:
1.本发明属于食品安全检测技术领域,具体涉及一种基于磁性固相萃取同时检测食用油中3-氯丙醇酯和缩水甘油酯的方法。
背景技术:
2.3-氯丙醇酯(3-mcpdes)和缩水甘油酯(ges)是植物油在精炼除臭阶段产生的污染物。这两类污染物经人体摄入后,代谢水解分别产生3-氯丙醇和缩水甘油。实验表明,这两类代谢物对人体具有肾毒性、生殖毒性和致癌性,故对3-氯丙醇酯和缩水甘油酯的检测意义重大。通常对3-mcpdes和ges的检测方法有两种:直接法与间接法。间接法的代表是aocs标准方法cd 29a-13,通过一系列反应将3-氯丙醇酯和缩水甘油酯转化为3-氯丙醇和3-溴丙醇,经衍生化后再进行gc-ms分析。
3.间接法由于其经济(无需过多标准品),能定量分析目标物总含量等优点已成为检测3-mcpdes和ges的最普遍方法,其前处理步骤涵盖ges的溴化,酯交换,净化和衍生化这几步。但其主要存在以下三个问题:1.前处理时植物油中的甘油单酯(mag)和甘油二酯(dag)会转化为3-溴丙醇酯,造成ges测量值偏高;2.传统的净化过程某些油脂基质组分无法被去除,会造成后续仪器污染,且会显著降低仪器灵敏度;3.间接法常采用选择选择离子扫描(sim)的方法,检出限较高,无法实现痕量检测。
4.样品前处理方面,针对油脂组分,氨基固相萃取小柱可以对其中极性较高的mag和dag进行吸附,将固相萃取法(spe)应用于油脂中mag和dag的去除具有良好前景。硼酸材料可以在较高ph值下(ph>pka)与顺式二羟基化合物特异性结合,形成五元或六元环酯,并在一定条件下(ph<pka)可以解离。功能化硼酸材料具有特异性强,比表面积大和稳定等优点,将其用于磁性固相萃取(mspe)可以实现对顺式二羟基化合物的快速高效分析,该方法已经在生物大分子富集方面受到广泛关注和应用。将此方法代替间接法中传统的净化过程,能很好解决仪器污染等问题。在气质联用分析方面,多反应监测(mrm)相比sim法,具有更高的灵敏度,更低的基质干扰,非常适合于痕量目标物的检测。因此,结合上述技术,开发新方法对植物油中3-mcpdes和ges的检测,具有实际意义。
技术实现要素:
6.鉴于目前解决现有技术中存在的方法灵敏度低,污染严重等问题,本发明将spe净化,mspe和gc-ms/ms技术相结合,提供一种基于磁性固相萃取同时检测植物油中3-氯丙醇酯和缩水甘油酯的方法,其具有灵敏度高,污染小等特点,该方法可对植物油中的3-mcpdes和ges进行高效准确的检测。
7.本发明技术方案步骤如下:
7.步骤1、制备fe3o4@sio
2-dffpba mnps,该步骤包括:制备fe3o
4 mnps;制备fe3o4@sio
2 mnps;制备fe3o4@sio
2-nh
2 mnps;制备fe3o4@sio
2-@dffpba mnps;
7.步骤2、样品添加内标,经spe净化除去mag和dag;
7.步骤3、净化后的样品经酸性溴化钠溶液处理,将所含缩水甘油酯转化为3-溴丙醇酯;
7.步骤4、样品经硫酸/甲醇溶液处理,将所含3-氯丙醇酯和3-溴丙醇酯转化成3-氯丙醇和3-溴丙醇;
7.步骤5、将上步所得溶液用制备的磁性纳米材料进行萃取;
7.步骤6、用苯硼酸对萃取后样品衍生化处理;
7.步骤7、将衍生化产物进行gc-ms/ms定量分析。
7.本发明中,具体参数可采用如下优选方式实现:
7.优选的,步骤1所述fe3o
4 mnps的制备中,无水fecl3的质量为1.0-2.0g,柠檬酸三钠的质量为0.2-0.8g,无水乙酸钠的质量为2.0-3.0g。反应条件为:在180-220℃条件下反应5-15h。
7.优选的,步骤1所述fe3o4@sio
2 mnps的制备中,teos的体积为1-3ml。
7.优选的,步骤1所述fe3o4@sio
2-nh
2 mnps的制备中,fe3o4@sio
2 mnps用量为0.1-0.25g,反应条件为:在80-110℃条件下反应8-15h。
7.优选的,步骤1所述fe3o4@sio
2-dffpba mnps的制备中,dffpba的质量为0.2-0.4g,nabh3cn的质量0.3-0.6g。。
7.优选的,所述步骤2中spe所采用的固相萃取柱是氨基固相萃取柱。
7.优选的,所述步骤3中,反应条件为:在40-60℃条件下反应10-30min。
7.优选的,所述步骤4中,反应条件为:在35-45℃条件下为10-20h。
7.优选的,所述步骤5中,磁性固相萃取的条件为:ph值为7-10下吸附5-20min,再在ph值为1-5下洗脱1-10min。
7.优选的,所述步骤6中,衍生化反应条件具体为:在40-80℃条件下反应10-30min。
7.优选的,所述步骤7中,气相色谱条件:进样口温度250℃,分流比50比1,色谱柱hp-5(30m
×
0.25mm
×
0.25m),程序升温:70℃保持1min,6℃/min升到178℃,再30℃/min升到280℃保持4min。质谱条件:电离源为ei源,源温230℃,ms接口温度为250℃,载气和碰撞气分别为氦气和氩气,质谱仪在70ev的电子电离模式下进行多反应监测(mrm)。
16.技术效果
17.本发明成功合成一种硼酸功能化磁性纳米材料用于磁性固相萃取,将其代替传统的净化步骤,由此成功建立了一种基于磁性固相萃取与gc-ms/ms联用,同时检测植物油中3-氯丙醇酯和缩水甘油酯的方法。本方法具有特异性强,灵敏度高,对仪器污染小等特点,已成功应用于植物油中3-氯丙醇酯和缩水甘油酯的检测,具有良好的应用和推广价值。
附图说明
18.为更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所要需要使用的附图简单地介绍,显然易见地,如下描述的附图仅为本发明的实例,对本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以通过附图获得其他的附图。
12.图1为本发明实施实例中(a)fe3o4,(b)fe3o4@sio2,(c)fe3o4@sio2-nh2和(d)fe3o4@sio2-dffpba的(a)红外光谱图,(b)x射线光电子能谱图,(c)x射线衍射图;(d)fe3o4
和fe3o4@sio2-dffpba的磁滞回线。
12.图2表示本发明实施实例中mspe条件的优化。研究了(a)吸附时间,(b)洗脱时间,(c)吸附ph值和(d)洗脱ph值对3-氯丙醇(3-mcpd)和3-溴丙醇(3-mbpd)萃取效率的影响。
12.图3为本方法实施实例中典型实际油样的色谱图。
12.图4为本方法实施实例中典型实际样品加标的色谱图。
具体实施方式
15.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
15.1实验部分
16.1.1 fe3o4@sio
2-dffpba mnps的制备
16.1.1.1 fe3o
4 mnps的制备
16.将1.4g无水fecl3和0.5g柠檬酸三钠加入40ml乙二醇里,溶解得到澄清黄色溶液,再加入2.4g无水乙酸钠并磁力搅拌30min,将混合液体转移至不锈钢反应釜的聚四氟乙烯内衬里,拧紧密闭后于烘箱中200℃加热反应12h。反应后冷却后磁分离黑色固体,水和乙醇交替洗涤3次后烘干即得fe3o
4 mnps。
16.1.1.2 fe3o4@sio
2 mnps的制备
16.取全部上述所得fe3o
4 mnps分散于200ml乙醇和50ml超纯水中,超声10min后再加入5ml质量分数为28%的氨水,再机械搅拌15min,再将2ml teos机械搅拌下逐滴加入上述混合液中,滴加完后搅拌反应8h。反应后磁分离,水和乙醇交替洗涤3次后烘干即得fe3o4@sio
2 mnps。。
16.1.1.3 fe3o4@sio
2-nh
2 mnps的制备
16.取0.15g上述所得fe3o4@sio
2 mnps分散于60ml无水甲苯,超声15min后,再将2ml aptes机械搅拌下逐滴加入,滴加完后110℃下回流反应12h,反应后磁分离,无水甲苯和甲醇交替洗涤3次后烘干即得fe3o4@sio
2-nh
2 mnps。
16.1.1.4 fe3o4@sio
2-dffpba mnps的制备
16.取全部上述所得fe3o4@sio
2-nh
2 mnps分散于30ml无水甲醇,依次加入0.3g dffpba和0.5g nabh3cn,超声15min后,机械搅拌反应72h。反应后磁分离,无水甲醇和去离子水交替洗涤3次后烘干即得fe3o4@sio
2-dffpba mnps。
16.1.2样品前处理
16.1.2.1 spe净化
16.用含有正己烷/乙酸乙酯(85/15,v/v)的3ml混合溶液a活化氨丙基spe小柱(500mg,6ml)。然后称取100.0mg样品,加入50μl混合内标溶液(0.5mg/kg pp-3-mcpd-d5和pp-gly-d5)后再溶解于1ml溶液a中,随后将其通过小柱。用5ml溶液a洗脱小柱,并用离心管收集。再次将收集的约6ml洗脱液通过小柱。然后,再次用5ml溶液a洗脱小柱,并收集到约11ml洗脱液。洗脱液在40℃氮气流下蒸发,所得残留物复溶于2ml四氢呋喃中。
16.1.2.2 ges转化为3-溴丙醇酯(3-mbpdes)
16.将30μl酸性溴化钠溶液(3.3mg/ml,硫酸体积分数为5%)添加到上一步所得的溶液中,并在50℃条件下反应15min进行ges的溴化。其后用3ml质量分数为0.6%的碳酸氢钠
溶液中止反应,并使用2ml正庚烷进行提取。提取物在40℃氮气流下蒸发,残留物复溶于1ml四氢呋喃中。
16.1.2.3酯交换反应
16.将1.8ml硫酸/甲醇(硫酸体积分数为1.8%)添加到上述所得溶液中,并在40℃条件下反应16h以进行酯交换反应。
16.1.2.4磁性固相萃取
16.上述反应用2ml质量分数为2.4%的碳酸氢钠溶液中止,加入适量的质量分数为28%的氨水将其ph值调至8。在此混合物中加入10mg fe3o4@sio
2-dffpba mnps后,涡旋10min以达到吸附平衡。再用1ml氨水(ph=8)洗涤后并磁分离后,将1ml乙酸水溶液(ph=2)加入fe3o4@sio
2-dffpba mnps中,涡旋5min以完成洗脱。
16.1.2.5衍生化反应
16.向上述洗脱液中加入2ml乙酸乙酯进行提取,其后向提取物中添加150μl质量分数为4%的苯硼酸丙酮溶液,并在70℃水浴下反应20min。然后将所得混合物在40℃下在氮气流下蒸发至干燥,再复溶于500μl异辛烷中。其后离心获得上清液,并储存在-18℃下3小时以沉淀多余的苯硼酸。最后上清液通过0.22μm ptfe滤膜过滤后待上机测试。
16.1.3 gc-ms/ms分析
16.gc-ms/ms分析在安捷伦8890gc串联安捷伦7010b三重四极质谱仪(安捷伦科技,美国)上进行。gc-ms/ms仪器条件为,气相色谱条件:进样口温度250℃,分流比50比1,进样量为1μl,色谱柱hp-5(30m
×
0.25mm
×
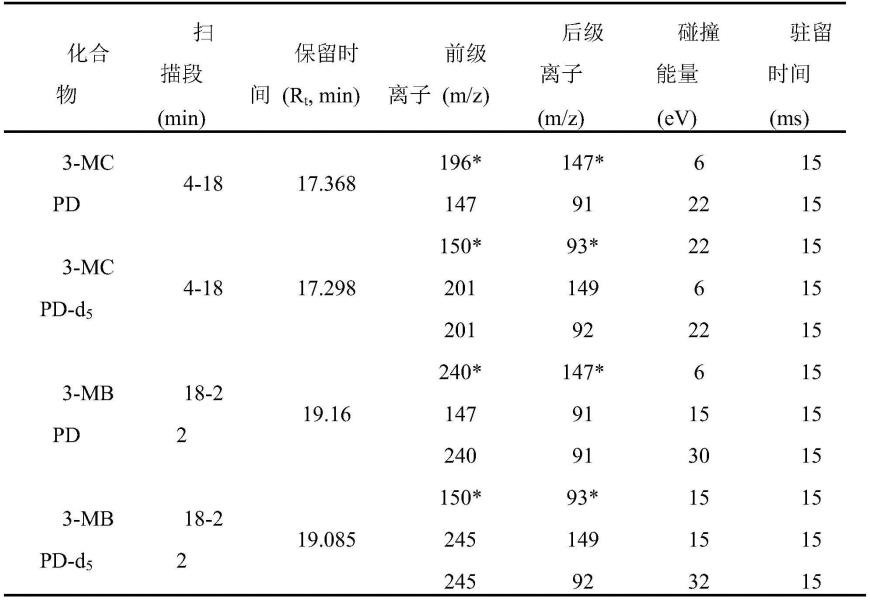
0.25m),程序升温:70℃保持1min,6℃/min升到178℃,再30℃/min升到280℃保持4min。质谱条件:电离源为ei源,源温为230℃,ms接口温度为250℃,载气和碰撞气分别为氦气和氩气,质谱仪在70ev的电子电离模式下进行多反应监测(mrm)。目标化合物的质谱条件如下表1所示。表1 3-mcpd,3-mbpd,3-mcpd-d5和3-mbpd-d5的衍生物的质谱条件。
*表示定量离子对
15.2结果与讨论
15.2.1 fe3o4@sio
2-dffpba mnps的表征
15.红外结果如图1a所示,最初580cm-1
的峰源于fe3o4中fe-o键的伸缩振动;包覆sio2后,可观察到1091cm-1
处出现的si-o-si的伸缩振动峰;其后观察到2923和2854cm-1
处出现的亚甲基的伸缩振动峰,证明aptes成功修饰上;对于最终产品,观察到1381cm-1
的峰归属于b
–
o键的振动,表明dffpba已成功修饰在mnps表面。x射线光电子能谱图如图1b所示,其与红外结果一致。si2p(102ev)、n1s(398ev)、f1s(698ev)和b1s(190ev)峰的出现分别表明在逐步修饰中成功地引入了sio2、-nh2和硼酸基团。
15.xrd图谱用于确定晶体结构,结果如图2c所示。在30.1
°
、35.3
°
、42.9
°
、53.8
°
、57.1
°
和62.8
°
处观察到2θ处的峰,分别对应于fe3o4的(220)、(311)、(400)、(422)、(511)和(440)晶面,表明fe3o4已成功合成,且在之后逐步修饰中材料晶型不变。
15.磁滞回线结果如图1d所示。所有磁滞回线显示矫顽力和剩磁几乎为零,表明相应的材料是超顺磁性的。经逐步反应后,fe3o4@sio
2-dffpba mnps仍展现出较大的最大饱和磁化强度(56.1emu/g),并可在15s内实现磁分离(图1d)。
15.2.2样品前处理的优化
15.2.2.1磁性固相萃取条件的优化
15.fe3o4@sio
2-dffpba mnps作为磁性吸附剂,用于mspe以代替间接法中传统的净化步骤。为获得最佳的实验参数,对mspe过程的吸附时间,洗脱时间,吸附ph值和洗脱ph值进行了优化。
15.实验考察了不同吸附时间(5,10,20,30和40min)对mspe效率的影响,结果如图2a所示。10min后3-mcpd和3-mbpd的峰面积几乎没有增加(p》0.05),表明吸附平衡可以在
10min内快速达成。选择10min进行吸附,因为它省时且确保了待测物和材料之间的充分相互作用。
15.实验考察了不同洗脱时间(2,5,10和15min)对mspe效率的影响。如图2b所示,随着时间的延长,3-mcpd的峰面积首先增加(图2b),但超过5min后差异不显著(p》0.05),表明5min足以洗脱3-mcpd。对于3-mbpd也有类似的结论,但洗脱时间减少到2min,比3-mcpd更快。考虑到需对目标化合物完全洗脱,选择5min作为3-mcpd和3-mbpd的洗脱时间。
15.实验考察了不同吸附ph值(7,8,9和10)对mspe效率的影响。由于氟原子的吸电子效应,dffpba具有较低的pka,甚至可以在中性环境中吸附3-mcpd和3-mbpd(图2c)。3-mcpd和3-mbpd的峰面积在ph值为8时达到最大值,然后随着ph值的增加而趋于减小,这可能是由于3-mcpd和3-mbpd在碱性条件下不稳定造成的,故选择ph值为8作为吸附ph值。
15.实验考察了不同洗脱ph值(2,3和4)对mspe效率的影响。如图2d所示,对于3-mcpd和3-mbpd,均在ph值为2时获得最大峰面积,其后随着ph值的增加而减小。这是因为dffpba的pka值较低,故也需较低的ph值进行洗脱,故选择ph值为2作为洗脱ph值。
15.最终确定mspe步骤的优化条件为:吸附时间10min,洗脱时间5min,吸附ph值为8及洗脱ph值为2。
15.2.2.2衍生化反应优化
15.考察了衍生化溶剂水相或有机相,超声或水浴对衍生化效率的影响。空白样品加标,分别进行了如下四组衍生化实验:(1)水相里超声5min;(2)水相里水浴加热20min;(3)有机相乙酸乙酯里超声5min;(4)有机相乙酸乙酯里水浴加热20min。实验结果表明,在乙酸乙酯里水浴加热20min下目标物响应最高,故选取此条件用于实验的衍生化反应。
15.2.3方法性能参数
15.如表2所示,总结了方法的线性,检出限(lod)和定量限(loq)。由表可知3-mcpdes和ges的线性范围为0.001-5mg/l,且线性良好(相关系数为0.9997)。3-mcpdes和ges的检出限分别为0.2和1.5μg/kg。虽然本实验为减少仪器污染,将传统的不分流方法调整为分流比50:1,但本实验的检出限值比传统方法仍低10倍左右。由此可见本方法解决了传统方法灵敏度低和仪器污染严重的问题。表2方法的线性,检出限(lod)和定量限(loq)。
15.进行了方法的准确度和精密度试验,结果如表3所示。两种目标物在三种加标水平下(0.2,1和2mg
·
kg-1
)的回收率在96.4-105.8%之间,对应日内和日间精密度实验的相对标准偏差分别1.7-5.9%和1.0-7.1%在范围内。表3方法的精密度和准确度实验结果。
15.2.4实际样品分析
15.为验证本方法的适用性,选取了两种植物油样品,采用本方法进行分析。结果测得样品1中3-mcpdes和ges的浓度分别为713.45和468.50μg/kg,样品2中3-mcpdes和ges的浓度分别为572.85和288.5μg/kg。图3为典型实际油样的色谱图,而图4为典型实际油样加标的色谱图。以上结果表明,本专利建立的方法可以准确可靠的对植物油中的3-氯丙醇酯和缩水甘油酯进行检测,并且与传统方法对比,能改善灵敏度低和仪器污染等问题。
15.以上所述的实施例仅为本发明的一种较佳的方案,然其并非用以限制本发明。有关技术领域的普通技术人员,在不脱离本发明的精神和范围的情况下,还可以做出各种替换和变型。如对于spe小柱,可选取填料,容量相近的小柱替换。因此凡采取等同替换或等效变换的方式所获得的技术方案,均落在本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。