
[0067]
在测量目标肽的肽水平,并且例如以概况或特征的形式提供此类肽水平数据之后,对肽水平进行分析或评估,以确定所述对象是否分型为具有继发性肝癌。这种分析涉及将测量的肽水平与同一肽的参考肽水平进行比较。
[0068]
术语“参考肽水平”表示可用于解释如本文所述在对象样品中测量的肽水平的标准化肽水平(或标准化肽水平概况或特征,或总标准化肽水平)。
[0069]
适用于本发明的分型的参考肽水平可由技术人员以多种替代方式设置,此类参考肽水平的设置属于技术人员的公知常识。例如,在本发明的分型方法中,参考肽水平可以是参考样品中所述肽的参考肽水平,优选基于参考样品获得。参考样品可以是来自任何个体的样品,例如健康或患病个体,但优选来自健康对象,优选健康人对象的样品。例如,样品可以是健康肾脏捐献者的(尿液)样品,优选在器官捐献之前获得。此类样品可以是来自未患癌症(如结直肠癌)且未患过癌症(如结直肠癌)的健康对象的样品。或者,此类样品可以是来自患有或患过原发性癌症的,优选原发性结直肠癌的对象的样品,其原发性癌症尚未发展为继发性肝癌。与患有未(尚未)发展为继发性肝癌的原发性癌症(优选原发性结直肠癌)的对象相比,患有继发性肝癌的对象的seq id no:1、seq id no:2或seq id no:4的肽水平优选增加。
[0070]
了解与继发性肝癌相关的肽水平方向,本领域技术人员可以通过常规地应用代表与继发性肝癌肽水平相似或不同的适当参考肽水平来执行如本文所述的分型方法。优选
地,在本文所述的分型方法中,当所述测量的肽水平相对于所述肽的参考肽水平(其中所述参考肽水平来自健康对象)增加时,所述对象或其样品被分型为具有继发性肝癌。或者,当所述测量的肽水平与所述肽的参考肽水平(其中所述参考肽水平为健康对象)相比降低或等同时,则所述对象或其所述样品被归类为未患继发性肝癌(即不存在)。
[0071]
参考样品也可以是来自多个个体的合并肽样品,例如如上所述的健康个体。所述样品可来自10个以上个体、20个以上个体、30个以上个体、40个以上个体或50个以上个体。
[0072]
另一个有益的参考肽水平是用于区分继发性肝癌和非继发性肝癌的绝对肽水平。设置这样的绝对阈值蛋白质水平是技术人员的常识。
[0073]
在本发明的分型方法中,可以以多种方式对对象进行分型。在一种方法中,确定一个系数,该系数是对目标样品(即待调查样品)中肽水平的相似性或相异性的量度。对象或样品的分型可基于其与单个参考概况模板或多个参考概况模板的相似(异)性。通过确定与概况模板的相关性,可以设置总体相似性评分。相似性评分是对对象样品中肽水平与参考概况模板的平均相关性的量度。所述相似性评分可以(但不必)是 1(表示肽水平和所述概况模板之间高度相关)和-1(表示反向相关性)之间的数值。然后可设置阈值,以区分被分型为继发性肝癌或非继发性肝癌的样品。所述阈值是允许区分继发性肝癌和非继发性肝癌样品的任意值。如果采用相似性阈值,则优选将其设置为一个值,在该值下,可接受数量的患有继发性肝癌对象将评分为假阴性,而可接受数量的无继发性肝癌的对象将评分为假阳性。优选地,在用户界面、计算机可读存储介质、或本地或远程计算机系统计算机可读系统上显示或输出相似性评分。
[0074]
当具有不同的预测因子时,计算相似性评分的经典方法是线性逻辑回归,但有更多的统计和数据挖掘分类法可供技术人员用于计算相似性评分。例如,一个非限制性的例子是支持向量机,这是一种用于建立分类模型的统计学习方法(cristianini等人,《支持向量机和其他基于核的学习方法简介》(an introduction to support vector machines and other kernel-based learning methods),2000,剑桥大学出版社;vapnik,《统计学习理论的本质》(the nature of statistical learning theory.),1995年,纽约斯普林格;zhang等人,bmc生物信息学,7:197(2006))。
[0075]
根据本发明分型的方法还可包括步骤:-在包含来自对象的蛋白质的样品中测量癌胚抗原(cea)蛋白质水平;-基于测得的肽水平和测得的cea蛋白水平,对所述对象进行是否存在继发性肝癌的分型。
[0076]
出人意料的是,当使用(i)如本文所述测得的肽水平和(ii)血液cea蛋白水平时,可以显著提高本发明分型方法的预测能力。
[0077]
cea是一种通常在健康个体血液中检测不到的蛋白质。cea由某些癌症类型产生,通常用于监测胃肠道(gi)癌症(如结直肠癌)患者,以筛查原发肿瘤切除后继发性肝癌的发生。
[0078]
本领域技术人员非常了解测量与继发性肝癌相关的cea蛋白水平的合适方法和手段。本领域技术人员可采用上文所述的与肽水平测量相关的基于ms的蛋白质测量技术。此外,本领域技术人员可以使用可供临床使用的标准免疫分析,包括基于抗体或适配体的蛋白质定量分析(例如酶联免疫吸附分析(elisa)分析,例如多重或夹心elisa分析、蛋白质印迹、基于facs的蛋白质分析等)。用于检测cea蛋白水平的商业试剂盒是一般可得的。例如,
abcam,plc.出售“人类癌胚抗原elisa试剂盒(cd66e)(ab183365)”,这是一种用于定量夹心elisa分析的试剂盒。这种分析方法可以测量血液样品(如血清或血浆样品)中的cea蛋白水平。
[0079]
优选地,当测量cea蛋白水平时,对象的样品是血液样品,更优选血清样品。本领域技术人员理解,在根据本发明的分型方法中,可以获得对象的两个样品,例如用于测量本文所述肽的肽水平的第一尿液样品,以及用于测量cea蛋白水平的第二血液样品。
[0080]
在本发明的一种分型方法中,其中测量本文所述肽的肽水平并测量cea蛋白水平,可通过使用各种公式计算继发性肝癌的概率,其中一个可选且非限制性示例如下所示:
[0081][0082]
其中,“gnd”是seq id no:1的肽或包含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的测量肽水平,其中“gnd”表示成质谱测量的峰(曲线)下面积;且“cea”是测得的cea蛋白水平,以ng cea/ml血清计。为完整起见,该非限制性示例性公式写为1/(1 e-1x(-24.1476 3.0365xgnd 3.4647xcea)
)。该公式给出的输出值介于0和1之间。在技术人员的常规能力范围内,将阈值或截留值设置在0和1之间,以便区分健康和患病对象。可用于区分健康对象和患病对象的一个合适阈值或截留值为0.439。得分低于0.439的样品被视为健康样品,得分高于0.439的样品被视为患病样品。同样,技术人员有许多可供选择的常规方法和手段来计算这样的可能性值。以相同的方式,当测量seq id no:4的肽或包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的肽水平,并且测量cea蛋白质水平时,可通过使用各种公式计算继发性肝癌的概率,其中一个可选的非限制性示例如下所示:
[0083][0084]
其中,“ger”是seq id no:4的肽或包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的测量肽水平,其中“ger”表示成质谱测量的峰(曲线)下面积;“cea”是测得的cea蛋白水平,以ng cea/ml血清计。为完整起见,该非限制性示例性公式写为1/(1 e-1x(-20.62 3.05xcea 2.49xger)
)。
[0085]
本发明的分型方法还可包括以下步骤:-将所述测量的蛋白质水平与参考cea蛋白质水平进行比较;和-根据(i)测量的肽水平和参考肽水平的比较,以及(ii)测量的cea蛋白水平和参考cea蛋白水平的比较,对所述对象是否存在继发性肝癌进行分型。
[0086]
可按照上述与参考肽水平相关的相同方式设置适当的参考cea蛋白水平。可在包含来自健康个体的参考对象的蛋白质样品中测量所述参考cea蛋白质水平。可在包含来自未患有或未患过癌症(包括结直肠癌)的参考对象的蛋白质样品中测量所述参考cea蛋白质水平。
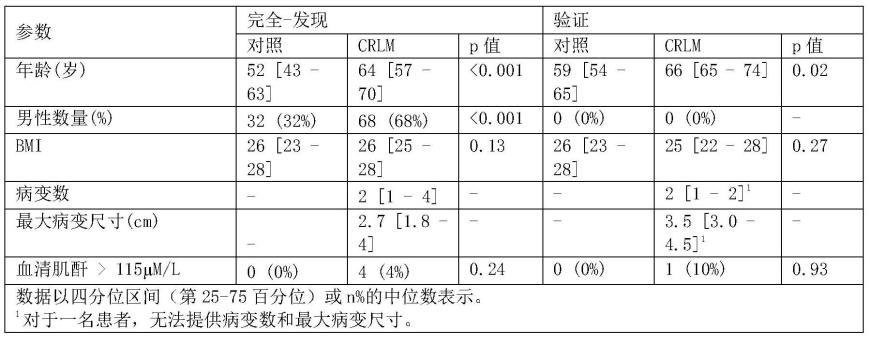
[0087]
本发明还提供了(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的用途,用于对对象是否患有继发性肝癌进行分型。本发明还提供了(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨
基酸序列的肽的用途,用于对对象是否患有继发性肝癌进行分型。优选地,这种用途是(i)如上所述的肽或肽水平与(ii)cea或cea蛋白质水平的组合用途。上述与分型方法有关的实施例也与本发明的用途有关。例如,所述用途中的肽优选如本文所述的肽。
[0088]
本发明还涉及根据本发明的分型方法定义的肽,包括包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽,或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽。优选地,序列相同性为至少91%、92%、93%、94%、95%、97%、98%或至少99%。优选地,包含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:1中定义的至少1、2、3、4、5或6个羟基化氨基酸残基。更优选地,包含与seq id no:1的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:1中定义的所有6个羟基化氨基酸残基,因此具有与seq id no:1中定义的相同羟基化模式。优选地,包含与seq id no:2的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:2中定义的至少1、2、3或4个羟基化氨基酸残基。更优选地,包含与seq id no:2的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:2中定义的所有4个羟基化氨基酸残基,因此具有与seq id no:2中定义的相同羟基化模式。优选地,包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:4中定义的至少1、2、3、4、5、6或7个羟基化氨基酸残基。更优选地,包含与seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽具有seq id no:4中定义的所有7个羟基化氨基酸残基,因此具有与seq id no:4中定义的相同羟基化模式。
[0089]
优选地,肽是分离的肽。肽优选至少部分纯化,并且可以具有至少40%、50%、60%、70%、80%、90%、91%、92%、93%、94%或至少95%的纯度。该肽还可以是化学合成的肽,任选地包含诸如荧光或稳定同位素标签(sil)之类的标签。
[0090]
本发明还提供了医疗方法,包括用于治疗根据上述分型方法被分型为患有继发性肝癌的对象的针对继发性肝癌的标准护理治疗剂。优选地,继发性肝癌是crlm。公开了与同样适用于该医疗用途的分型方法有关的实施方式。
[0091]
同样,本发明提供了一种针对继发性肝癌的标准护理治疗剂的用途,用于制造治疗患有继发性肝癌的对象的药物,其中根据本发明的分型方法,所述对象被分型为患有继发性肝癌。优选地,继发性肝癌是crlm。公开了与同样适用于该医疗用途的分型方法有关的实施方式。
[0092]
本发明还提供了一种治疗患有继发性肝癌的对象的方法,包括以下步骤—进行本发明的分型方法;和
[0093]-当所述对象被分型为患有继发性肝癌时,施用治疗有效量的针对继发性肝癌的标准护理治疗剂。优选地,继发性肝癌是crlm。公开了与同样适用于该医疗用途的分型方法有关的实施方式。
[0094]
本文所用的术语“标准护理治疗剂”是指治疗化合物或此类化合物的组合,其被执业医师视为适当、可接受和/或广泛用于继发性肝癌的特定类型的患者、疾病或临床情况。本领域提供对抗继发性肝癌的标准护理疗法。用于治疗继发性肝癌的标准护理治疗剂包括靶向剂,例如抗体,包括西妥昔单抗、贝伐单抗或帕尼单抗。另一种靶向剂是阿柏西普。用于治疗继发性肝癌的特定标准护理治疗剂还包括一种或多种化疗剂,例如folfox(叶酸、氟尿
嘧啶和奥沙利铂)或folfiri(叶酸、氟尿嘧啶和伊立替康)。
[0095]
术语“治疗有效量”指的是足以在用该药剂治疗的对象中实现所需效果的特定药剂的量。理想情况下,药物的治疗有效量是足以抑制或治疗疾病或状况而不会对对象造成实质性细胞毒性作用的量。药物的治疗有效量将取决于被治疗的对象、痛苦的严重程度和治疗剂的给药方式。技术熟练的从业者可以在其知识和能力范围内确定治疗有效的给药方案。
[0096]
本文所使用的术语“给药”是指使用本领域技术人员已知的各种方法和递送系统中的任何一种,向患有继发性肝癌的对象物理导入药剂或治疗性化合物。技术人员了解适当的给药方法和剂型。小分子给药通常可以通过非胃肠外给药,如口腔和肠内给药。基于蛋白质的药剂(如抗体)的优选给药途径为胃肠外给药,包括静脉内、肌肉内、皮下、腹膜内、脊髓或其他胃肠外给药途径,尤其是以溶液形式注射或输注。可以例如一次、多次和/或在一个或多个延长的时期内进行给药。
[0097]
本发明还提供了一种测量肽水平的方法,包括以下步骤:-可任选地,提供包含来自对象的肽的样品;-在包含来自对象的肽的样品中测量(i)包含seq id no:1、seq id no:2或seq id no:4的氨基酸序列的肽,或(ii)包含与seq id no:1、seq id no:2或seq id no:4的氨基酸序列具有至少90%序列相同性的氨基酸序列的肽的肽水平。优选地,用于测量肽水平的方法还包括以下步骤:-任选地,提供包含来自所述对象的蛋白质的样品;-在包含来自所述对象的蛋白质的样品中测量癌胚抗原(cea)蛋白质水平。优选地,所述包含肽的样品是尿液样品,所述包含蛋白质的样品是血液样品,优选血清或血浆样品。
[0098]
本文中描述的关于提供样品和测量肽或蛋白质水平的步骤的实施方式,也是本文中描述的测量肽(和蛋白质)水平的方法的实施方式。
[0099]
出于清楚和简洁的目的,各特征在本发明中被描述为相同或单独的实施方式的一部分,然而,应理解公开内容包括具有所有或一些所述特征的组合的实施方式。
[0100]
本文所指文献的内容通过引用并入本文。
[0101]
图示说明
[0102]
图1.流程图
[0103]
图1显示了本研究中使用的样品流程图,显示了发现队列1和验证队列2。
[0104]
图2.优化碰撞能量nop
[0105]
图2列出了nop agp、gpp和gnd的最佳碰撞能量等特征。
[0106]
图3.散点图最佳lrm(gnd cea)和旧lrm(agp cea)
[0107]
散点图显示了使用新的生物标志物组合(gnd cea;最佳lrm)(散点图的左半部分)和已知的生物标志物组合(agp cea;旧lrm)(散点图的右半部分)预测crlm。条纹线代表每个模型的最佳截留线。
[0108]
序列表
[0109]
seq id no:1:羟基化gnd肽
[0110]
gndgargsdgqpgpp(-oh)gp(-oh)p(-oh)gtagfp(-oh)gsp(-oh)gak(-oh)gevgp
[0111]
seq id no:2:羟基化gpp肽
[0112]
gppgeagk(-oh)p(-oh)geqgvp(-oh)gdlgap(-oh)gp
[0113]
seq id no:3:羟基化agp肽
[0114]
agpp(-oh)geagkp(-oh)geqgvp(-oh)gdlgap(-oh)gp
[0115]
seq id no:4:羟基化ger肽
[0116]
gergsp(-oh)ggp(-oh)gaagfp(-oh)garglp(-oh)gpp(-oh)gsngnpgpp(-oh)gp(-oh)。
[0117]
在seq id no中,(-oh)表示前面的氨基酸残基是羟基化的。例如,p(-oh)g表示p是羟基化的。
[0118]
实施例
[0119]
实施例1.
[0120]
材料和方法
[0121]
实验设计和统计原理
[0122]
本研究由erasmus mc伦理审查委员会(mec-2008-062)批准,并根据赫尔辛基宣言进行。用质谱法交替测量健康肾脏捐献者(对照)和crlm患者的尿液样品。
[0123]
尿液中新胶原nop的鉴定基于鉴定尿液中所有的nop(发现组1:对照,n=40;crlm,n=40)。在先前的研究中,每组25个样品的样品规模足以鉴定组织中自下而上蛋白质组学中基于肽的标志物(van huizen等人,《生物化学杂志》294:281-9(2018))。然而,在尿液中观察到的nop水平差异较小。功效分析(α=0.05,β=0.20)所用的平均值和标准差(sd)是根据五名crlm患者和五名对照患者的尿液样品中对数转化显著上调的胶原肽的总体数据计算得出的(对照组平均值=6.76,crlm平均值=6.98,sd
合并
=0.75)。功效分析结果显示每组的样品规模为40个。
[0124]
在发现组1和另一个尿液样品组(发现组2:对照,n=60;crlm,n=60)上对感兴趣的nop进行靶向分析。lalmahomed等,am j cancer res.,6:321-30(2016)中描述了本文中用于发现的发现组1和2。对独立采集的尿液样品进行验证(对照,n=12;crlm,n=10)(broker等人,plos one;8:e70918(2013))。所用样品的流程图如图1所示。
[0125]
自下而上的蛋白质组学用于鉴定新的nop。在自下而上的蛋白质组学数据中,通过排列测试确定偶然的显著nop数量的评估。
[0126]
选择了与三条最丰富的胶原α链相关的三个最显著的nop,它们在crlm组织中也比在健康肝组织中更强烈地上调(van huizen等人,《生物化学杂志》294:281-9(2018))。由于自下而上的蛋白质组学是一种半定量技术,因此开发了一种靶向定量质谱方法(平行反应监测,prm)来验证这些发现。
[0127]
开发的prm法符合分析实验验证的第3级(共3级)(carr等人,mol cell proteomics;13:907-17(2014)),这意味着该分析是一种靶向发现分析。prm方法应用于完整发现组和验证组。为了确定最佳模型,拟合了一个逻辑回归模型(lrm),其中包含nop(由三个字母代码表示)和cea。通过反向消除包含所有分子标志物(agp、gnd、gpp和cea)的lrm中的预测因子,拟合最佳lrm。在验证组上对优化模型进行验证。对发现组2以及发现组1和2和组合(完整发现组)进行统计分析。然而,组合发现组1和发现组2创建了一个依赖性数据组,因为发现组1已经用于自下而上的蛋白质组学。然而,组合会产生更高的统计能力。在测量所有样品并优化lrm后,我们选择了三个样品,其中最优lrm中存在低、中、高水平的预测因子。由于sil肽并不适用于所有预测因子,因此这三个样品被处理五次,以获得再现性的估值。
[0128]
化学品
[0129]
超高压液相色谱级溶剂来自biosolve(荷兰瓦尔肯斯瓦德)。从pepscan(荷兰莱利斯塔德)获得agpp(-oh)geagk(sil)p(-oh)geqgvp(-oh)gdlgap(-oh)gp的稳定同位素标记的(sil)肽,赖氨酸用
13c615
n2标记。使用hplc-uv和esi-ms对该sil肽进行了表征。其它肽是gppgeagk(-oh)p(-oh)geqgvp(-oh)gdlgap(-oh)gp和gndgargsdgqpgpp(-oh)gp(-oh)p(-oh)gtagfp(-oh)gsp(-oh)gak(-oh)gevgp。
[0130]
这三种尿液nop将由前三种氨基酸(分别为agp、gpp和gnd)缩写表示。
[0131]
所有其他化学品均从sigma aldrich(荷兰zwijndrecht)获得。
[0132]
样品筛选
[0133]
重新分析了lalmahomed等人,am j cancer res.,6:321-30(2016)和broker等人,plos one;8:e70918(2013)的研究中描述的组的样品(图1)。收集后,将第1组和第2组的样品储存在-80℃的聚丙烯试管中。一个crlm样品被排除在当前研究的验证组中,因为相应的cea值未知。由于broker等人,2013年研究的验证组的cea水平未知,因此排除了这组样品。
[0134]
通过t检验计算对照组和crlm患者之间的年龄和bmi差异,通过卡方检验计算性别和高于115μm/l的血清肌酐水平方面的的差异。p值低于0.05/4=0.0125(bonferroni校正以校正多重检验)被认为是显著的。
[0135]
样品制备
[0136]
根据lalmahomed等人,《美国癌症研究杂志》6:321-30(2016)的描述,从尿液中分离出用于自下而上蛋白质组学和靶向质谱分析的nop。简言之,通过mrp c-18高回收蛋白质柱(4.6
×
50mm)(荷兰阿姆斯特丹安捷伦)从小分子、盐和蛋白质中分离nop,该柱安装在配备在线分馏器的ultimate 300 lc系统(dionex,荷兰阿姆斯特丹)中。分离后,收集nop组分,干燥,重建,并用质谱分析。
[0137]
自下而上蛋白组学
[0138]
对于nop的鉴定,我们采用了标准的自下而上lc-ms/ms方法,如van huizen等人,j biol chem.94:281-9(2018)所述。简而言之,将ultimate 3000nano rslc系统(thermo fischer scientific,德国热尔梅林)在线偶联到orbitrap fusion lumos tribrid质谱仪(thermo fischer scientific,美国加利福尼亚州圣何塞)上。捕获注入的样品并在捕集柱上清洗(c18 pepmap,300μm id x 5mm,2μm粒径,孔径;thermo fisher scientific,荷兰)。洗涤后,在质谱分析之前,将捕集柱切到与分析柱(pepmap c18,75μm id x 250mm,2μm粒径,孔径;thermo fisher scientific,荷兰)连接,进行肽分离。我们改变了van huizen等人2018年的方案,即在进行质谱分析之前,既没有在测试hplc系统上分析样品,也没有对注射体积定标。对于每个样品,注入2μl的体积。自下而上的蛋白组学数据上传至pride档案(pxd013533)。
[0139]
自下而上数据分析
[0140]
mgf峰值列表文件由proteowizard(v3.0.9166)从原始文件中提取。mgf峰值列表文件使用mascot搜索引擎(v2.3.2,英国伦敦matrix science inc.)和uniprot/swissprot数据库(20194个条目)进行搜索。以下设置用于数据库检索:酶被设置为开放,因为我们分析了nop;肽质量的质量耐受性设置为10ppm,片段质量的质量耐受性设置为0.5da。作为可变修饰,选择脯氨酸、赖氨酸的羟基化和甲硫氨酸的氧化( 16da);未添加固定修饰。mascot
标识被导入scaffold(v4.6.2,美国俄勒冈州波特兰)。在scaffold中,蛋白质置信水平设置为1%的伪发现率(fdr),每个蛋白质至少2个肽,肽水平为1%的fdr。通过包含由mascot生成的诱饵数据库检索估计fdr。将原始文件与progenesis qi(v4,nonlinear dynamics,泰恩河畔纽卡斯尔,英国)中scaffold导出的识别列表比对并合并,然后将标准化丰度导出到excel 2010(美国华盛顿州雷德蒙微软公司)。一式两份的特征强度被求和。使用excel、graphpad prism(v5.01,美国加利福尼亚州拉霍亚)和r(v3.3.1,奥地利维也纳)进一步处理数据。在
10
log转换之前,添加了一个值“10”,目的是包含缺失的值,以便进行进一步的数据分析。
10
log转换后,假设数据为正态分布。采用不等方差独立样品t检验,检验对照组和crlm之间的nop显著性。低于0.05的p值被认为是显著的。
[0141]
仅考虑了胶原α链,其也被发现在crlm组织和正常肝组织之间存在差异(van huizen等人,j biol chem.294:281-9(2018))。构建了一个nop分子面板,由九个nop组成,即来自最丰富的三条胶原α链中最重要的三个nop。除了这九种nop,早期报道的nop命名为agp(lalmahomed等人,《美国癌症研究杂志》,6:321-30(2016);broker等人,plos one;8:e70918(2013)),包含在靶向质谱法中。
[0142]
排列测试是根据作为van huizen等人,j biol chem.294:281-9(2018)的补充文件的r-script进行的。简而言之,数据在肽水平上随机分为两组;使用wilcoxon符号秩检验确定两组之间的显著差异。每个排列的显著差异(p值《0.05)相加,取
10
log。假设
10
log加和的显著p值分布为正态。如果真实数据组的值大于排列测试的均值加上两倍标准差(p《0.05),则认为差异显著。
[0143]
靶向质谱分析
[0144]
在用于自下而上蛋白质组学的相同nanolc esi-orbitrap-lumosfusion上进行靶向质谱测量。为了测量样品,开发了一种具有优化的碰撞能量的prm法。排除了无法确定最佳碰撞能量或识别信号强度过低的nop。图2中列出了一张表格,以及优化碰撞能量等特征。
[0145]
在不同时间测量完整的发现组和验证组,以提高有效性。使用其他数据组的对照组均值,比对数据组与发现组1。这仅对没有产生sil肽的nop有必要。靶向质谱数据被上传到pride档案库(pxd013705)。
[0146]
靶向数据分析
[0147]
质谱仪产生的原始文件被导入skyline(maclean等人,生物信息学;26:966-8 9(2010))。对于每条肽,我们选择了最多五个高强度跃迁,且相邻峰没有明显干涉。使用skyline的gnd和gpp肽峰面积,agp使用与sil肽的比率。
[0148]
逻辑回归模型
[0149]
统计分析是在r(版本3.3.1,维也纳,奥地利)(r核心团队,统计计算r基金会,奥地利,维也纳,可得自https://www.r-project.org/.(2016))。分别在发现组2(独立数据组)和完整发现组(依赖性数据组)上进行预测因子筛选。如果用于选择预测因子的数据组(完整发现组或发现组2)未显示不同的预测因子选择,则使用完整发现组进行分析以防功能丧失。为了选择相关的预测因子来拟合最佳逻辑回归模型,使用显著性水平0.05。临界p值根据预测因子数量或测试比较的数量进行了bonferroni校正。
[0150]
目前的分子面板由agp和cea组成,并用新识别的nop(gpp和gnd)进行了扩充。为了使新模型与分子标志物相匹配,对这些标志物在患者特征、个体显著性和多重共线性之间
的任何关系上进行了测试。患者特征“年龄”、“性别”、“bmi”、“血清肌酐》115μm/l”之间的关系是通过拟合线性模型来确定的,该线性模型预测每个患者特征的个体分子标志物和预测因子“组(健康/疾病)”。与患者特征显著相关的分子标志物被排除在进一步分析之外。通过将lrm与单个预测因子进行拟合,对所有剩余的个体预测因子进行显著性检验。个体预测因子的显著性基于wald统计。通过计算方差膨胀因子(vif),对选出的显著预测因子进行多重共线性评估。假设vif》10时存在多重共线性;如果必要的话,为了防止多重共线性,弃去预测因子。
[0151]
所选的预测因子适合于组合lrm(完全lrm)。通过从完全-lrm中反向消除非显著预测因子形成最佳lrm(最佳-lrm)。
[0152]
通过拟合线性模型,测试最佳-lrm中的分子标志物与最大肿瘤尺寸以及肿瘤数量之间的关系。个体预测因子的显著性基于wald统计。
[0153]
库克距离测试用于检查数据是否存在异常值和/或杠杆点。用公式4/(n-k-1)计算怀疑为异常值/杠杆点的阈值,其中n=样品数,k=预测因子数。通过手动检查样品识别的异常值和/或杠杆点已从数据集中移除。
[0154]
我们先前的逻辑回归模型(旧-lrm)包含agp和cea。在比旧-lrm和最佳-lrm之前,计算了两者之间的皮尔逊相关性。为了选择预测能力最高的lrm,需要将最佳-lrm的性能与旧-lrm的性能进行比较。如果存在嵌套,则将预测能力与“方差分析(anova)”函数进行比较,否则使用德隆检验来比较auc。
[0155]
结果
[0156]
患者特征
[0157]
表1概述了患者的基本特征。对照和crlm患者的年龄和性别差异显著。四名患者的血清肌酐水平高于115μm/l,表明存在肾损害。
[0158]
表1,患者特征
[0159][0160]
自下而上质谱
[0161]
在发现组1中共鉴定出1683个nop,属于175种蛋白质。三种最常见的蛋白质是1(i)型胶原(n=183nop)、1(iii)型胶原(n=157nop)和尿调节蛋白(n=84nop)。453个nop(27%)属于13种胶原α链(表2)。对照和crlm之间有406个nop(24%)存在显著差异,其中118个属于胶原(表2)。
[0162]
表2.每个胶原α链鉴定出的nop数
[0163][0164][0165]
靶向质谱
[0166]
通过包括agp(lalmahomed等人,am j cancer res.,6:321-30(2016);broker等人,plos one;8:e70918(2013))和三条最丰富的胶原α链中最显著不同的三种nop构建了尿nop面板。无法确定七种尿nop的最佳碰撞能量。三种剩余的尿nop是agpp(-oh)geagkp(-oh)geqgvp(-oh)gdlgap(-oh)gp,gppgeagk(-oh)p(-oh)geqgvp(-oh)gdlgap(-oh)gp,和
[0167]
gndgargsdgqpgpp(-oh)gp(-oh)p(-oh)gtagfp(-oh)gsp(-oh)gak(-oh)gevg p。agp和gpp来源于胶原α链1(i),gnd来源于胶原α链1(iii)。
[0168]
对逻辑回归模型
[0169]
在发现组2和完全发现组上进行预测因子选择过程。在拟合完全-lrm之前,测试了分子标志物(agp、gpp、gnd和cea)与任何患者特征(年龄、性别、bmi和血清肌酐水平)的线性关系。未发现显著线性关系。通过拟合每个标志物的lrm,还测试了个体分子标志物的独立显著性。结果如表3所示。单独来看,所有的分子标志物都显示出显著性,并被纳入完全-lrm。分子标志物之间不存在多重共线性。因此,所有分子标志物都包含在完整发现组和发现组2中的完全-lrm中。在完全发现组中,任何个体分子标志物之间以及任何分子标志物与最大肿瘤尺寸和肿瘤数量之间均不存在显著的线性关系。
[0170]
最佳-lrm是通过反向消除非显著预测因子形成的。对于这两个数据组,这导致了一个包含gnd和cea(最佳-lrm)的模型。预测因子的选择与完整发现组或发现组2的使用无
关。因此,剩下的分析仅使用完整发现组进行,以防止统计能力丧失。公式1显示了预测个体患crlm可能性的公式。gnd 95%置信区间的or为21[8.5-60],cea为32[10-129]。
[0171]
公式1:
[0172][0173]
计算库克距离是为了确保该公式不受异常值/杠杆点的严重影响。14个数据点高于阈值,并进行了人工检查。没有一个似乎是错误的测量值,因此没有一个被删除。
[0174]
在重新测量时,来自旧-lrm和新最佳-lrm数据组的agp值高度相关(相关性=0.89,p值《2.2*10-16
)。旧agp值(agp_旧)与当前agp值之间的线性关系为:agp=0.9 1.57*agp_旧。使用德隆检验比较了旧-lrm和最佳-lrm的auc。旧-lrm的auc为0.8824,这与最佳-lrm的auc为0.9256显著不同(p值=0.032)。包含最佳-lrm和旧-lrm值的散点图如图3所示。
[0175]
根据最佳-lrm计算值的roc曲线,选择了0.439的截留值。该截留值导致完全发现组的灵敏度和特异性分别为86%和84%,验证组的灵敏度和特异性分别为92%和90%(表4)。
[0176]
为了估计样品处理对于gnd值的再现性,我们测量了三个样品,在所有测量值的较低、中和高范围内选择五次。测量了以下样品,括号内为面积的
10
log和%cv:vms-248(低,6.1
±
1.4%)、vms-253(中,6.9
±
1.8%)和vms-163(高,7.6
±
1.4%)。
[0177]
表3预测因子筛选,显著预测因子用粗体标记
[0178][0179][0180]
表4gnd和cea值概述以及获得的灵敏度和特异性。
[0181][0182]i平均[第一四分位数-第三四分位数]
[0183]
实施例2.
[0184]
该实施例是实施例1的补充。
[0185]
材料和方法
[0186]
如实施例1所述的程序用于鉴定和测试尿液中的另一种天然发生肽(nop),也称为“ger”,其氨基酸序列为gergsp(-4hyp)ggp(-4hyp)gaagfp(-4hyp)garglp(-4hyp)garglp(-4hyp)gpp(-4hyp)gsngnpgpp(-4hyp)gp(-4hyp)。“p(-4hyp)”是指氨基酸脯氨酸(p)被修饰成4-羟脯氨酸。这种天然发生肽(nop)来源于胶原蛋白α-1(iii)(col3a1,蛋白质编码uniprot/swissprot=p02461)。下文简要介绍了该程序。
[0187]
为了发现新nop ger,我们得到了大量健康对照尿液(n=100)和患有crlm的患者(n=100)的尿液样品。样品组以40:60比分开。nop ger作为crlm新标志物的鉴定是基于使用无偏半定量蛋白质组学法对40个对照和40个crlm尿液进行分析。通过在全样品组(对照n=100,crlm n=100)上使用靶向定量质谱法进一步验证nop ger的值。nop gnd与血清癌胚抗原(cea)一起形成了一组符合逻辑回归模型(lrm-gnd)的标志物。从lrm-gnd开始,nop gnd被nop ger(lrm-ger)取代。首先,根据瓦尔德统计(p值《0.05)和优势比的95%置信区间(ci)(不与1重叠),测试nop ger是否对模型有显著贡献。其次,通过使用德隆检验比较roc曲线的曲线下面积(auc)来比较lrm-gnd和lrm-ger的预测能力,低于0.05的p值被认为是显著的。
[0188]
结果
[0189]
与nop gnd一样,nop ger被确定为继发性肝癌的生物标志物。此外,表5显示了lrm-ger的结果,显示了nop ger在模型中的重要性。nop ger对模型有显著贡献,p值为3.60*10-7
,优势比的95%ci(不与1重叠)也证实了这一点。
[0190]
表5.天然发生肽ger在预测继发性肝癌的逻辑回归模型中的意义。
[0191][0192]
ci 置信区间
[0193]
cea 癌胚抗原
[0194]
col3a1胶原α-1(iii)
[0195]
用于根据lrm-ger计算患者患继发性肝癌几率的示例性公式(公式2)为:
[0196][0197]
根据roc曲线的auc比较lrm-gnd和lrm-ger的预测能力。lrm-ger的auc为0.9079,lrm-gnd为0.9256。auc没有显著差异(p=0.28),表明两个模型具有相似的预测能力。与nop gnd相似,nop ger和血清cea的组合比血清cea本身具有更高的预测能力,并且与nop gnd和cea的组合相似。尿液浓度校正的标准是尿液中的肌酐水平。在模型中添加尿肌酐水平不会对nop gnd或nop ger的预测能力产生负面影响(数据未显示)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。