1.本发明涉及吲哚哌啶嘧啶类衍生物在制备新型冠状病毒抑制剂中的应用,属于药物技术领域。
背景技术:
2.新型冠状病毒,是引起不明原因肺炎流行的病原体。2020年2月11日,国际病毒学分类委员会将其命名为sars-cov-2,同日,世界卫生组织(who)将其引发的肺炎疾病命名为covid-19。sars-cov-2 疫苗虽然在抗击疫情中功不可没,但是能否有效应对sars-cov-2病毒的快速变异不得而知。国际上普遍认为,仅靠特异性针对已知病原的疫苗难以彻底终结covid-19疫情,小分子抗病毒药物则被寄予厚望。
3.目前sars-cov-2小分子抑制剂的研究正在进行中,有两种口服抗病毒药物饱受人们的关注。第一种是由默沙东公司研发的molnupiravir,其临床数据显示:在第一组临床实验中,与服用安慰剂相比,服用molnupiravir 的参与者的住院率和死亡率降低了一半。但在第二组中,服用抗病毒药物的人与服用安慰剂的人在结果上几乎没有差异。这个结果使得molnupiravir 失去了光环。第二种是辉瑞(pfizer)公司研发的新冠口服药paxlovid,试验结果显示,如果高风险成年人在出现首次症状后的几天内服用该口服药,能降低住院或死亡率达89%。此外,辉瑞表示,该口服药对快速传播的omicron 毒株也仍然有效。2021年12月22日,fda给予了paxlovid(nirmatrelvir [pf-07321332]片剂和利托那韦片剂,共同包装口服使用)紧急使用授权。
[0004]
目前,尚未有关吲哚哌啶嘧啶类衍生物可用于制备新型冠状病毒抑制剂的相关报道。
技术实现要素:
[0005]
本发明的目的是克服现有技术的不足,提供吲哚哌啶嘧啶类衍生物在制备新型冠状病毒抑制剂中的应用。
[0006]
本发明解决上述技术问题的技术方案如下:吲哚哌啶嘧啶类衍生物在制备新型冠状病毒抑制剂中的应用。
[0007]
本发明的吲哚哌啶嘧啶类衍生物在制备新型冠状病毒抑制剂中的应用的原理是:
[0008]
目前,尚未有关吲哚哌啶嘧啶类衍生物可用于制备新型冠状病毒抑制剂的相关报道。本发明的抗sars-cov-2活性测试结果表明,吲哚哌啶嘧啶类衍生物对sars-cov-2具有明显的抑制作用,抑制活性达到微摩尔水平,因此,可用于制备新型冠状病毒抑制剂。
[0009]
本发明的吲哚哌啶嘧啶类衍生物在制备新型冠状病毒抑制剂中的应用的有益效果是:
[0010]
本发明首次发现吲哚哌啶嘧啶类衍生物可以用于制备新型冠状病毒抑制剂,既开拓了吲哚哌啶嘧啶类衍生物新的应用领域,也开拓了新的新型冠状病毒抑制剂,具有重要的社会意义和医学价值。
[0011]
在上述技术方案的基础上,本发明还可以做如下改进。
[0012]
进一步,所述吲哚哌啶嘧啶类衍生物的化学结构式如式ⅰ所示:
[0013][0014]
其中,r为甲基、氯、溴、氟、三氟甲基和氰基中的任意一种。
[0015]
采用上述进一步的有益效果是:上述结构的吲哚哌啶嘧啶类衍生物具有优良的新型冠状病毒抑制活性,可用于制备新型冠状病毒抑制剂。
[0016]
更进一步,所述r为2-氯、3-氯、2-溴、3-溴、3-氟、3-三氟甲基、4
‑ꢀ
三氟甲基、4-氰基和3,5-二甲基-4-氯中的任意一种。
[0017]
采用上述更进一步的有益效果是:上述取代基对应的吲哚哌啶嘧啶类衍生物对sars-cov-2具有低的抑制活性(ec
50
)、高的半细胞毒性浓度(cc
50
)。
[0018]
更进一步,所述r为3-氟、3-三氟甲基和4-三氟甲基中的任意一种。
[0019]
采用上述更进一步的有益效果是:上述取代基对应的吲哚哌啶嘧啶类衍生物对sars-cov-2具有极低的抑制活性(ec
50
)、高的半细胞毒性浓度(cc
50
)。
[0020]
更进一步,所述r为2-氯、3-氯、2-溴、3-溴、3-氟、3-三氟甲基、4
‑ꢀ
三氟甲基、4-氰基和3,5-二甲基-4-氯中的任意一种。
[0021]
采用上述更进一步的有益效果是:上述取代基对应的吲哚哌啶嘧啶类衍生物具有较高的半细胞毒性浓度(cc
50
)。
[0022]
更进一步,所述r为2-氯、3-氯、2-溴、3-溴、3-氟、3-三氟甲基、4
‑ꢀ
三氟甲基和3,5-二甲基-4-氯中的任意一种。
[0023]
采用上述更进一步的有益效果是:上述取代基对应的吲哚哌啶嘧啶类衍生物对sars-cov-2具有极低的抑制活性(ec
50
)和很高的半细胞毒性浓度 (cc
50
)。
[0024]
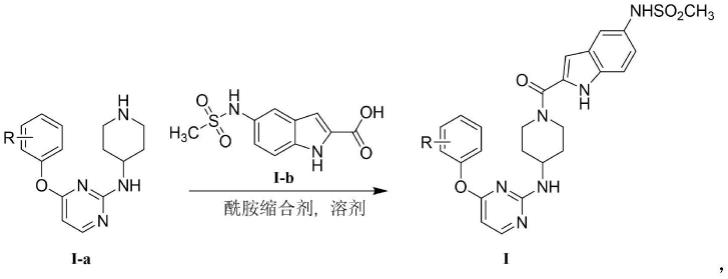
进一步,所述吲哚哌啶嘧啶类化合物的制备方法如下:
[0025]
在惰性气体保护下,将式i-b所示的5-(甲基磺酰氨基)-1h-吲哚-2-羧酸和酰胺缩合剂溶于溶剂中,搅拌后加入式
ⅰ‑
a所示的原料,室温搅拌至各原料消耗完全,分离纯化得到如式ⅰ所示的吲哚哌啶嘧啶类衍生物,其反应通式如下:
[0026][0027]
其中r具有与上述相同的定义。
[0028]
采用上述进一步的有益效果是:采用上述制备方法,可以得到本发明的吲哚哌啶嘧啶类化合物。
[0029]
更进一步,所述酰胺缩合剂为n,n-羰基二咪唑、n,n-二异丙基碳二亚胺、 n,n-二环己基碳二亚胺、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、 2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯和苯并三氮唑
ꢀ‑
n,n,n',n'-四甲基脲六氟磷酸盐中的任意一种或几种的混合。
[0030]
更进一步,所述溶剂为二氯甲烷、氯仿、四氯化碳、1,2-二氯乙烷、1,4
‑ꢀ
二氧六环、四氢呋喃、n,n-二甲基甲酰胺和二甲基亚砜中的任意一种或几种的混合。
[0031]
进一步,所述新型冠状病毒抑制剂包括吲哚哌啶嘧啶类衍生物和药学上可接受的载体。
[0032]
采用上述进一步的有益效果是:吲哚哌啶嘧啶类衍生物可以和药学上可接受的载体一起,制成新型冠状病毒抑制剂。
[0033]
更进一步,所述药学上可接受的载体为缓释剂、赋形剂、填充剂、粘合剂、润湿剂、崩解剂、吸收促进剂、吸附载体、表面活性剂和润滑剂中的任意一种或两种以上的混合物。
[0034]
采用上述更进一步的有益效果是:载体能改变抑制剂进入人体的方式和在体内的分布、控制抑制剂的释放速度并将抑制剂输送到靶向器官的体系。各种抑制剂载体释放和靶向系统能够减少抑制剂降解及损失,降低副作用,提高生物利用度,控制抑制剂释放速度,减少或避免血药浓度的“峰谷”波动,使抑制剂较平稳地持续发挥疗效。
[0035]
更进一步,所述新型冠状病毒抑制剂为外用制剂、口服制剂和或注射制剂中的任意一种。
[0036]
采用上述更进一步的有益效果是:吲哚哌啶嘧啶类衍生物可以制成多种剂型的药物,适用于多种给药途径,如外用制剂、口服制剂或注射制剂,注射给药可以为皮内、皮下、肌内、局部或静脉内给药。
[0037]
更进一步,所述外用制剂为喷雾剂或气雾剂。
[0038]
采用上述更进一步的有益效果是:外用制剂为喷雾剂或气雾剂,使用方便,奏效迅速;能保持抑制剂清洁和无菌状态,并能提高抑制剂的稳定性。
[0039]
更进一步,所述口服制剂为颗粒剂、胶囊剂、片剂和囊泡剂中的任意一种。
[0040]
采用上述更进一步的有益效果是:口服制剂是最常用的途径,具有安全、方便和经济的特点。
[0041]
更进一步,所述注射制剂由吲哚哌啶嘧啶类衍生物、主溶剂和助溶剂组成。
[0042]
采用上述更进一步的有益效果是:药效迅速、剂量准确、作用可靠,可适用于不宜口服给药的患者和不宜口服的药物,可发挥局部定位作用。
[0043]
更进一步,所述主溶剂为注射用水或者0.9%氯化钠溶液。
[0044]
更进一步,所述助溶剂为吐温-80、丙二醇、甘油、乙醇和peg-400中的任意一种或多种。
具体实施方式
[0045]
以下结合具体实施例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0046]
实施例1:目标化合物(
ⅰ‑
1)的合成
[0047][0048]
称取
ⅰ‑
b(1mmol)和2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(1mmol)溶于二氯甲烷(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a1(1mmol)的二氯甲烷(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
1,收率33.33%(以i-b计)。hrms(esi )m/z:541.4151(m h)
。1h nmr(400 mhz,dmso-d6)δ(ppm):11.62(s,1h,indnh),9.37(s,1h,s-nh),8.21(s, 1h,pyrh),7.59(s,1h,arh),7.30-7.48(m,6h,arh and nh),7.11(d, 1h,pyrh),6.67(m,1h,indch),6.24(s,1h,arh),4.38(m,2h,piph), 4.01(s,1h,piph),2.87(s,3h,s-ch3),1.78-1.98(m,3h,piph),1.40(m, 2h,piph),1.16(t,1h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm):170.41, 168.69,161.74,161.50,148.28,133.72,131.07,130.50,130.26, 128.54,127.12,127.03,126.38,124.58,119.60,114.51,112.59, 103.67,59.83,38.41,31.61,20.81,14.13。
[0049]
实施例2:目标化合物(
ⅰ‑
2)的合成
[0050]
[0051]
称取
ⅰ‑
b(1mmol)和苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐(1 mmol)溶于氯仿(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a2(1mmol) 的氯仿(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
2,收率38.23%(以i-b 计)。hrms(esi )m/z:541.4152(m h)
。1h nmr(400mhz,dmso-d6)δ (ppm):11.63(s,1h,indnh),9.37(s,1h,s-nh),8.21(s,1h,pyrh), 7.09-7.48(m,8h,arh and nh),6.74(s,1h,pyrh),6.21(s,1h,indch), 4.38(s,2h,piph),4.02(t,1h,piph),2.87(s,3h,s-ch3),1.98(s,1h, piph),1.89(s,2h,piph),1.42(s,2h,piph),1.16(t,1h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm):170.37,169.00,161.72,161.45,153.14, 133.70,133.45,131.06,130.97,130.49,127.01,125.38,122.32, 120.81,119.56,114.45,112.56,103.63,59.79,38.38,31.51,20.78, 14.11。
[0052]
实施例3:目标化合物(
ⅰ‑
3)的合成
[0053][0054]
称取
ⅰ‑
b(1mmol)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐 (1mmol)溶于四氯化碳(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a3 (1mmol)的四氯化碳(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
3,收率 26.13%(以i-b计)。hrms(esi )m/z:585.0756(m h)
。1h nmr(400mhz, dmso-d6)δ(ppm):11.62(s,1h,indnh),9.37(s,1h,s-nh),8.20(s,1h, pyrh),7.72(s,1h,arh),7.09-7.48(m,7h,arh and nh),6.73(d,1h, pyrh),6.22(m,1h,indch),4.38(m,2h,piph),4.02(m,1h,piph), 2.88(s,3h,s-ch3),1.80-1.98(m,3h,piph),1.41(m,2h,piph),1.16(t, 1h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm):169.10,162.17,161.91, 160.65,149.94,134.14,133.68,131.52,130.91,129.53,127.81, 127.45,125.00,120.03,116.52,114.94,113.00,104.07,60.23,38.82, 31.87,21.22,14.54。
[0055]
实施例4:目标化合物(
ⅰ‑
4)的合成
[0056][0057]
称取
ⅰ‑
b(1mmol)和n,n-二环己基碳二亚胺(1mmol)溶于1,2-二氯乙烷(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a4(1mmol)的1,2-二氯乙烷(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
4,收率29.6%(以i-b 计)。hrms(esi )m/z:585.0754(m h)
。1h nmr(400mhz,dmso-d6)δ (ppm):11.62(s,1h,indnh),9.36(s,1h,s-nh),8.22(s,1h,pyrh), 7.09-7.47(m,8h,arh and nh),6.74(s,1h,pyrh),6.21(s,1h,indch), 4.38(s,2h,piph),4.02(m,1h,piph),2.87(s,3h,s-ch3),1.99(s,1h, piph),1.88(s,2h,piph),1.42(s,2h,piph),1.17(m,1h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm):169.01,161.73,161.45,160.52,153.18, 133.70,131.31,131.09,130.48,128.26,127.02,125.11,121.17, 119.58,114.48,112.56,103.63,59.79,48.07,38.40,31.57,20.79, 14.12。
[0058]
实施例5:目标化合物(
ⅰ‑
5)的合成
[0059][0060]
称取
ⅰ‑
b(1mmol)和n,n-二异丙基碳二亚胺(1mmol)溶于1,4-二氧六环(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a5(1mmol)的1,4-二氧六环(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
5,收率43.5%(以i-b 计)。hrms(esi )m/z:525.1573(m h)
。1h nmr(400mhz,dmso-d6)δ (ppm):11.58(s,1h,indnh),9.33(s,1h,s-nh),8.20(s,1h,pyrh), 7.04-7.48(m,8h,arh and nh),6.74(s,1h,pyrh),6.18(s,1h,indch), 4.37(d,2h,piph),4.02(m,1h,piph),2.88(s,3h,s-ch3),1.88-1.98(m, 3h,piph),1.43(m,2h,piph),1.15-1.22(m,1h,piph)。
[0061]
实施例6:目标化合物(
ⅰ‑
6)的合成
[0062][0063]
称取
ⅰ‑
b(1mmol)和n,n-羰基二咪唑(1mmol)溶于四氢呋喃(6ml) 中,室温搅拌1h后,加入含有
ⅰ‑
a6(1mmol)的四氢呋喃(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
6,收率35.15%(以i-b计)。hrms(esi )m/z: 575.1525(m h)
。1h nmr(400mhz,dmso-d6)δ(ppm):11.59(s,1h,indnh), 9.34(s,1h,s-nh),8.22(s,1h,pyrh),7.37-7.68(m,6h,arh),7.17(s, 1h,nh),7.10(d,1h,arh),6.72(s,1h,pyrh),6.25(s,1h,indch), 4.35(d,2h,piph),4.03(m,1h,piph),2.87(s,3h,s-ch3),1.84-1.98(m, 3h,piph),1.15-1.40(m,3h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm): 169.40,162.16,161.85,160.59,153.12,134.14,131.49,131.35, 130.91,127.43,126.74,125.55,122.43,120.02,119.34,114.92, 112.99,104.04,60.22,48.42,38.85,31.91,21.20,14.53。
[0064]
实施例7:目标化合物(
ⅰ‑
7)的合成
[0065][0066]
称取
ⅰ‑
b(1mmol)和n,n-羰基二咪唑 n,n-二异丙基碳二亚胺 (0.5mmol 0.5mmol)溶于n,n-二甲基甲酰胺(6ml)中,室温搅拌1h 后,加入含有
ⅰ‑
a7(1mmol)的n,n-二甲基甲酰胺(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
7,收率33.56%(以i-b计)。hrms(esi )m/z:575.1524 (m h)
。1h nmr(400mhz,dmso-d6)δ(ppm):11.60(s,1h,indnh),9.35(s, 1h,s-nh),8.24(s,1h,pyrh),7.82(s,2h,arh),7.36-7.47(m,4h,arh), 7.18(s,1h,nh),7.10(d,1h,arh),6.73(m,1h,pyrh),6.27(s,1h, indch),4.02-4.37(m,3h,piph),2.87(s,3h,s-ch3),1.87-1.98(m,3h, piph),1.15-1.40(m,3h,piph)。
13
c nmr(100mhz,dmso-d6)δ(ppm): 169.27,162.17,161.89,160.68,155.98,134.14,131.52,130.93, 127.44,126.33,125.96,123.15,119.99,114.89,112.98,104.01,60.20, 38.86,31.92,21.20,18.99,14.53。
[0067]
实施例8:目标化合物(
ⅰ‑
8)的合成
[0068][0069]
称取
ⅰ‑
b(1mmol)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐 n,n-羰基二咪唑(0.5mmol 0.5mmol)溶于二甲基亚砜(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a8(1mmol)的二甲基亚砜(6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
8,收率33.33%(以i-b计)。hrms(esi )m/z: 541.4151(m h)
。1h nmr(400mhz,dmso-d6)δ(ppm):11.62(s,1h,indnh), 9.37(s,1h,s-nh),8.21(s,1h,pyrh),7.59(s,1h,arh),7.30-7.48 (m,6h,arh and nh),7.11(d,1h,pyrh),6.67(m,1h,indch),6.24(s, 1h,arh),4.38(m,2h,piph),4.01(s,1h,piph),2.87(s,3h,s-ch3), 1.78-1.98(m,3h,piph),1.40(m,2h,piph),1.16(t,1h,piph)。
13
c nmr (100mhz,dmso-d6)δ(ppm):170.41,168.69,161.74,161.50,148.28, 133.72,131.07,130.50,130.26,128.54,127.12,127.03,126.38, 124.58,119.60,114.51,112.59,103.67,59.83,38.41,31.61,20.81, 14.13。
[0070]
实施例9:目标化合物(
ⅰ‑
9)的合成
[0071][0072]
称取
ⅰ‑
b(1mmol)和n,n-二异丙基碳二亚胺 2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(0.5mmol 0.5mmol)溶于二甲基亚砜(6ml)中,室温搅拌1h后,加入含有
ⅰ‑
a9(1mmol)的四氢呋喃 (6ml)溶液,室温搅拌8h,薄层色谱监测反应,反应完全后,减压蒸馏反应液,得到油状物,柱层析分离纯化得
ⅰ‑
9,收率38.23%(以i-b计)。 hrms(esi )m/z:569.1569(m h)
。1h nmr(400mhz,dmso-d6)δ(ppm): 11.62(s,1h,indnh),9.36(s,1h,s-nh),8.17(s,1h,pyrh),7.41(s, 2h,arh),7.38(d,1h,arh),7.06-7.16(m,4h,arh and nh),6.74(s,1h, indch),6.11-6.17(m,1h,pyrh),4.37(s,2h,piph),4.02(m,1h,piph), 2.87(s,3h,s-ch3),2.33(s,6h,ch3),1.88-1.98(m,3h,piph),1.41(m, 2h,piph),1.17(m,1h,piph)。
[0073]
实验例
[0074]
采用如下方法对9个吲哚哌啶嘧啶类衍生物进行抗sars-cov-2生物活性测试:
[0075]
体外细胞水平的抗sars-cov-2病毒活性测定,主要包括对sars-cov-2 感染的vero-e6细胞的抑制活性及细胞毒性两方面。方法描述如下:使化合物在sars-cov-2感染的vero-e6细胞中,用核酸定量法测定药物对 sars-cov-2诱变的细胞病变的保护作用,计算使50%的细胞免于sars-cov-2 诱导的细胞病变所需的半数有效浓度(ec
50
),毒性测定与抗sars-cov-2活性实验平行进行,也是在vero-e6细胞培养中,用cck-8法测定使50%的未感染细胞发生细胞病变的浓度(cc
50
)。
[0076]
材料与方法:各化合物的抗sars-cov-2活性由药物对sars-cov-2在细胞中引起的细胞病变的抑制作用效率来监控。采用vero-e6细胞进行细胞培养。采用的病毒株为sars-cov-2病毒株。
[0077]
具体操作如下:
[0078]
1.细胞传代:提前一天将vero-e6细胞传代于48孔板中,5
×
104个/孔。
[0079]
2.药物稀释:初始药物用二甲基亚砜(dmso)溶解至0.1m浓度,而后用 dmem梯度稀释至测试浓度。
[0080]
3.细胞感染:感染前1h细胞培养基更换为含有测试浓度的化合物的培养基,而后细胞培养板转移至bsl3实验室,进行病毒感染。感染滴度为 0.01moi,所以每孔加入病毒量为500tcid
50
,用含同样浓度的dmso的培养基作为对照,每个化合物每个浓度重复三个孔。37℃孵育1h后,弃掉上清,用dmem洗一遍细胞,每孔加250μl含梯度浓度化合物的培养基,感染24h 后收感染上清进行病毒核酸拷贝数定量。
[0081]
4.感染后24h收感染感染上清,140μl感染上清加入560μl avl裂解液中,轻轻混匀,裂解15min后加入560μl无水乙醇,颠倒混匀,裂解病毒带出bsl3实验室,依照试剂盒(qiagen)说明书提取病毒核酸。
[0082]
5.病毒拷贝数定量:提取核酸上游引物 rbd-qf1:5'-caatggtttaacaggcacagg-3'(seq id no.1)、下游引物 rbd-qr1:5'-ctcaagtgtctgtggatcacg-3'(seq id no.2),进行荧光定量pcr,定量试剂盒为vazyme(ii),依据标准曲线计算每个反应中的病毒核酸拷贝数,而后计算化合物对病毒的抑制效果。药物抑制浓度以药物对病毒细胞病变作用产生50%抑制作用而同时对细胞无直接毒性的浓度 (cc
50
)。需要强调的是,当化合物水溶性较差,需要用dmso才能溶解时, dmso比浓度相对于水来讲,一般低于10%,dmso在vero-e6细胞培养介质中最终浓度小于2%。dmso最终浓度(1/1000)远远低于影响sars-cov-2在 vero-e6细胞中复制所需的浓度。
[0083]
表1中列出了9个吲哚哌啶嘧啶类衍生物对sars-cov-2抑制活性ec
50
和cc
50
。瑞德西韦的活性代谢物gs-441524也被作为参考药物同时测试。
[0084]
表1吲哚哌啶嘧啶类衍生物对sars-cov-2的抑制活性与细胞毒性
[0085][0086]
由表1可知,所测试的9个吲哚哌啶嘧啶类衍生物均具有sars-cov-2 抑制活性,且化合物的毒性小,选择性高,可用于制备新型冠状病毒抑制剂。
[0087]
所述新型冠状病毒抑制剂包括吲哚哌啶嘧啶类衍生物和药学上可接受的载体。
[0088]
所述药学上可接受的载体为缓释剂、赋形剂、填充剂、粘合剂、润湿剂、崩解剂、吸收促进剂、吸附载体、表面活性剂和润滑剂中的任意一种或两种以上的混合物。
[0089]
所述新型冠状病毒抑制剂为外用制剂、口服制剂和或注射制剂中的任意一种。
[0090]
所述外用制剂为喷雾剂或气雾剂。
[0091]
所述口服制剂为颗粒剂、胶囊剂、片剂和囊泡剂中的任意一种。
[0092]
所述注射制剂由吲哚哌啶嘧啶类衍生物、主溶剂和助溶剂组成。
[0093]
所述主溶剂为注射用水或者0.9%氯化钠溶液。
[0094]
所述助溶剂为吐温-80、丙二醇、甘油、乙醇和peg-400中的任意一种或多种。
[0095]
综上,本发明首次发现吲哚哌啶嘧啶类衍生物可以用于制备新型冠状病毒抑制剂,既开拓了吲哚哌啶嘧啶类衍生物新的应用领域,也开拓了新的新型冠状病毒抑制剂,具有重要的社会意义和医学价值。
[0096]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
