一种(bipy)cu
ii-tempo/有机碱催化体系催化氧化的反应机理研究分析方法
技术领域
1.本发明涉及一种量子化学计算方法,具体是针对(bipy)cu
ii-tempo/有机碱催化体系催化 氧化的反应机理研究分析方法。
背景技术:
2.醇的选择性氧化在有机合成中是非常重要的反应之一。目前,人们对于醇氧化的研究主 要集中在醇的绿色有氧氧化技术上。目前研究主要集中在cu-tempo类催化体系上。该类体 系对于一级醇的有氧氧化是非常有效的,并且由于该类体系使用氧气或空气作为氧化剂,其 氧化产物为醛和水,对环境没有污染,因此受到了人们的广泛关注。到目前为止,已经报道 了一系列的cu-tempo催化体系及反应机理的相关研究方法。例如,1984年,semmelhack 小组首次提出的cucl/tempo/dmf催化体系,能够有效地将苄基醇和烯丙基醇催化氧化成 相应的醛。但是,该催化体系对脂肪醇无活性。2003年,sheldon小组报道的 cubr2/bipy/tempo/kotbu催化体系能够以乙腈/水作为溶剂,在室温常压下,使用氧气为氧 化剂,将苄基醇和烯丙基醇氧化为相应的醛,值得一提的是该体系还成功地将脂肪族醇正辛 醇氧化成相应的醛。但是,正辛醇在常温下的氧化是缓慢的,但当反应体系的温度提升到40℃ 时,正辛醇就能完全转化了。
3.最近,kumpulainen小组在sheldon催化体系的基础上,用dbu或者nmi取代了有机碱 kotbu,并使用乙腈作为溶剂,发展出了(dbu/nmi)cu
ii
/bipy/tempo催化体系。该催化体系 在常温常压下,以氧气为氧化剂,不仅能将苄基醇和烯丙基醇氧化为相应的醛,而且对脂肪 醇的氧化也是非常有效的。根据实验数据可知,dbu作为碱时,反应体系有较强的活性,而 nmi作为碱时,反应活性则降低。但有趣的是,对于一些脂肪醇(例如: (ch3)2c=ch(ch2)2(ch3)c=chch2oh),使用dbu碱时没有活性,而使用nmi为碱时则反应 有很好的活性。而对于另外一些脂肪醇(例如:c9h
19
ch2oh),仅使用dbu碱或仅使用nmi 碱时反应均没有活性,dbu碱和nmi碱同时使用时,反应有很好的活性。因此,我们推测dbu 或nmi在cu
ii
/bipy/tempo催化体系催化氧化醇中起着非常重要的作用。然而,在该体系中仍 然存在许多没有解决的问题,例如nmi和dbu作为有机碱在cu
ii
/bipy/tempo催化体系中的具 体作用;cui氧化的具体的反应步骤;阴离子-otf在该催化系统中的作用以及反应中间体,过 渡态的结构;nmih
-otf抑制作用等等。
4.发明人在先专利cn1088256286b研究了以水为唯一溶剂的cu
ii-tempo催化体系催化氧 化醇成醛的反应机理,但是对于其他溶剂(如有机溶剂)中的cui/ii/氮氧自由基类仿生催化体 系,由于二者催化体系不同,所需溶剂不同、泛函方法、建模参数、过渡态计算方法、tof 计算公式等均有所不同,且需要考虑dbu/nmi和-otf存在着的协同作用。
5.因此,针对(bipy)cu
ii-tempo/有机碱催化体系催化氧化反应机理,需要建立一种高效准 确的反应机理研究方法,采用量子化学计算方法对该体系的反应机理进行分子水平的计算分 析,确定体系中间体、过渡态等情况,得到最优化的反应路径,cui氧化的具体
的反应步骤; 阴离子-otf在该催化体系中的作用;nmih
-otf抑制作用;以及nmi和dbu在催化体系 中的作用。
技术实现要素:
6.(bipy)cu
ii-tempo/有机碱(nmi、dbu)催化体系,是基于cu
ii
/bipy/tempo催化体系 中,nmi(n-甲基咪唑)、dbu(1,8-二氮杂二环[5.4.0]十一碳-7-烯)作为有机碱可以夺取醇 上的质子,反应式为(2-1):
[0007][0008]
反应生成的nmih
和dbuh
分别与-otf结合生成共轭碱酸nmih
-otf和 dbuh
-otf,它们的pka值分别为14.3和24(在乙腈中)。
[0009]
对于cu
ii
/bipy/tempo催化体系中,hoover通过动力学实验发现,cu
ii
/bipy/tempo/dbu 催化体系:反应对o2浓度呈一级动力学,对cu浓度呈一级和二级混合动力学,对tempo 浓度呈零级动力学,对醇的浓度成饱和动力学。
[0010]
cu
ii
/bipy/tempo/nmi催化体系:反应对o2浓度呈零级动力学,对cu浓度呈一级动力 学,对tempo浓度呈一级动力学,对醇的浓度成饱和动力学。同时hoover根据反应对cu 浓度呈一级和二级混合动力学的结果推测出反应依据以下方式进行:
[0011][0012]
该反应机理表明,cui的氧化过程需要经过两步反应,第一步为氧气氧化cui为cu
ii
oo
·-结构,该过程是一个可逆过程。第二步为cu
ii
oo
·-结构氧化另一个cui生成双核cu
ii
oocu
ii
中间体。
[0013]
为了进一步了解nmi和dbu作为有机碱在cu
ii
/bipy/tempo催化体系中的具体作用; cui氧化的具体的反应步骤;阴离子-otf在该催化系统中的作用以及反应中间体,过渡态的 结构;nmih
-otf抑制作用等等。本发明拟采用量子化学计算方法对(bipy)cu
ii-tempo/有机 碱催化体系催化氧化的反应机理进行研究。
[0014]
本发明提供(bipy)cu
ii-tempo/有机碱催化体系催化氧化的反应机理研究分析方法,通过 构建催化剂活性中心结构的计算模型选择最优模型;设计反应路径,通过反应中间体和过渡 态建模,得到正确的过渡态和反应路径;然后分析反应过程键长、键角、配位体结构等相关 情况,最终计算催化剂的转化频率tof值并与实验相比较,验证本分析方法的准确性。
[0015]
本发明所有的计算都是在gaussian 16程序下完成。在几何优化过程中采用更高精度的高 质量的dft积分格点,积分格点从fine级别增加到ultrafine级别。
[0016]
在反应模型中,本发明采用苯甲醇为反应物,乙腈(介电常数为35.688)为反应溶剂。 使用smd溶剂化模型(隐式溶剂化模型),在b3lyp-d3(bj)/def2svp/smd(acetonitrile)水平 下对所有中间体和过渡态进行几何优化并计算频率。
[0017]
优化计算的输入文件如下,中间体优化输入文件:#opt freq ub3lyp/def2svp nosymmscrf=(smd,solvent=acetonitrile)em=gd3bj;过渡态优化输入文件:#opt=(calcfc,ts,noeigen,gdiis) freq ub3lyp/def2svp nosymm scrf=(smd,solvent=acetonitrile)em=gd3bj;所有中间体没有虚频, 过渡态有且仅有一个虚频,并使用
shermo软件提取优化结束的输出文件*.log文件中吉布斯 热力学校正值,为了进一步细化能量,在m06/6-311 g(d,p)/smd(acetonitrile)水平下计算中间 体和过渡态的单点能量。
[0018]
单点能量计算输入文件如下:#um06/6-311 g**scrf=(smd,solvent=acetonitrile) geom=allcheck guess=read sp int=ultrafine,gaussian计算单点的输出文件*.log,读取log文件 中scf done:e(um06)后的能量为单点能量;将单点能量加上吉布斯热校正值得到每个结构的 吉布斯自由能量。
[0019]
在整个计算过程中,对于2:1变换使用-2.6kcal/mol的校正来减少熵贡献的影响,文中 势能面上的能量都是校正之后的能量。反应过程涉及两态反应,所以对于势能面交叉位置。 本发明采用卢天等人修改的sobmecp程序在b3lyp/def2svp计算水平下计算了最小能量交叉 点(sobmecp)。
[0020]
为确保所得结论不受计算方法的影响,关键步骤的中间体和过渡态均采用m06和tpssh 泛函方法重新计算。
[0021]
内禀反应坐标理论(irc)得到最小能量路径用于确定过渡态的准确性。irc方法被用来 确认过渡态;对于反铁磁耦合的cu配合物来说,即开壳层单重态,使用guess=mix和 guess=(mix,always)来获得稳定的波函数;由于一些反铁磁耦合的开壳层计算单线态导致一 定程度的自旋污染,根据下面公式进行能量校正:
[0022]
δst=2(es–et
)/2
–
《s2》s,
[0023]
其中es是开壳层单重态能量,e
t
是三重态能量,《s2》s是开壳层单重态的自旋污染,下 标s表示开壳层单重态,t表示三重态。
[0024]
本发明采用密度泛函方法对设计的各条反应路径上反应物、中间体、过渡态和产物,基 于化学键的生成和断裂形式以及关键配体的配位情况进行几何结构调整,然后在 b3lyp/def2svp计算水平下进行几何优化并得到各基元反应的活化能和反应焓变,绘出相应 的势能面图。
[0025]
在计算时要进行波函数稳定性测试,并采用对称性破损方法计算开壳层单重态的中间体 和过渡态的几何结构,并进行能量校正;从热力学和动力学角度分析得到势能面图,确定最 佳反应路径及其速控步骤,通过与实验结果比较进行可靠性分析后确定反应机理和催化剂最 优活性中心结构,并采用“真实溶剂模型”计算乙腈参与下的反应势能面。
[0026]
(1)构建催化剂活性中心结构的计算模型
[0027]
基于以(bipy)cu
ii-tempo/有机碱为催化剂进行苯甲醇有氧氧化成苯甲醛实验提供 的反应条件信息,构建催化剂活性中心的计算模型;采用密度泛函方法进行几何优化, 选择最优模型;
[0028]
所述有机碱选自dbu或nmi。
[0029]
(2)设计反应路径
[0030]
根据步骤(1)中选择的最优模型进行可能的反应路径设计;然后进行反应中间体 和过渡态建模:
[0031]
(a)构建c
α-h键断裂的中间体和过渡态:
[0032]
中间体建模:二配位的主配体和底物醇盐构成cu中心的三配位结构,向三配位的 cu中心加上tempo自由基构成cu中心的四配位结构,设置关键键长进行几何优化, 得到对应中间体结构;
[0033]
过渡态建模:采用柔性扫描方法,通过对关键化学键改变过程中可能的结构改变的 分析,进一步计算每一步结构的能量,获得关键化学键改变过程势能曲线;将该势能曲 线上能量最高点的对应结构作为初始过渡态结构完成初步建模;对过渡态模型进行几何 优化,获得相应结构,并对其进行hessian矩阵计算,获得频率计算结果,且频率计算证 明该过渡态结构有且仅有一个虚频,且虚频振动方向模式符合关键化学键生成或断裂规 律,过渡态模型建立正确;
[0034]
(b)构建o-h键断裂的中间体和过渡态:
[0035]
中间体建模:主配体、有机碱和-otf构成cu中心的四配位结构,调整关键键长: cu-n
有机碱
、cu-o
otf
和cu-n
ligand
键长,向四配位的cu中心加上底物醇,调整cu-o
sub
的 键长以及底物醇oh上的h与有机碱上的n的距离;进行几何优化,得到对应中间体结 构;
[0036]
过渡态建模:调整cu-n
有机碱
键、cu-o
otf
键、cu-n
ligand
键以及底物醇上c
α-h键键 长;设置底物醇oh上的h与有机碱上的n的距离,进行过渡态几何优化得到优化结构, 且频率计算结果有且仅有一个虚频;且分析该虚频的振动方向符合o
sub-h-n
有机碱
,证明 过渡态寻找正确;
[0037]
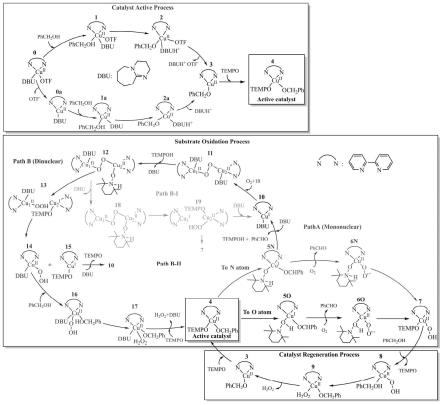
(c)构建oo-h键生成的中间体和过渡态:
[0038]
中间体建模:主配体、o2和tempoh构成cu中心的四配位结构,调整cu-oo
·-键、cu-o
tempoh
键、cu-n
ligand
键键长,设置tempoh-oo
·-距离;进行几何优化,得到 对应中间体结构;
[0039]
过渡态建模:调整cu-oo
·-键、cu-n
ligand
键、tempoh上n-h的键、tempoh-oo
·-键键长,进行过渡态几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一 步分析该虚频的振动方向符合oo
·-‑
h-n
tempo
,证明过渡态寻找正确;
[0040]
(d)构建h2o2生成的中间体和过渡态:
[0041]
中间体建模:主配体和ooh构成cu中心的三配位结构,底物醇配位到cu上形成 四配位结构;调整cu-ooh键、cu-n
ligand
键、cu-o
sub
键键长,设置phch2oh-ooh距 离,进行几何优化,得到对应中间体结构;
[0042]
过渡态建模:调整关键键长,调整phch2oh-ooh键、cu-n
ligand
键、设置phch2o-h 距离;将该过渡态模型进行几何优化和频率计算,得到有且只有一个虚频的过渡态结构; 进一步分析该虚频的振动方向符合phch2o-h-ooh,证明过渡态寻找正确;
[0043]
(3)分析反应路径和反应机理
[0044]
(4)tof计算
[0045]
使用能量跨度模型来分析整体的吉布斯自由能曲线,转化效率tof由autof程序 计算,通过下面公式计算催化剂的转化频率tof值,验证本分析方法的准确性:
[0046][0047]
其中:kb是玻尔兹曼常数,h是普朗克常数,r为理想气体常数,t为反应温度,δe 为反应路径的能量跨度,rdz区域指中间体(tdi)和决速过渡态(tdts)之间的区域, 该区域对应实验动力学分析结果。
[0048]
具体来说,对于(bipy)cu
ii-tempo/dbu催化体系催化氧化的反应机理研究分析方法,具 体包括如下步骤:otempoh
键、cu-n
ligand
键键长,设置tempoh-oo
·-距离;进行几何优化,得到对应中间 体结构。
[0066]
过渡态建模:调整cu-oo
·-键、cu-n
ligand
键、tempoh上n-h的键、tempoh-oo
·-键键长,进行过渡态几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分 析该虚频的振动方向符合oo
·-‑
h-n
tempo
,证明过渡态寻找正确。
[0067]
(d)构建h2o2生成的中间体和过渡态:
[0068]
中间体建模:主配体和ooh构成cu中心的三配位结构,底物醇配位到cu上形成四配 位结构;调整cu-ooh键、cu-n
ligand
键、cu-o
sub
键键长,设置phch2oh-ooh距离,进行 几何优化,得到对应中间体结构。
[0069]
过渡态建模:调整关键键长,调整phch2oh-ooh键、cu-n
ligand
键、设置phch2o-h距 离;将该过渡态模型进行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一 步分析该虚频的振动方向符合phch2o-h-ooh,证明过渡态寻找正确。
[0070]
对于dbu的双核路径,oo-h键生成与h2o2生成与单核路径有所不同。
[0071]
dbu的双核路径pathb-i:采用步骤(e)替换patha的步骤(c):
[0072]
(e)构建oo-h生成的中间体和过渡态:
[0073]
中间体建模:主配体、o2、tempoh和两分子cu构成cu中心的四配位双核结构 [(bipy
(1)
cu
(1)ii
(oo)cu
(2)ii tempoh(bipy
(2)
)]
2
,tempoh配位在cu2中心,调整cu
1-oo
·-键、 调整cu
2-oo
·-键、cu-o
tempoh
键、cu-n
ligand
键键长以及tempoh-oo
·-距离;设置结束后 进行几何优化,得到对应中间体结构。
[0074]
过渡态建模:调整关键键长:调整cu
1-oo
·-键、cu
2-oo
·-键、cu-n
ligand
键、tempoh 上n-h的键长以及tempoh-oo
·-键键长;将调整好的过渡态模型进行过渡态几何优化和频 率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合 oo
·-‑
h-n
tempo
,证明过渡态寻找正确。
[0075]
dbu的双核路径pathb-ii:采用步骤(f),(g)分别替换patha的步骤(c),(d):
[0076]
(f)构建oo-h键生成的中间体和过渡态:
[0077]
中间体建模:主配体、o2和tempoh、dbu和两分子cu构成cu中心的四配位双核结 构[(bipy
(1)
(dbu)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
,dbu和tempoh分别配位在两个 cu中心,调整cu
1-oo
·-键、cu
2-oo
·-键、cu-o
tempoh
键、调整cu
1-n
dbu
键长、cu-n
ligand
键 键长、以及tempoh-oo
·-距离;设置结束后进行几何优化,得到对应中间体结构;
[0078]
过渡态建模:调整关键键长:调整cu
1-oo
·-键、cu
2-oo
·-键、cu
1-n
dbu
键、cu-n
ligand
键、tempoh上n-h的键长以及tempoh-oo
·-键键长;将调整好的过渡态模型进行过渡态 几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向 符合oo
·-‑
h-tempo,证明过渡态寻找正确;
[0079]
(g)构建h2o2生成的中间体和过渡态:
[0080]
中间体建模:主配体、ooh和dbu构成cu中心的四配位结构,底物醇通过氢键连接 配位到cu的第二配位层;调整cu-ooh键、cu-n
ligand
键、cu-o
sub
键键长、phch2oh-ooh 距离以及cu-n
dbu
键长,设置结束后进行几何优化,得到对应中间体结构;
[0081]
过渡态建模:调整关键键长:phch2oh-ooh键、cu-n
ligand
键键长以及phch2o-h距离; 将该过渡态模型进行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分 析该虚频的振动方向符合phch2o-h-ooh,证明过渡态寻找正确。
位结构;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键长为 phch2oh-ooh距离为设置结束后进行几何优化,得到对应中间体结构;
[0111]
过渡态建模:调整关键键长,调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确。
[0112]
同理dbu碱源的催化体系,对于nmi的催化体系的反应机理,双核反应机理pathd-i 和pathd-ii除了包括单核反应机理的一些中间体和过渡态的建模过程,还包含以下不同的反 应中间体和过渡态建模。
[0113]
对于nmi的催化体系的双核路径pathd-i,采用步骤(e)替换pathc的步骤(c):
[0114]
(e)构建oo-h生成的中间体和过渡态:
[0115]
中间体建模:主配体、o2、tempoh和两分子cu构成cu中心的四配位双核结构[(bipy
(1)
cu
(1)ii
(oo)cu
(2)ii tempoh(bipy
(2)
)]
2
,tempoh配位在cu2中心,调整cu
1-oo
·-键长 为调整cu
2-oo
·
键长为cu-o
tempoh
键长为cu-n
ligand
键长为 tempoh-oo
·-距离为设置结束后进行几何优化,得到对应中间体结构;
[0116]
过渡态建模:调整关键键长:调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为 cu-n
ligand
键长从调整到tempoh上n-h的键长从1.062调整到 tempoh-oo
·-键键长从调整到将调整好的过渡态模型进行过渡态几何优化 和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合 oo
·-‑
h-tempo,证明过渡态寻找正确。
[0117]
对于nmi的双核路径pathd-ii,采用步骤(f),(g)分别替换pathc的步骤(c),(d):
[0118]
(f)构建oo-h键生成的中间体和过渡态:
[0119]
中间体建模:主配体、o2和tempoh、nmi和两分子cu构成cu中心的四配位双核结 构[(bipy
(1)
(nmi)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
,nmi和tempoh分别配位在两个cu 中心,调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为cu
2-o
tempoh
键长为调整cu
1-n
nmi
键长为cu-n
ligand
键长为tempoh-oo
·-距离为设置 结束后进行几何优化,得到对应中间体结构;
[0120]
过渡态建模:调整关键键长:调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为 cu
1-n
nmi
键长从调整到cu-n
ligand
键长从调整到tempoh 上n-h的键长从1.064调整到tempoh-oo
·-键键长从调整到将调 整好的过渡态模型进行过渡态几何优化和频率计算,得到有且只有一个虚频的过渡态结构; 进一步分析该虚频的振动方向符合oo
·-‑
h-tempo-,证明过渡态寻找正确;
[0121]
(g)构建h2o2生成的中间体和过渡态:
[0122]
中间体建模:主配体、ooh和nmi构成cu中心的四配位结构,底物醇通过氢键连接配 位到cu的第二配位层;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键 长为phch2oh-ooh距离为cu-n
nmi
键长为设置结束后进行几何 优化,得
到对应中间体结构;
[0123]
过渡态建模:调整关键键长:调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确。
[0124]
(3)分析反应机理和路径;
[0125]
分析各基元反应中过渡态及中间体的键长、键角及中心金属离子周围的立体化学变化, 总结出反应过程中心金属离子的配位数,配体的空间构型,配体与中心金属离子的键长、键 角,及相应的结构参数的变化规律。
[0126]
(4)tof计算验证
[0127][0128]
为了获得完整催化循环的更多信息,我们使用能量跨度模型来分析整体的吉布斯自由能 曲线,转化效率(“tof”)由autof程序计算。
[0129]
对于cu
ii
/bipy/tempo/nmi体系来说,对于pathc、pathd-i和pathd-ii,autof程序计 算的tof决速中间体(tdi)和决速过渡态(tdts)均相同,对应的能量跨度δe为16.7kcal/mol, tof=1.5
×
104h-1
,该体系的速控步骤是底物氧化步骤中的tempo夺取底物醇盐的h原子生 成产物醛的过程,计算结果符合实验上的动力学依赖。
[0130]
本发明取得的有益效果:
[0131]
本发明通过量子化学技术手段从原子水平上获得反应机理和催化剂构效关系,并最终建 立了(bipy)cu
ii-tempo/有机碱为催化剂进行苯甲醇有氧氧化成苯甲醛的催化机理进行研究进 而评价催化剂活性,为设计绿色环保的新型仿生催化剂提供基础数据和理论指导,帮助缩短 催化剂的研发周期和降低经费投入。
[0132]
本发明通过采用密度泛函理论(dft)对以(bipy)cu
ii-tempo/有机碱(dbu或nmi)为 催化剂进行苯甲醇有氧氧化成苯甲醛的催化机理进行研究,对于(bipy)cu
ii-tempo/dbu催 化体系,提出了三条反应路径,其中pathb-i(双核)是优势路径(δe=11.2kcal mol-1
;tof= 1.4
×
108h-1
h-1
)。在该体系中,评估了阴离子-otf的作用,通过本发明的分析方法及计算结 果表明-otf能够帮助dbu夺氢,说明阴离子-otf在该过程中起到一定的贡献。
[0133]
在(bipy)cu
ii-tempo/nmi催化体系中,我们将(bipy)cu
ii-tempo/dbu催化体系中的dbu 用nmi取代来评估该体系的反应机理,通过计算,该体系的速控步骤是底物氧化步骤中的 tempo夺取底物醇盐的h原子生成产物醛的过程,计算结果也符合实验上的动力学依赖。 在该体系中,我们也评估了阴离子-otf的作用,通过本发明的分析方法及计算结果表明-otf 能够帮助nmi夺氢,说明阴离子-otf在该过程中起到一定的贡献。
[0134]
通过本发明的分析方法,可以看出(bipy)cu
ii-tempo/dbu催化体系的反应势垒比 (bipy)cu
ii-tempo/nmi催化体系的反应能量势垒低,说明(bipy)cu
ii-tempo/dbu催化体系的 反应活性比(bipy)cu
ii-tempo/nmi催化体系的反应活性高,并且nmih
-otf对反应体系有抑 制作用,这与实验结果一致。说明本发明的分析方法准确性高。
[0135]
本发明还探究了nmih
-otf对反应体系的抑制作用,nmi碱源的(bipy)cu
ii-tempo
催化体系的速控步骤在底物氧化过程,涉及了共轭酸nmih
-otf,nmih
-otf在乙腈中的pka=14.3,它容易提供氢质子可以促进cu
ii-or分解,平衡向左进行会抑制生成活性催化剂,因此加入nmih
-otf会对体系的产生抑制作用,在dbu碱源的(bipy)cu
ii-tempo催化体系的速控步骤在催化剂再生过程,速控步骤不涉及共轭酸dbuh
-otf和cu
ii-or基团,且dbuh
-otf在乙腈中的pka=24,不易提供氢质子分解cu
ii-or基团,因此加入dbuh
-otf不会对催化体系的反应活性产生影响。
附图说明
[0136]
图1cu
ii
/bipy/tempo/dbu催化体系一级醇氧化反应可能的机理(patha、pathb-i和pathb-ii);
[0137]
图2cu
ii
/bipy/tempo/nmi催化体系一级醇氧化反应可能的机理(pathc、pathd-i和pathd-ii);
[0138]
图3patha的吉布斯自由能曲线图;
[0139]
图414
→1ts
4-5_n
/1ts
4-5_o
的氢原子转移的分子轨道图;
[0140]
图5pathb-i的吉布斯自由能曲线图;
[0141]
图6pathb-ii的吉布斯自由能曲线图;
[0142]
图7pathc的吉布斯自由能曲线图;
[0143]
图8pathd-i的吉布斯自由能曲线图;
[0144]
图9pathd-ii的吉布斯自由能曲线图。
具体实施方式
[0145]
下面结合具体实施例对本发明作进一步说明,但本发明并不限于以下实施例。
[0146]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0147]
起始底物确定
[0148]
在cu
ii
/bipy/tempo催化体系中,存在多种物质:底物醇、tempo、o2、dbu(nmi)、cu(-otf)2等。因为cu是以 2价存在的,所以我们选取了3种可能的起始物:[(bipy)cu
ii
(-otf)]
,[(bipy)cu
ii
(dbu/nmi)]
2
和[(bipy)cu
ii
(dbu/nmi)(-otf)]
。通过计算,我们发现[(bipy)cu
ii
(dbu/nmi)(-otf)]
的能量最低(如表1)。
[0149]
进一步地,[(bipy)cu
ii
(dbu/nmi)(-otf)]
的能量最低,而只存在有机碱dbu/nmi或是只存在-otf的起始底物能量均比[(bipy)cu
ii
(dbu/nmi)(-otf)]
高,说明有机碱dbu/nmi和-otf存在着一定的协同作用。
[0150]
因此,我们选择[(bipy)cu
ii
(dbu/nmi)(-otf)]
作为cu
ii
/bipy/tempo催化体系催化剂的起始结构。
[0151]
反应机理
[0152]
在不同碱源dbu和nmi的cu
ii
/bipy/tempo体系中,我们分别都探索了三种不同的反应路径,所有的这些路径考虑了完整的催化循环,即催化剂的活化,底物的氧化和催化剂的再生过程三部分。
[0153]
表1
cu-o
sub
的键长为底物醇oh上的h与dbu上的n的距离设置为设置结 束后进行几何优化,得到对应中间体结构;
[0169]
过渡态建模:调整关键键长,将cu-n
dbu
键键长从调整到将cu-o
otf
键键长从调整到cu-n
ligand
键长为从调整到将底物醇上c
α-h 键键长从调整到底物醇oh上的h与dbu上的n的距离设置由调整为将调整好的过渡态模型进行过渡态几何优化得到优化结构,且频率计算结果 有且仅有一个虚频;且分析该虚频的振动方向符合o
sub-h-n
dbu
,证明过渡态寻找正确;
[0170]
(c)构建oo-h键生成的中间体和过渡态:
[0171]
中间体建模:主配体、o2和tempoh构成cu中心的四配位结构,调整cu-oo
·-键长为 cu-o
tempoh
键长为cu-n
ligand
键长为tempoh-oo
·-距离为设置结束后进行几何优化,得到对应中间体结构;
[0172]
过渡态建模:调整关键键长,调整cu-oo
·-键长从调整到cu-n
ligand
键 长从调整到tempoh上n-h的键长从1.054调整到tempoh-oo
·-键键长从调整到将调整好的过渡态模型进行过渡态几何优化和频率计算, 得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合oo
·-‑
h-n
tempo
, 证明过渡态寻找正确;
[0173]
(d)构建h2o2生成的中间体和过渡态:
[0174]
中间体建模:主配体和ooh构成cu中心的三配位结构,底物醇配位到cu上形成四配 位结构;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键长为 phch2oh-ooh距离为设置结束后进行几何优化,得到对应中间体结构;
[0175]
过渡态建模:调整关键键长,调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确;
[0176]
对于dbu的催化体系的反应机理,双核反应机理pathb-i和pathb-ii除了包括单核反应 机理的一些中间体和过渡态的建模过程,还包含以下不同的反应中间体和过渡态建模。
[0177]
对于dbu的双核路径pathb-i,其他步骤与单核patha相同,仅是步骤c)替换为如下中 间体和过渡态建模的步骤e):
[0178]
(e)构建oo-h生成的中间体和过渡态:
[0179]
中间体建模:主配体、o2、tempoh和两分子cu构成cu中心的四配位双核结构 [(bipy
(1)
cu
(1)ii
(oo)cu
(2)ii tempoh(bipy
(2)
)]
2
,tempoh配位在cu2中心,调整cu
1-oo
·-键长 为调整cu
2-oo
·-键长为cu-o
tempoh
键长为cu-n
ligand
键长为 tempoh-oo
·-距离为设置结束后进行几何优化,得到对应中间体结构;
[0180]
过渡态建模:调整关键键长:调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为 cu-n
ligand
键长从调整到tempoh上n-h的键长从1.062调整到
ꢀ
tempoh-oo
·-键键长从调整到将调整好的过渡态模型进行过渡态几何优化 和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合 oo
·-‑
h-n
tempo
,证明过渡态寻找正确。
[0181]
对于dbu的双核路径pathb-ii,其他步骤与单核patha相同,仅是采用步骤(f),(g)替换 步骤(c),(d):
[0182]
(f)构建oo-h键生成的中间体和过渡态(替换单核patha中的步骤c):
[0183]
中间体建模:主配体、o2和tempoh、dbu和两分子cu构成cu中心的四配位双核结 构[(bipy
(1)
(dbu)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
,dbu和tempoh分别配位在两个 cu中心,调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为cu
2-o
tempoh
键长为 调整cu
1-n
dbu
键长为cu-n
ligand
键长为tempoh-oo
·-距离为距离为设置结束后进行几何优化,得到对应中间体结构;
[0184]
过渡态建模:调整关键键长:调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为 cu
1-n
dbu
键长从调整到cu-n
ligand
键长从调整到tempoh上 n-h的键长从1.069调整到tempoh-oo
·-键键长从调整到将调整 好的过渡态模型进行过渡态几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进 一步分析该虚频的振动方向符合oo
·-h-tempo,证明过渡态寻找正确;
[0185]
(g)构建h2o2生成的中间体和过渡态(替换单核patha的步骤d):
[0186]
中间体建模:主配体、ooh和dbu构成cu中心的四配位结构,底物醇通过氢键连接 配位到cu的第二配位层;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键 长为phch2oh-ooh距离为cu-n
dbu
键长为设置结束后进行几何 优化,得到对应中间体结构;
[0187]
过渡态建模:调整关键键长:调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确。
[0188]
(3)分析反应机理和路径;
[0189]
分析各基元反应中过渡态及中间体的键长、键角及中心金属离子周围的立体化学变化, 总结出反应过程中心金属离子的配位数,配体的空间构型,配体与中心金属离子的键长、键 角,及相应的结构参数的变化规律;
[0190]
单核patha:
[0191]
o-h键断裂过程中,cu-n
dbu
键键长从增加到cu-o
otf
键键长从增加到将底物醇上o-h键键长从增加到底物醇oh上的h与dbu 上的n的距离设置由减小为中心金属cu
ii
周围为二配位的主配体、dbu配 体和-otf及底物醇;经过o-h键断裂后形成以二配位的主配体、dbuh
和-otf和醇盐配位 的四面体形式配位的cu
ii
活性中心结构;随后,c
α-h键断裂过程cu-o
phch2o
键长从增加到醇盐上c
α-h键键长从增加到tempo-h键键长从减少到
经过c
α-h键断裂过程后形成以二配位的主配体、产物苯甲醛和tempoh配 位的cui活性中心结构;随后,oo-h键生成过程中从cu-oo
·-键长从减小到 cu-n
ligand
键长从减小到tempoh上n-h的键长从增加到 tempoh-oo
·-键键长从减小到经过oo-h键生成过程后形成以二配位的主 配体、tempo和ooh配位的cu
ii
活性中心结构;最后,h2o2生成过程中phch2oh-ooh 键长从减小到cu-n
ligand
键长从减小到phch2o-h距离从 增加到经过h2o2生成过程后形成以二配位的主配体、醇盐和h2o2配位的 cu
ii
活性中心结构。
[0192]
双核pathb-i:
[0193]
pathb-i与patha仅是催化剂的再生过程不同(oo-h键生成过程不同),其他部分与patha 相同,具体来说:o-h键断裂过程中,cu-n
dbu
键键长从增加到cu-o
otf
键 键长从增加到将底物醇上o-h键键长从增加到底物醇oh 上的h与dbu上的n的距离设置由减小为中心金属cu
ii
周围为二配位的 主配体、dbu配体和-otf及底物醇;经过o-h键断裂后形成以二配位的主配体、dbuh
和 ‑
otf和醇盐配位的四面体形式配位的cu
ii
活性中心结构;随后,c
α-h键断裂过程cu-o
phch2o
键长从增加到醇盐上c
α-h键键长从增加到tempo-h键 键长从减少到经过c
α-h键断裂过程后形成以二配位的主配体、产物苯甲醛 和tempoh配位的cui活性中心结构;随后,oo-h键生成过程中cu
1-oo
·-键长从调整到cu
2-oo
·-键长从调整到cu-n
ligand
键长从调整到 tempoh上n-h的键长从1.062增加到tempoh-oo
·-键键长从减小到 经过oo-h键生成过程后形成以二配位的主配体、tempo和ooh和两分子cu配 位的cui和cu
ii
活性中心的四配位双核结构[(bipy
(1)
cu
(1)i
(ooh)cu
(2)ii
(tempo)(bipy
(2)
)]
2
最 后,h2o2生成过程中phch2oh-ooh键长从减小到cu-n
ligand
键长从键长从减小到phch2o-h距离从增加到经过h2o2生成过程后形成以二 配位的主配体、醇盐和h2o2配位的cu
ii
活性中心结构。
[0194]
双核pathb-ii:
[0195]
pathb-i与patha仅是催化剂的再生过程(即oo-h键生成过程过程和h2o2生成过程) 不同,其他部分与patha相同,具体分析如下:o-h键断裂过程中,cu-n
dbu
键键长从键键长从增加到cu-o
otf
键键长从增加到将底物醇上o-h键键长从h键键长从增加到底物醇oh上的h与dbu上的n的距离设置由减小为中心金属cu
ii
周围为二配位的主配体、dbu配体和-otf及底物醇;经过o-h键断裂后形成 以二配位的主配体、dbuh
和-otf和醇盐配位的四面体形式配位的cu
ii
活性中心结构;随后, c
α-h键断裂过程cu-o
phch2o
键长从增加到醇盐上c
α-h键键长从增 加到tempo-h键键长从减少到经过c
α-h键断裂过程后形成以二 配位的主配体、产物苯甲醛和tempoh配位的cui活性中心结构(这里的红字和单核一样); 随后,oo-h键生成过程中cu
1-oo
·-键长从调整为cu
2-oo
·-键长从调整为
cu
1-n
dbu
键长从减小到cu-n
ligand
键长从调整到 tempoh上n-h的键长从1.069增加到tempoh-oo
·-键键长从减 小到经过oo-h键生成过程后形成以二配位的主配体、tempo、dbu、ooh和两 分子cu配位的cui和cu
ii
活性中心的四配位双核结构[(bipy
(1)
(dbu)cu
(1)ii
(ooh)cu
(2)i tempo(bipy
(2)
)]
2
。最后,h2o2生成过程中phch2oh-ooh键长从调整到 cu-n
ligand
键长从调整到phch2o-h距离从调整到经过h2o2生成过程后形成以二配位的主配体、醇盐、dbu和h2o2配位的cu
ii
活性中心结构。
[0196]
根据反应路径和绘制势能图,分析得到单核path a反应原理:
[0197]
1)催化剂活化
[0198]
催化剂活化过程包含两个步骤,质子转移步骤和活性催化剂形成的步骤(图1)。有机 碱dbu/nmi和-otf存在着一定的协同作用。在碱源dbu的(bipy)cu
ii-tempo催化体系中, 起始结构为(bipy)cu
ii
(-otf)(dbu)(标记为0)。底物醇phch2oh配位到20上形成结构21, 随后,dbu夺取phch2oh上的质子,形成了22结构。该dbu夺取质子的过程的反应势垒 为18.0kcal/mol,22结构中的dbuh
和-otf解离形成23结构,然后tempo配位到23上形 成活性催化剂14。催化剂活化步骤(
20→14)需吸热5.1kcal/mol。
[0199]
另外,-otf是否能够帮助dbu夺取phch2oh上的质子?我们需要评估-otf的作用, 在没有-otf的结构中,重新标记为20a,21a,2ts
1a-2a
,22a。21a
→22a的过程为有-otf参与 的dbu夺取phch2oh上的质子的过程。在此过程中,需要经过能量势垒为21.1kcal/mol的 2
ts
1a-2a
。通过对比21a
→22a和
21→22的反应过程,我们发现,有-otf参与的反应过程
21→22 的能量势垒比没有-otf参与的反应过程21a
→22a的能量势垒低3.1kcal/mol。因此,我们认为 阴离子-otf参与了dbu夺氢的过程,能够降低质子转移过程的能量势垒,帮助dbu夺氢。
[0200]
2)底物氧化
[0201]
催化剂活化过程之后,进行底物氧化过程,在4结构中,由于tempo有一个成单电子, 所以结构4有两种自旋态
1,3
4,14比34低0.5kcal/mol。由于b3lyp泛函计算的误差在 3-5kcal/mol之内,而14和34的能量差在其误差范围之内,因此两态反应均需要考虑。底物 醇上的c
α-h键上的h原子转移到tempo上形成产物醛。至今,h原子转移到tempo的的 n原子上还是o原子上仍然存在争议,在本体系中用乙腈作溶剂时,h原子是转移到哪里尚 未可知,因此对于这两种情况都进行了考虑(到n上:4
→
ts
4-5n
;到o上:4
→
ts
4-5o
图3)。 如图3所示,h原子转移到n原子的势垒比o原子低5.0kcal/mol。为了验证该能量是否受使 用的泛函的影响,本文使用m06和tpssh两个泛函重新计算关键中间体4和过渡态(ts
4-5n
和ts
4-5o
)(表2),即使用不同的dft方法,对于这个h原子转移步骤仍然会得到一致的 结论。为验证这一的结论,采用m06泛函和tpssh泛函将活性催化剂4和过渡态ts
4-5o
和 ts
4-5n
重新进行了计算;计算结果显示,对于h转移步骤,即使采用不同的dft方法,该h 原子转移步骤仍然会得到一致的结论,综上,该催化体系中底物醇上的c
α-h键上的h原子 转移到tempo的n原子上。从配合物34到
1u
ts
4-5n
的h转移步骤的能垒为9.0kcal mol-1
, 随后产生产物苯甲醛和tempoh。
[0202]
表2
[0203][0204]
为了进一步了解h转移到n原子上的能量更低的原因,本发明讨论了 14→
1u
ts
4-5n
/
1u
ts
4-5o
→15n/15o期间的电子转移过程。在14
→
1u
ts
4-5n
/
1u
ts
4-5o
→15n/15o过程 中,cu、tempo和och2ph基团的自旋密度cu:( 0.46
→
0.35/ 0.21
→
0.00/0.00);och2ph: ( 0.15
→‑
0.21/-0.15
→
0.00/0.00);tempo:(-0.77
→‑
0.23/-0.13
→
0.00/0.00)。结合分子 轨道分析(图4),对于14
→
1u
ts
4-5o
过程,两个键(cu-och2ph和och2ph基团中c
α-h键) 部分断裂,一个键(otempo-h)部分形成。cu-och2ph键的均裂产生一小部分β-电子迁 移到cu中心,导致cu中心的β-自旋数增加(ts
4-5o
:cu:9.3%
→
67.6%)。相应的α-自旋 电子迁移到och2ph基团,导致och2ph基团的α-自旋密度增加(14.4%
→
26.6%)。c
α-h键 的均裂裂解提供了一小部分β-电子给och2ph基团,这导致och2ph基团上的β-自旋密度 增加(ts
4-5_o
:1.4%
→
11.8%)。相应的α-电子伴随着h原子迁移到tempo且ts
4-5o
部分 形成otempo-h键以增加tempo上的α-自旋密度(ts
4-5o
:6.1%
→
21.3%),而对于 14→
1u
ts
4-5n
,cu-och2ph键没有发生均裂,只有-och2ph基团中的c
α-h键部分均裂,形成 一个键(ntempo-h)。如果该过程发生了cu-och2ph键的均裂,则必会增加och2ph基 团上的α-自旋密度。然而,och2ph基团上的α自旋密度减少(14.4%
→
9.7%),显然,在 14→
1u
ts
4-5n
期间没有发生cu-och2ph键断裂。wbi键极和键长变化数据(表3)进一步证 实了这一点。显然,破坏一个化学键所需的能量少于破坏两个化学键所需的能量。这是底物 醇上的c
α-h键上的h原子转移到tempo的n原子上具有较低势垒的主要原因。
[0205]
表3
[0206][0207]
[0208]
3)催化剂的再生
[0209]
在底物氧化过程中形成产物phcho后,活性催化剂4需要再生以继续催化循环。假设4 的再生是由o2辅助的。如图3所示,产物5n中的phcho被o2取代以产生中间体
1,3
6n(36n: 基态)。在36n
→37过程中,我们观察到自旋密度变化(ρcu: 0.53
→
0.52;ρl: 0.15
→
0.18; ρo2:-0.82
→
0.27;ρtempoh: 0.14
→
1.04)。这些自旋密度数据表明,36n中的
·
oo-片 段从tempoh中夺取h原子,在37中形成tempo和ooh基团(36n
→37: (tempoh)(l)cu
ii-(
·
oo-)
→
(tempo)(l)cu
ii-(ooh))。随后,37结构中tempo被底物醇替代 形成28,28中的醇提供质子到
–
ooh片段到29中形成h2o2。随后29结构中h2o2解离形成中 间体3,然后tempo配位到3上形成活性催化剂4。那么,整个催化过程就构成了一个闭环。
[0210]
用相同的分析研究方法对双核路径path b-i和path b-ii进行反应机理分析(path b-i 如图5和path b-ii如图6):
[0211]
由于双核反应机理与单核反应机理的不同在于催化剂再生过程不同,因此,我们仅对催 化剂再生过程进行讨论。对于15n,dbu取代产物phcho和tempoh生成110 ([(bipy)cui(dbu)]
)。随后,两分子110和一分子o2复合生成了双核化合物11,对于11 结构,我们考虑了三种可能的结构,cu(ⅱ)-1,2-μ-peroxo、cu(iii)-bis(μ-oxo)、cu(ⅱ)-μ-η2:η 2-peroxo:11(a)、11(b)、11(c)。通过计算,11(a)的能量最低。因此,后续过程只考虑11(a) 中间体。
[0212][0213]
11(a)有两种自旋态
1,3
11(a),且三重态为基态,311(a)呈[(bipy
(1)
(dbu)cu
(1)ii
(oo)cu
(2)ii
(dbu)(bipy
(2)
)]
2
结构。tempoh取代dbu配位到311(a)结构上形成中间体312[(bipy
(1) (dbu)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
,在该过程tempoh配位时,由于tempoh 存在两种构型(h在n上和h在o上),我们对这两种情况都进行了考虑,通过计算配位 tempoh(h在n上)的能量更低,因此后续只考虑配位tempoh(h在n上)的过程。从312 结构开始我们考虑了两种可能性,设计出了两条双核路径path b-i和path b-ii。
[0214]
path b-i:
[0215]
生成312结构后,312结构解离dbu形成318后进行h原子转移过程(图4),318
→119 是h原子转移的过程,tempoh上的h原子转移到o2上生成tempo自由基和ooh的过 程,同时,cu
(2)ii
还原成cu
(2)i
,在该过程涉及到了s=1
→
s=0的自旋交叉,即两态反应,为 了阐明h原子转移过程中发生了自旋翻转的过程,由卢天等人修改的sobmecp程序计算了 最小能量交叉点,得到了mecp的精确结构,能量为-17.2kcal/mol,该h原子转移过程需要 克服-16.1kcal/mol的势垒。119呈[(bipy
(1)
cu
(1)i
(ooh)cu
(2)ii
(tempo)(bipy
(2)
)]
2
结构。随 后,119结构解离后伴随着dbu配位形成110和37,37结构继续反应可以生成活性催化剂14, 因此,整个过程也形成了一个催化循环。
[0216]
path b-ii:
[0217]
生成312结构后,可以直接进行h原子转移过程(312
→313)(图5),tempoh上的 h原
子转移到o2上形成313结构[(bipy
(1)
(tempo)cu
(1)i
(ooh)cu
(2)ii
(dbu)(bipy
(2)
)]
2
, 同时,cu
(2)ii
还原成cu
(2)i
,该过程需要克服-22.9kcal/mol的势垒。随后313结构解离成214 ([(bipy)cu
ii
(dbu)(ooh)]
)和215([(bipy)cui(tempo)]
)两个结构,dbu取代215中的 tempo回到了110结构,底物醇配位到214上形成216结构,216
→217过程是质子转移过程, 2
16中的ooh基团夺取底物醇上的质子形成h2o2,生成217结构 ([(bipy)cu
ii
(dbu)(phch2o)(h2o2)]
),该过程需克服-18.0kcal/mol的势垒。tempo取代217 中的dbu和h2o2形成活性催化剂14,因此,该过程就形成了一个催化循环。
[0218]
(4)tof计算验证
[0219][0220]
为了获得完整催化循环的更多信息,我们使用能量跨度模型来分析整体的吉布斯自由能 曲线,转化效率(“tof”)由autof程序计算,对于dbu碱源的cu
ii
/bipy/tempo催化体 系来说,tof计算中20结构不能被忽略,即使20是一个催化循环之外的中间体,由于20与 活性催化循环(14
→14)处于快速平衡状态,并且他它将大部分催化剂浓度吸收到自身中,因 此在tof的计算中予以考虑。dbu碱源的cu
ii
/bipy/tempo催化体系在不同路径的tof结 果如下表4所示。
[0221]
表4有机碱源dbu的催化体系在不同路径下的tof计算
[0222][0223]
由此可见,对于cu
ii
/bipy/tempo/dbu体系来说,在patha中,autof程序计算的tof 决速中间体(tdi)是36n,决速过渡态(tdts)为2ts
8-9
,对应的能量跨度δe为13.9kcal/mol, tof=1.4
×
106h-1
,对于pathb-i,110为决速中间体(tdi),3ts
18-19
为决速过渡态(tdts), 对应的能量跨度δe为11.2kcal/mol,tof=1.4
×
108h-1
,对于pathb-ii,313为决速中间体,
1u
ts
4-5n
为决速过渡态,对应的能量跨度δe为14.6h-1
,tof=2.7
×
105h-1
,计算结果表明,pathb-i的 能量势垒最低,pathb-i为优势路径,且该路径也符合实验上的动力学依赖。
[0224]
而对于pathb-i和pathb-ii来说,两条路径的区别在于从312结构开始后续的反应过程不 同,pathb-i的后续过程是312结构解离dbu生成318结构后进行h原子转移的过程,318结 构的能量是-21.8kcal/mol,pathb-ii的后续过程是生成312结构后进行h原子的转移,3ts
12-13
的能量为-22.9kcal/mol,对比两条路径,pathb-ii的后续过程过渡态3ts
12-13
的能量比pathb-i 的后续过程318中间体的能量还低,仅从局部热力学考虑后续过程应按照pathb-ii来进行, 但是从整个催化循环来考虑,pathb-i的能垒比pathb-ii的能垒低,转化效率也更高,因此更 容易发生,且pathb-ii也不符合实验上的动力学依赖。pathb-i为优势路径。
[0225]
实施例2(nmi有机碱)
[0226]
将碱源dbu用nmi替换,cu
ii
/bipy/tempo/nmi催化体系的反应机理研究分析,具体 包括如下步骤:
[0227]
(1)构建催化剂活性中心结构的计算模型;
[0228]
基于以(bipy)cu
ii-tempo/nmi为催化剂进行苯甲醇有氧氧化成苯甲醛实验提供的反应条件信息,构建催化剂活性中心的计算模型;采用密度泛函方法进行几何优化,根据计算结果综合考虑构型稳定性、能量高低、计算结果和计算成本来选择最优模型;
[0229]
(2)设计反应路径;
[0230]
根据步骤(1)中选择的最优模型进行可能的反应路径设计;然后进行反应中间体和过渡态建模:nmi的pathc(单核)除了构建o-h键断裂的中间体和过渡态(步骤b)与dbu的patha(单核)不同,其他都相同。
[0231]
单核pathc
[0232]
(a)构建c
α-h键断裂的中间体和过渡态:
[0233]
中间体建模:二配位的主配体和底物醇盐构成cu中心的三配位结构,向三配位的cu中心加上tempo自由基构成cu中心的四配位结构,设置cu-n
ligand
的键长为cu-o
tempo
的键长为cu-o
phch2o
的键长为底物醇盐上的c
α-h键的键长为设置结束后进行几何优化,采用如下输入进行中间体模型几何优化:#optfrequb3lyp/def2svpnosymmscrf=(smd,solvent=acetonitrile)em=gd3bj;得到对应中间体结构;
[0234]
过渡态建模:采用柔性扫描方法,通过对关键化学键改变过程中可能的结构改变的分析,进一步计算每一步结构的能量,获得关键化学键改变过程势能曲线。将该势能曲线上能量最高点的对应结构作为初始过渡态结构完成初步建模。随后,对过渡态模型进行几何优化,获得相应结构,并对其进行hessian矩阵计算,获得频率计算结果,且频率计算证明该过渡态结构有且仅有一个虚频,且虚频振动方向模式符合关键化学键生成或断裂规律,方可证明该过渡态模型建立正确。
[0235]
具体来说,用gaussview软件通过柔性扫描的方法寻找过渡态,将c
α-h键键长从以的增幅增大到计算每一步的能量,找到能量最高点的对应结构,然后调整建立模型中底物醇上的c
α-h及n
tempo-h的距离,分别设置在和
[0236]
采用如下输入进行过渡态模型的几何优化:#opt=(calcfc,ts,noeigen,gdiis)frequb3lyp/def2svpnosymmscrf=(smd,solvent=acetonitrile)em=gd3bj;几何优化后得到优化结构,频率计算结果也只有一个虚频;且分析该虚频的振动方向符合c
α-h-n
tempo
,证明过渡态寻找正确;
[0237]
(b)构建o-h键断裂的中间体和过渡态:
[0238]
中间体建模:主配体、nmi和-otf构成cu中心的四配位结构,调整cu-n
nmi
键长为cu-o
otf
键长为cu-n
ligand
键长为向四配位的cu中心加上底物醇,cu-o
sub
的键长为底物醇oh键上的h与nmi上的n的距离设置为设置结束后进行几何优化,得到对应中间体结构;
[0239]
过渡态建模:调整关键键长,将cu-n
nmi
键键长从调整到将cu-o
otf
键键长从调整到cu-n
ligand
键长为从调整到将底物醇上o-h键键长从调整到底物醇oh上的h与nmi上的n的距离设置由底物醇oh上的h与nmi上的n的距离设置由调整为将调整好的过渡态模型进行过渡态几何优化得到优化结构,且频率计算结 果有
且仅有一个虚频;且分析该虚频的振动方向符合o
sub-h-n
nmi
,证明过渡态寻找正确;
[0240]
(c)构建oo-h键生成的中间体和过渡态:
[0241]
中间体建模:主配体、o2和tempoh构成cu中心的四配位结构,调整cu-oo
·-键长为 cu-o
tempoh
键长为cu-n
ligand
键长为tempoh-oo
·-距离为设置结束后进行几何优化,得到对应中间体结构;
[0242]
过渡态建模:调整关键键长,调整cu-oo
·-键长从调整到cu-n
ligand
键 长从调整到tempoh上n-h的键长从1.054调整到tempoh-oo
·-键键长从调整到将调整好的过渡态模型进行过渡态几何优化和频率计算, 得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合oo
·-‑
h-n
tempo
, 证明过渡态寻找正确;
[0243]
(d)构建h2o2生成的中间体和过渡态:
[0244]
中间体建模:主配体和ooh构成cu中心的三配位结构,底物醇配位到cu上形成四配 位结构;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键长为 phch2oh-ooh距离为设置结束后进行几何优化,得到对应中间体结构;
[0245]
过渡态建模:调整关键键长,调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确;
[0246]
同理dbu碱源的催化体系,对于nmi的催化体系的双核反应机理pathd-i和pathd-ii 除了包括单核反应机理的一些中间体和过渡态的建模过程,还包含以下不同的反应中间体和 过渡态建模。
[0247]
对于nmi的催化体系的双核路径pathd-i,采用步骤(e)替换pathc的步骤(c):
[0248]
(e)构建oo-h生成的中间体和过渡态:
[0249]
中间体建模:主配体、o2、tempoh和两分子cu构成cu中心的四配位双核结构 [(bipy
(1)
cu
(1)ii
(oo)cu
(2)ii tempoh(bipy
(2)
)]
2
,tempoh配位在cu2中心,调整cu
1-oo
·-键长 为调整cu
2-oo
·-键长为cu-o
tempoh
键长为cu-n
ligand
键长为 tempoh-oo
·-距离为设置结束后进行几何优化,得到对应中间体结构;
[0250]
过渡态建模:调整关键键长:调整cu
1-oo
·
键长为调整cu
2-oo
·-键长为 cu-n
ligand
键长从调整到tempoh上n-h的键长从1.062调整到 tempoh-oo
·-键键长从调整到将调整好的过渡态模型进行过渡态几何优化 和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方向符合 oo
·-‑
h-tempo,证明过渡态寻找正确。
[0251]
对于nmi的双核路径pathd-ii,采用步骤(f),(g)分别替换pathc的步骤(c),(d):
[0252]
(f)构建oo-h键生成的中间体和过渡态:
[0253]
中间体建模:主配体、o2和tempoh、nmi和两分子cu构成cu中心的四配位双核结 构[(bipy
(1)
(nmi)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
,nmi和tempoh分别配位在两个cu 中
心,调整cu
1-oo
·-键长为调整cu
2-oo
·-键长为cu
2-o
tempoh
键长为调整cu
1-n
nmi
键长为cu-n
ligand
键长为tempoh-oo
·-距离为设置 结束后进行几何优化,得到对应中间体结构;
[0254]
过渡态建模:调整关键键长:调整cu
1-oo
·-键长为调整cu
2-oo
·
键长为 cu
1-n
nmi
键长从调整到cu-n
ligand
键长从调整到tempoh 上n-h的键长从1.064调整到tempoh-oo
·-键键长从调整到将调 整好的过渡态模型进行过渡态几何优化和频率计算,得到有且只有一个虚频的过渡态结构; 进一步分析该虚频的振动方向符合oo
·-‑
h-tempo-,证明过渡态寻找正确;
[0255]
(g)构建h2o2生成的中间体和过渡态:
[0256]
中间体建模:主配体、ooh和nmi构成cu中心的四配位结构,底物醇通过氢键连接配 位到cu的第二配位层;调整cu-ooh键长为cu-n
ligand
键长为cu-o
sub
键 长为phch2oh-ooh距离为cu-n
nmi
键长为设置结束后进行几何 优化,得到对应中间体结构;
[0257]
过渡态建模:调整关键键长:调整phch2oh-ooh键长从调整到cu-n
ligand
键长从调整到phch2o-h距离从调整到将该过渡态模型进 行几何优化和频率计算,得到有且只有一个虚频的过渡态结构;进一步分析该虚频的振动方 向符合phch2o-h-ooh,证明过渡态寻找正确。
[0258]
(3)分析反应机理和路径;
[0259]
分析各基元反应中过渡态及中间体的键长、键角及中心金属离子周围的立体化学变化, 总结出反应过程中心金属离子的配位数,配体的空间构型,配体与中心金属离子的键长、键 角,及相应的结构参数的变化规律;
[0260]
单核pathc:
[0261]
o-h键断裂过程中,将cu-n
nmi
键键长从调整到将cu-o
otf
键键长从调整到cu-n
ligand
键长为从调整到将底物醇上o-h键键长 从调整到底物醇oh上的h与nmi上的n的距离设置由调整为 中心金属cu
ii
周围为二配位的主配体、nmi和-otf及底物醇;经过o-h键断裂后 形成以二配位的主配体、nmih
和-otf和醇盐配位的四面体形式配位的cu
ii
活性中心结构; 随后,c
α-h键断裂过程cu-o
phch2oh
键长从增加到醇盐上c
α-h键键长从h键键长从增加到tempo-h键键长从减少到经过c
α-h键断裂过程后形成 以二配位的主配体、产物苯甲醛和tempoh配位的cui活性中心结构;随后,oo-h键生成 过程中从cu-oo
·-键长从减小到cu-n
ligand
键长从减小到 tempoh上n-h的键长从增加到tempoh-oo
·-键键长从减小到 经过oo-h键生成过程后形成以二配位的主配体、tempo和ooh配位的cu
ii
活性 中心结构;最后,h2o2生成过程中phch2oh-ooh键长从减小到cu-n
ligand
键长从减小到phch2o-h距离从增加到经过h2o2生成过 程后形成以二配位的主配体、醇盐和h2o2配位的cu
ii
活性中心结构。
[0262]
双核pathd-i:
[0263]
o-h键断裂过程中,将cu-n
nmi
键键长从调整到将cu-o
otf
键键长从调整到cu-n
ligand
键长为从调整到将底物醇上o-h键键长 从调整到底物醇oh上的h与nmi上的n的距离设置由调整为 中心金属cu
ii
周围为二配位的主配体、nmi和-otf及底物醇;经过o-h键断裂后 形成以二配位的主配体、nmih
和-otf和醇盐配位的四面体形式配位的cu
ii
活性中心结构; 随后,c
α-h键断裂过程cu-o
phch2oh
键长从增加到醇盐上c
α-h键键长从h键键长从增加到tempo-h键键长从减少到经过c
α-h键断裂过程后形成 以二配位的主配体、产物苯甲醛和tempoh配位的cui活性中心结构;随后,oo-h键生成 过程中cu
1-oo
·-键长从调整到cu
2-oo
·-键长从调整到 cu-n
ligand
键长从调整到tempoh上n-h的键长从1.062增加到 tempoh-oo
·-键键长从减小到经过oo-h键生成过程后形成以二配位的主 配体、tempo和ooh和两分子cu配位的cui和cu
ii
活性中心的四配位双核结构 [(bipy
(1)
cu
(1)i
(ooh)cu
(2)ii
(tempo)(bipy
(2)
)]
2
最后,h2o2生成过程中phch2oh-ooh键长 从减小到cu-n
ligand
键长从减小到phch2o-h距离从h距离从增加到经过h2o2生成过程后形成以二配位的主配体、醇盐和h2o2配位的cu
ii
活 性中心结构。
[0264]
双核pathd-ii:
[0265]
o-h键断裂过程中,将cu-n
nmi
键键长从调整到将cu-o
otf
键键长从调整到cu-n
ligand
键长为从调整到将底物醇上o-h键键长 从调整到底物醇oh上的h与nmi上的n的距离设置由调整为 中心金属cu
ii
周围为二配位的主配体、nmi和-otf及底物醇;经过o-h键断裂后 形成以二配位的主配体、nmih
和-otf和醇盐配位的四面体形式配位的cu
ii
活性中心结构; 随后,c
α-h键断裂过程cu-o
phch2oh
键长从增加到醇盐上c
α-h键键长从h键键长从增加到tempo-h键键长从减少到经过c
α-h键断裂过程后形成 以二配位的主配体、产物苯甲醛和tempoh配位的cui活性中心结构;随后,oo-h键生成 过程中cu
1-oo
·-键长从调整为cu
2-oo
·-键长从调整为 cu
1-n
nmi
键长从调整到cu-n
ligand
键长从调整到tempoh 上n-h的键长从1.064调整到tempoh-oo
·-键键长从调整到经过 oo-h键生成过程后形成以二配位的主配体、tempo、nmi、ooh和两分子cu配位的cui和cu
ii
活性中心的四配位双核结构[(bipy
(1)
(nmi)cu
(1)ii
(ooh)cu
(2)i tempo(bipy
(2)
)]
2
。最后, h2o2生成过程中phch2oh-ooh键长从调整到cu-n
ligand
键长从调 整到phch2o-h距离从调整到经过h2o2生成过程后形成以二配位 的主配体、醇盐、nmi和h2o2配位的cu
ii
活性中心结构。
[0266]
根据反应路径和绘制势能图,分析得到单核path c反应原理:
[0267]
在单核路径中,nmi和dbu仅在底物醇质子转移过程不同,因此我们只对这一过程
进 行讨论(图7)。在该体系中,起始结构为220结构(bipy)cu
ii
(-otf)(nmi)。底物phch2oh 配位到220上形成221结构。对于221结构,nmi能像dbu夺氢过程一样,能够夺取phch2oh 上的质子,在222结构中形成nmih
,该质子转移过程需要克服12.4kcal/mol的能垒。
[0268]
在碱源dbu的cu
ii
/bipy/tempo催化体系中,我们发现阴离子-otf在dbu夺氢过程中 起到了一定的作用,因此,在该过程中,我们也将评估阴离子-otf的作用,因此,在没有-otf 参与的去质子化步骤的过程重新标记为220a
→222a,底物醇配位到220a上形成了221a。随后 nmi夺取底物phch2oh的质子形成nmih
,此过程需要经过能量势垒为17.5kcal/mol的 2
ts
21a-22a
。通过对比220a
→222a和220
→222的反应过程,我们发现,没有-otf参与的反应过 程220a
→222a的能量势垒比有-otf参与的反应过程220
→222的能量势垒高5.1kcal/mol。因 此,我们认为阴离子-otf参与了nmi夺氢的过程,并且能够帮nmi夺氢,且阴离子-otf 在nmi夺氢过程比在dbu夺氢过程中的作用更强。
[0269]
双核pathd反应路径和反应原理:
[0270]
与dbu碱源的催化机理类似,nmi碱源的双核反应机理仅在催化剂再生过程与单核路 径不同,因此我们仅讨论催化剂的再生过程。对于15n,nmi取代产物phcho和tempoh 生成123([(bipy)cui(nmi)]
)。两分子123和一分子o2复合生成了双核化合物24,对于24 结构,我们同样也考虑了三种可能的结构,cu(ⅱ)-1,2-μ-peroxo、cu(iii)-bis(μ-oxo)、cu(ⅱ)-μ-η 2:η2-peroxo:24(a)、24(b)、24(c)。通过计算,24(a)的能量最低。因此,后续讨论我们只考 虑了24(a)中间体。
[0271][0272]324(a)呈[(bipy
(1)
(nmi)cu
(1)ii
(oo)cu
(2)ii
(nmi)(bipy
(2)
)]
2
结构。tempoh取代nmi 配位到324(a)结构上形成中间体325[(bipy
(1)
(nmi)cu
(1)ii
(oo)cu
(2)ii
(tempoh)(bipy
(2)
)]
2
, 同样的,生成325结构后有两种可能性。因此设计了两条双核路径path d-i和path d-ii。
[0273]
双核path d-i
[0274]
对于nmi碱源的cu
ii
/bipy/tempo/nmi体系生成325结构解离nmi形成318后进行h 原子转移过程与dbu碱源的后续反应过程基本相同(图8)。318
→119是h原子转移的过程, tempoh上的h原子转移到o2上生成tempo自由基和ooh的过程,同时,cu
(2)ii
还原成 cu
(2)i
,在该过程涉及到了s=1
→
s=0的自旋交叉,即两态反应,为了阐明h原子转移过程中 发生了自旋翻转的过程,由卢天等人修改的sobmecp程序计算了最小能量交叉点,得到了 mecp的精确结构,能量为-17.2kcal/mol,该h原子转移过程需要克服-16.1kcal/mol的势垒。 1
19呈[(bipy
(1)
cu
(1)i
(ooh)cu
(2)ii
(tempo)(bipy
(2)
)]
2
结构。随后,119结构解离后伴随着 nmi配位形成123和37,37结构继续反应可以生成活性催化剂14,因此,整个过程也形成了 一个催化循环。
[0275]
双核path d-ii
[0276]
形成325结构后进行h原子转移过程(图9),325中o2夺取tempoh上的h原子形 成326结构[(bipy
(1)
(tempo)cu
(1)i
(ooh)cu
(2)ii
(nmi)(bipy
(2)
)]
2
,cu
(2)ii
还原成cu
(2)i
, 该过程需
要克服-14.7kcal/mol的势垒。随后,中间体227([(bipy)cu
ii
(nmi)(ooh)]
)和215 ([(bipy)cui(tempo)]
)从326结构解离,nmi取代215中的tempo形成了123结构,底物 醇配位到227上形成228结构,228
→229过程是质子转移过程,228中的ooh基团夺取底物 醇上的质子形成229结构([(bipy)cu
ii
(nmi)(phch2o)(h2o2)]
),该过程需克服-9.0kcal/mol 的势垒,tempo取代229中的nmi和h2o2形成活性催化剂14结构,因此,该过程就形成 了一个催化循环。
[0277]
(4)tof计算验证
[0278][0279]
为了获得完整催化循环的更多信息,我们使用能量跨度模型来分析整体的吉布斯自由能 曲线,转化效率(“tof”)由autof程序计算,对于碱源nmi的cu
ii
/bipy/tempo催化体 系来说,tof计算中220结构不能被忽略,即使220是一个催化循环之外的中间体,由于220 与活性催化循环(14
→14)处于快速平衡状态,并且他它将大部分催化剂浓度吸收到自身中, 因此在tof的计算中予以考虑。碱源nmi的cu
ii
/bipy/tempo催化体系在不同路径的tof 结果如下表5所示:
[0280]
表5有机碱nmi的催化体系在不同路径下的tof计算
[0281][0282]
由此可见,对于cu
ii
/bipy/tempo/nmi体系来说,对于pathc、pathd-i和pathd-ii,autof 程序计算的tof决速中间体(tdi)是220,决速过渡态(tdts)为
1u
ts
4-5n
,对应的能量跨 度δe为16.7kcal/mol,tof=1.5
×
104h-1
,该体系的速控步骤是底物氧化步骤中的tempo夺取底 物醇盐的h原子生成产物醛的过程,计算结果符合实验上的动力学依赖。同时,dbu碱源的 (bipy)cu
ii-tempo催化体系比nmi碱源的(bipy)cu
ii-tempo催化体系的反应能垒低,说明dbu 碱源的催化体系比nmi碱源的催化体系的反应活性高,这与实验结果一致。并且nmih
-otf 对反应体系有抑制作用。因此,我们还探究了nmih
-otf对反应体系的抑制作用,nmi碱源 的(bipy)cu
ii-tempo催化体系下的速控步骤在底物氧化过程220
→1ts
4-5n
的过程,涉及了共轭 酸nmih
-otf,nmih
-otf在乙腈中的pka=14.3,它容易提供氢质子可以促进cu
ii-or分解, 平衡向左进行会抑制生成活性催化剂4,因此加入nmih
-otf会对体系的产生抑制作用,在 dbu碱源的(bipy)cu
ii-tempo催化体系的速控步骤在催化剂再生步骤的110
→3ts
18-19
过程, 速控步骤不涉及共轭酸dbuh
-otf和cu
ii-or基团,且dbuh
-otf在乙腈中的pka=24,不易 提供氢质子分解cu
ii-or基团,因此加入dbuh
-otf不会对催化体系的反应活性产生影响。
[0283]
综上所述,本发明通过密度泛函方法研究了两种碱源dbu和nmi的cu
ii
/bipy/tempo 催化体系催化氧化一级醇的反应机理。我们针对这两种碱源分别提供了三种可能的反应路径, 单核反应机理patha(pathc)及双核反应机理pathb-i(pathd-i)和pathb-ii(pathd-ii),通过构建 催化剂活性中心结构的计算模型选择最优模型,设计反应路径,通过反应中间体和过渡态建 模,得到正确的过渡态和反应路径,分析催化体系的反应机理。催化循环
由三部分组成,即 催化剂的活化、底物的氧化和催化剂的再生过程。最终计算催化剂的转化频率tof值并与实 验相比较,计算结果表明,对于碱源dbu的催化体系来说,在获得整个催化循环的基础上, 通过能量跨度模型分析,pathb-i是优势路径,且pathb-i也满足实验上的动力学依赖。在该 体系中,我们评估了-otf在催化体系中的作用,发现有-otf参与的dbu夺氢过程比没有-otf 参与的反应过程能量势垒低3.1kcal/mol,说明-otf参与了dbu夺氢过程,能够降低该过程 的能量势垒,帮助有机碱dbu夺氢。在碱源nmi的cu
ii
/bipy/tempo催化体系中,我们发 现该催化体系的速控步骤在底物氧化过程(220
→1ts
4-5n
的过程),这一结果也与实验一致, 并满足实验上的动力学依赖。在该体系中,发现-otf也能够帮助有机碱nmi夺氢,且与有 机碱dbu夺氢过程相比作用更强。同时,我们发现,dbu碱源的(bipy)cu
ii-tempo催化体 系比nmi碱源的催化体系反应能垒低,说明dbu碱源的(bipy)cu
ii-tempo催化体系比nmi 碱源的(bipy)cu
ii-tempo催化体系的反应活性高,并且nmih
-otf对反应体系有抑制作用, 这与实验结果一致。并且探究了nmih
-otf的抑制作用,nmi碱源的(bipy)cu
ii-tempo催 化体系下的速控步骤在底物氧化过程,涉及了共轭酸nmih
-otf,nmih
-otf在乙腈中的 pka=14.3,它容易提供氢质子可以促进cu
ii-or分解,平衡向左进行会抑制生成活性催化剂4, 因此加入nmih
-otf会对体系的产生抑制作用。在dbu碱源的(bipy)cu
ii-tempo催化体系 的速控步骤在催化剂再生过程,速控步骤不涉及共轭酸dbuh
-otf和cu
ii-or基团,且 dbuh
-otf在乙腈中的pka=24,不易提供氢质子分解cu
ii-or基团,因此加入dbuh
-otf 不会对催化体系的反应活性产生影响。通过分析底物氧化步骤中tdts(1ts
4-5n
和1ts
4-5o
) 的几何结构和电子结构,我们发现底物醇上c
α-h键的上的h原子转移到n原子上而不是o 原子上,因为断开一个化学键(1ts
4-5n
:och2ph基团中的c
α-h键)比断开两个化学键(1ts
4-5o
: cu-och2ph和och2ph基团中的c
α-h键)所需要的能量更少。本发明通过探索该催化体系 的反应机理,希望这项研究可以为反应过程提供有用的见解,并为设计出新型的高效的催化 剂提供理论基础。
[0284]
本领域的技术人员应当理解,上述实施方式仅仅是为了清楚地说明本公开,而并非是对 本公开的范围进行限定。对于所属领域的技术人员而言,在上述公开的基础上还可以做出其 它变化或变型,并且这些变化或变型仍处于本公开的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。