1.本发明涉及一种液质联用快速测定动物性食品中海南霉素残留的方法,属于分析化学技术领域。
背景技术:
2.海南霉素(c
47h79o15
na)是我国养禽业常用的抗生素,它是一元酸多醚类抗生素,能与碱性金属阳离子形成盐,其游离酸的溶解度几乎与盐类相同,通常以其钠盐使用,是一种广泛应用于养殖场的新型聚醚抗生素。中国食品安全国家标准gb 31650兽药残留中禁用海南霉素,因此,非常有必要开发一种适用于动物组织中海南霉素残留检测的前处理和检测方法。
3.目前,海南霉素的检测分析方法有紫外分光光度法、飞行时间(tof)质谱法、高效液相色谱-蒸发光散射检测法和高效液相色谱-串联质谱,飞行时间(q-tof)质谱只能分析海南霉素的分子量,紫外分光光度法和高效液相色谱-蒸发光散射只能定量高浓度的海南霉素标准溶液(分别为100,000
–
1,400,000ng/ml和500,000
–
2,000,000ng/ml),目前研究中的液相色谱-串联质谱(lc-ms/ms)可以检测鸡血中的低浓度海南霉素(1-500ng/ml),其甲醇乙腈直接提取后检测的前处理方法不适用于动物组织,因为血浆主要成分是水和蛋白质,而动物组织成分脂类含量高,如果提取之后不净化不能达到在动物性食品中检测低浓度海南霉素的要求,其lc-ms/ms的流动相等度分离条件分离度低,可耐受压力低,也不能满足多种复杂基质与海南霉素的有效快速分离。目前尚未有对动物组织中海南霉素的检测进行的研究,因此,迫切需要一种可靠、简单、准确的分析方法来监测畜禽动物组织中海南霉素的浓度,目前常用的兽药残留前处理有固相萃取、固相微萃取、液液萃取等,固相萃取操作繁琐,耗时长,成本高,固相微萃取对一部分兽药的回收率低,急需开发一种适用于海南霉素的提取净化、高度分离的快速检测方法。
技术实现要素:
4.【技术问题】
5.海南霉素是一种新型聚醚抗生素。毒理学研究表明海南霉素对动物和人类有不利影响。目前,没有检测畜禽动物食用组织中海南霉素残留的研究。因此,对海南霉素的快速和准确的检测方法至关重要。但是由于鸡蛋、鸡肝、肉等基质中含有大量的脂肪、蛋白质等,如果直接使用溶剂提取不加以净化,杂质会干扰样品检测,还会损害仪器和色谱柱,也会对其灵敏度有很大的影响,等度洗脱也无法有效分离杂质和海南霉素。为了消除基体对测定的干扰,提高方法的选择性使被测组分从复杂的样品中分离出来,因此样品在进行液质分析前非常有必要进行前处理以达到净化,探索优化梯度洗脱条件,提高方法的灵敏度与选择性,保护分析仪器及测试系统,以免影响仪器性能和使用寿命,从而建立稳定、高效分离的超高效液相色谱串联质谱检测的新方法。
6.【技术方案】
7.针对现有的技术问题,本发明旨在提供一种超高效液相色谱-高分辨质谱法快速检测动物性食品中海南霉素残留量的方法,以检测多种动物性食品中残留的海南霉素,包括牛奶,鸡蛋,脂肪,肾,肌肉和鸡,牛肉和羊肉。由于基质更为复杂,需要对现有预处理进行改进才能获得较好的效果,参考目前现有的文献方法:以甲醇-乙腈进行提取后直接检测,无法达到1ng/g的检测限,由于动物性食品基质更为复杂,杂质较多,文献记载的预处理方法无法达到相当效果,通过溶剂的选择以及前处理条件的选择、流动相的优化等,对现有检测方法进行改进优化才能获得较好的检测效果,才能够很好的将海南霉素洗脱并在低浓度时有良好的信号强度,前处理使用乙腈有效地提取了海南霉素,乙腈饱和正己烷除去脂肪和其他亲脂性化合物,检测限和定量限分别为0.4和0.8ng/g,远低于国标中其它聚醚类药物(1-150μg/kg)的最大残留限量。此方法对海南霉素的回收率范围为71.6%至101.2%,相对标准偏差范围为3.3%至14.9%。研究结果证明,该方法在检测动物性食品中残留海南霉素可靠简单的,并且在各种基质中具有高精度和准确性。
8.本发明的第一个目的是提供一种提取动物性食品中海南霉素的预处理方法,所述方法包括以下步骤:
9.(1)提取:向样品中加入乙腈、无水硫酸镁、无水乙酸钠进行提取,离心取上清液,得到提取液;
10.(2)净化:向提取液中加入乙腈饱和正己烷进行萃取,离心后取下层溶液。
11.在本发明的一种实施方式中,所述动物性食品包括牛奶,鸡蛋,脂肪,肾,肌肉和鸡,牛肉和羊肉。
12.在本发明的一种实施方式中,所述步骤(1)中乙腈添加量为3-5ml/g样品,无水硫酸镁添加量为0.3-0.5g/g样品,无水乙酸钠添加量为0.1-0.2g/g样品。
13.在本发明的一种实施方式中,所述步骤(1)中提取时间为1-3min。
14.在本发明的一种实施方式中,乙腈饱和正己烷添加量为1-2ml/ml提取液。
15.本发明的第二个目的是提供一种动物性食品中海南霉素含量的测定方法,所述方法包括以下步骤:
16.(1)样品预处理:先向样品中加入乙腈、无水硫酸镁、无水乙酸钠进行提取,离心取上清液,得到提取液;再向提取液中加入乙腈饱和正己烷进行萃取,离心后取下层溶液,氮吹、过膜;
17.(2)超高效液相色谱-高分辨质谱法测定样品中海南霉素的含量:采用高效液相色谱与串联质谱联用测定动物性食品中海南霉素的含量;所述液相色谱采用acquity uplc beh c18柱色谱柱,流动相a为乙腈,流动相b为甲酸-乙酸铵水溶液。
18.在本发明的一种实施方式中,所述甲酸-乙酸铵溶液中乙酸铵浓度为5mmol/l,甲酸浓度为0.1%,v/v。
19.在本发明的一种实施方式中,液相色谱采用acquity uplc beh c18柱(2.1mm
×
50mm,2.5μm)色谱柱,流动相为乙腈(a)和0.1%甲酸水溶液(含5mmol乙酸铵)(b);柱温为40℃;进样体积为3μl;流速为0.45ml/min;运行梯度为0-0.5min,60%a;0.5-4min,60%-2%a;4-6min,2%a;6-6.1min,2-60%a。
20.在本发明的一种实施方式中,超高效液相色谱-高分辨质谱法检测条件为:acquity uplc h-class(uhplc)超高效液相色谱和ab sciex三重四极5500系统进行检测;
其中,质谱采用电喷雾(esi )离子源,多反应监测(mrm)模式检测扫描目标分析物;质谱条件:离子源为esi( ),喷雾电压5500.0v,气帘气压力30.0psi,碰撞室压力13.0pis,离子源温度550℃。
21.在本发明的一种实施方式中,所述方法包括下述步骤:
22.(1)样品预处理
23.(a)称取2g试样,加入8ml乙腈,0.8g无水硫酸镁,0.2g无水乙酸钠,剧烈震摇1min,振荡提取2min,超声10min,8000r/min下离心5min,转移上清液至另一离心管中。
24.(b)准确取步骤(a)的待净化液2ml提取液于离心管中,加入2ml乙腈饱和正己烷,涡旋1min,8000r/min离心5min,弃去正己烷层,下层溶液于40℃氮气吹干,准确加入1ml乙腈溶液涡旋溶解,过0.22μm有机滤膜,供lc-ms/ms分析。
25.(2)lc-ms/ms测定样品中海南霉素的含量:
26.液相色谱采用acquity uplc beh c18柱(2.1mm
×
50mm,2.5μm)色谱柱,流动相为乙腈(a)和0.1%甲酸水(含5mmol乙酸铵)(b);柱温为40℃;进样体积为3μl;流速为0.45ml/min;运行梯度为0-0.5min,60%a;0.5-4min,60%-2%a;4-6min,2%a;6-6.1min,2-60%a。
27.超高效液相色谱-高分辨质谱法检测条件为:acquity uplc h-class(uhplc)超高效液相色谱和ab sciex三重四极5500系统进行检测;其中,质谱采用电喷雾(esi )离子源,多反应监测(mrm)模式检测扫描目标分析物;质谱条件:离子源为esi( ),喷雾电压5500.0v,气帘气压力30.0psi,碰撞室压力13.0pis,离子源温度550℃。
28.在本发明的一种实施方式中,海南霉素一级质谱扫描和二级质谱扫描,确定定量离子为845.6m/z,定性离子为863.6m/z,碰撞能量均为48ev,去簇电压为80.0v。
29.本发明的第三个目的是提供一种上述预处理方法和检测方法在检测动物食品品质方面的应用。
30.本发明的有益效果:
31.与现有技术相比,本发明具有以下优点:
32.1.本发明首次建立了动物性食品中海南霉素液质联用检测法,能高选择性检测动物组织和鸡蛋、牛奶等10种不同基质样本中海南霉素,填补畜禽可食组织中海南霉素检测的空白,为畜牧业养殖环境监管提供有力的法律依据和技术支撑,对促进我国畜牧业健康、快速、稳定、协调发展,以及促进我国的经济发展都具有深远的意义。
33.2.本发明方法检测动物组织和鸡蛋中海南霉素具有高灵敏度、重复性好等优势,回收率在71.6%至101.2%,相对标准偏差范围为3.3%至14.9%,检出限低至0.4μg/kg,远低于国标中其它聚醚类药物(1-150μg/kg)的最大残留限量。
附图说明
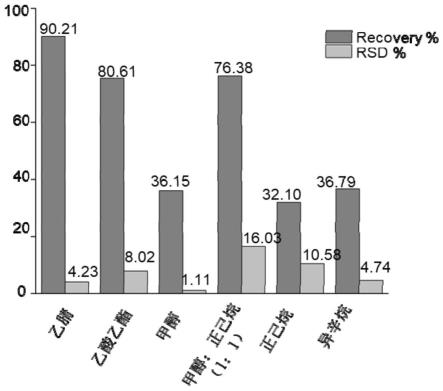
34.图1不同试剂提取海南霉素回收率。
35.图2提取过程中添加不同盐时海南霉素提取回收率(%)。
36.图3不同净化方式下海南霉素回收率(%)。
37.图4海南霉素在不同基质校准曲线。
38.图5 10种基质的基质效应评估(%)。图6在0.8ng/g添加回收浓度下,海南霉素标准物质总离子流图。
39.图7在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白鸡蛋总离子流图,b为空白鸡蛋加标后的总离子流图。
40.图8在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白牛奶总离子流图,b为空白牛奶加标后的总离子流图。
41.图9在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白鸡肉总离子流图,b为空白鸡肉加标后的总离子流图。
42.图10在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白鸡肝总离子流图,b为空白鸡肝加标后的总离子流图。
43.图11在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白牛肉总离子流图,b为空白牛肉加标后的总离子流图。
44.图12在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白牛肝总离子流图,b为空白牛肝加标后的总离子流图。
45.图13在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白羊肉总离子流图,b为空白羊肉加标后的总离子流图。
46.图14在0.8ng/g添加回收浓度下,海南霉素在不同基质中的总离子流图,其中,a为空白羊肝总离子流图,b为空白羊肝加标后的总离子流图。
47.图15 1ng/ml海南霉素在450,550,600℃离子源温度下信号强度。
48.图16海南霉素(1ng/ml)在流动相为a:乙腈和0.1%甲酸和b:乙腈and 0.1%甲酸(5mmol/l乙酸铵)是的离子流图。
具体实施方式
49.以下对本发明的优选实施例进行说明,应当理解实施例是为了更好地解释本发明,不用于限制本发明。
50.实施例1:
51.试样处理
52.取适量新鲜或冷藏空白或供试鸡蛋、动物组织,混合均匀。
53.‑‑
取均质后的供试样品,作为供试试料。
54.‑‑
取均质后的空白样品,作为空白试料。
55.‑‑
取均质后的空白样品,添加适宜浓度的标准工作液,作为空白添加试料。
56.试料的保存:-20℃以下保存
57.样品前处理:
58.(1)样品预处理
‑‑
鸡蛋、牛奶、鸡肝、鸡肉、牛肉、牛肝、羊肉、羊肝、脂肪、肾。
59.(a)称取2g试样,加入8ml试剂,0.8g无水硫酸镁,0.2g无水乙酸钠,剧烈震摇1min,振荡提取2min,超声10min,8000r/min下离心5min,转移上清液至另一离心管中。
60.(b)准确取步骤(a)的待净化液2ml提取液于离心管中,于40℃氮气吹干,加入2ml乙腈复溶和2ml乙腈饱和正己烷,涡旋1min,8000r/min离心5min,弃去正己烷层,下层溶液于40℃氮气吹干,准确加入1ml乙腈溶液涡旋溶解,过0.22μm有机滤膜,供lc-ms/ms分析。
61.海南霉素的液质联用检测方法,包括下述步骤:
62.仪器:ab sciex三重四极5500系统,含串联(tq)四极杆mass检测器和analyst软
件;waters2695hplc高效液相色谱仪;色谱柱:waters acquity uplc beh c18(2.5μm,2.1mm
×
50mm);电子天平:me204e电子天平(梅特勒-托利多仪器有限公司,上海)milli-q超纯水系统(millipore公司,美国);涡旋混合器:si可调速漩涡混合器,上海思伯明仪器设备有限公司;超声清洗:;试剂:海南霉素购自中国兽药检验所(中国北京)。
63.乙酸乙酯和甲醇、乙腈为色谱纯级,三者均购自sigma sigma-aldrich corporation(美国密苏里州圣路易斯);正己烷为分析纯级,购自sinopharm chemical reagent co.,ltd(国药集团化学试剂有限公司,上海);无水硫酸镁(分析级,99%,北京伊诺凯),无水乙酸钠(分析级,99%,北京伊诺凯))。
64.标准溶液配置:准确称取100mg海南霉素标准品,用甲醇水溶液溶解并定容至100ml,配置成浓度为1mg/ml的海南霉素标准储备液,放置-20℃储存六个月。
65.海南霉素工作液:准确吸取适量海南霉素标准储备液,用甲醇稀释成10μg/ml的标准工作液,4℃储存2周
66.液相条件:色谱柱:beh c18,(2.5μm,2.1mm
×
50mm),或性能相当者;流动相:a-乙腈,b-0.1%甲酸水溶液(含5mmol/l乙酸铵),洗脱程序如下表。
67.表1梯度洗脱程序
[0068][0069]
进样体积:3μl;质谱条件:扫描方式:正离子扫描;检测方式:mrm;检测器:串联四极杆;喷雾电压:5500.0v;去簇电压dp:80.0v;射入电压ep:10.0v;碰撞室射出电压cxp:13.0v;气帘气压力:30.0psi;离子源温度:550.0℃;柱温:40℃;流速:0.45ml/min;
[0070]
海南霉素钠相对分子式c
47h80o15
na,准确分子质量为907.12,采用电喷雾离子源正模式扫描,主要离子峰质荷比为907.5,二级质谱碎片863.5、845.5,以845.5为定量离子。
[0071]
基质匹配标准曲线
[0072]
准确移取海南霉素标准工作溶液适量,用乙腈稀释配置成浓度系列为0.5ng/ml、1ng/ml、5ng/ml、20ng/ml、50ng/ml的海南霉素标准溶液,空白样品经预处理,在复溶时,用海南霉素标准溶液复溶,制得浓度梯度为0.5ng/ml、1ng/ml、5ng/ml、20ng/ml、50ng/ml海南霉素基质匹配标准系列溶液,过0.22μm滤膜后,供lc-ms/ms分析,在0.2~50ng/ml浓度间,相关系数>0.99,线性良好。
[0073]
图4显示了从10种不同基质中获得的海南霉素的校准曲线。证明了该方法对基质成分具有选择性,适用于多种类型的基体。使用双尾t检验,通过t检验比较斜率表明,对于所有基质,校准曲线在α=0.05处具有统计差异,这表明存在绝对基质效应。因此,有必要在基质匹配的校准曲线中进行分析和定量。海南霉素的基质效应为-7.8%~-43.9%(图5),均为基质抑制效应。
[0074]
检出限(lod)与定量限(loq)的确定
[0075]
在空白基质中添加一定量的海南霉素标准品,样品经前处理后,用lc-ms/ms进行测定,当检测样品中色谱峰的信噪比大于等于3时,添加海南霉素的浓度即为检出限,样品中海南霉素的峰的信噪比大于等于10时,添加海南霉素的浓度即为定量限。该海南霉素分析方法对鸡、牛、羊的肌肉及其肝脏组织、牛奶和鸡蛋中检出限为0.4μg/kg、定量限为1μg/kg。
[0076]
回收率和精密度
[0077]
每种基质取空白样品,添加海南霉素0.8、2、10μg/kg 3个不同浓度,每个浓度平行6份,样品前处理后,在规定的测试条件下进行检测,计算回收率和实验室内变异系数。
[0078]
将样品前处理步骤(a)添加试剂调整为乙腈,步骤(b)调整为:(a)的待净化液2ml提取液于离心管中,加入2ml乙腈饱和正己烷,涡旋1min,8000r/min离心5min,弃去正己烷层,下层溶液于40℃氮气吹干,准确加入1ml乙腈溶液涡旋溶解,过0.22μm有机滤膜,供lc-ms/ms分析。其他条件或者参数与实施例1一致。
[0079]
当在基质中添加海南霉素0.8ng/g时,海南霉素在不同基质中的总离子流图如图6-14所示,无明显杂质干扰,证明检测方法的可靠性与准确性。
[0080]
由表2可知,海南霉素回收率在71.6%至101.2%,满足《实验室质量控制规范食品理化检测》(gb 27404-2008)附录f.1关于被测组分含量<0.1mg/kg时回收率范围在60%~120%的要求;批间相对标准偏差在3.3%-14.5%之间,满足《实验室质量控制规范食品理化检测》(gb 27404-2008)附录f.2关于被测组分含量<1μg/kg时实验室内变异系数<30%的要求。
[0081]
表2海南霉素检测方法的回收率和批间相对标准偏差
[0082]
[0083]
[0084]
[0085][0086]
实施例2:提取试剂的选择
[0087]
使用各种萃取溶剂评估提取效率以检测鸡蛋中的海南霉,以提高提取效率和回收率:
[0088]
(1)乙酸乙酯,(2)甲醇,(3)甲醇:乙腈(v/v=50/50),(4)乙腈,(5)异辛烷和(6)正己烷,其他条件同实施例1。图1展示了海南霉素的回收率和相对标准偏差(rsd)(%)的提取效率的比较。发现乙腈的提取效率(90.21%)高于乙酸乙酯(81.61%),乙酸乙酯用于提取鸡蛋、牛奶时极易发生乳化,阻碍提取的进行,乙腈的适用的范围更广,因此选用乙腈作为提取溶剂。
[0089]
实施例3:盐的选择
[0090]
前处理时加入盐能够促进有机相和水相分离,盐可以改变有机相在水中的溶解度,盐析作用还可以促进蛋白质沉淀,防止蛋白质乳化,调节ph,并增加了兽药的稳定性。通过筛选不同的盐来优化提取效率,分别使用了氯化钠、硫酸镁、硫酸镁和无水乙酸钠、硫酸钠、硫酸镁和柠檬酸钠、硫酸镁和氯化钠,其他条件或者参数与实施例1一致。并对比不使用盐的效果,如图2所示,结果发现,使用硫酸镁和无水乙酸钠的回收率最高(91.36%)。因此选用无水硫酸镁、无水乙酸钠辅助提取,乙腈作为萃取溶剂。
[0091]
实施例4:净化方法的选择
[0092]
样品制备是任何分析方法的关键方面。有效的样品预处理可以促进目标海南霉素的可靠分离,并用于减少基质效应。目前许多文献已经开发了各种预处理方法,用于监测非法使用的抗球虫药。但是由于基质的复杂性以及真实样品中的痕量水平的存在,这些前处理方法仍不能完全除去盐和内源化合物,这将会导致基质效应,大多数清洁方法基于固相微萃取(dspe)和固相萃取spe或液液萃取(lle)。
[0093]
为了找到合适的前处理方法,除去在提取海南霉素时的杂质,减少对仪器的损伤和检测的精确度,以鸡肝为处理对象,使用固相微萃取(dispersive solid phase extraction d-spe dspe1和dspe2)和液液萃取利用c18、乙腈饱和正己烷除去脂肪降低基质效应,以提高鸡肝中的海南霉素的回收率,其中,dspe1:25mg c18,150mg mgso4;dspe2:50mg psa,50mg c18,7.5mg gcb 150mg mgso4,具体操作步骤如下:
[0094]
将样品前处理步骤(b)调整为:(a)的待净化液2ml提取液于离心管中,分别加入2ml乙腈饱和正己烷、quechers dspe1、quechers dspe2,涡旋1min,8000r/min离心5min,弃去正己烷层、弃去下层quechers颗粒,将溶液于40℃氮气吹干,准确加入1ml乙腈溶液涡旋溶解,过0.22μm有机滤膜,供lc-ms/ms分析,其他条件或者参数与实施例1一致。
[0095]
图3呈现了使用液液萃取(乙腈饱和正己烷)和固相微萃取的回收率的比较,液液萃取(98.3%)在明显优于固相微萃取(dspe1和dspe2分别为75.3%和62.7%)。因此,在本研究中采用乙腈饱和的正己烷作为净化方法。
[0096]
实施例5:
[0097]
为了优化质谱以提高分析物海南霉素的灵敏度,调节离子源温度(450℃、550℃和600℃)(1ng/ml注射三次),当温度从550℃降低到450℃、升高到600℃时,在图15中可以观察到峰面积有39%减少,离子源温度在450℃和600
°
℃之间没有显着差异。因此,选择550℃作为合适离子源温度。
[0098]
实施例6:洗脱条件的选择
[0099]
为了更好洗脱海南霉素分离杂质优化色谱分辨率,对洗脱条件进行优化,当使用a:乙腈,b:0.1%甲酸水进行梯度洗脱时,发现色谱峰有拖尾现象,如图16所示,当将5mm乙酸铵加入含有0.1%甲酸的水中,显著改善了色谱峰拖尾现象,在流动相为a:乙腈,b:0.1%甲酸水(5mm乙酸铵)时,能够在最快洗脱时间内洗脱并且干扰峰较小,具有更好的峰形状。
[0100]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
