nmibc预后预测分子标志物、筛选方法及建模方法
技术领域
1.本发明涉及泌尿肿瘤医学领域,特别是涉及基于转录组基因表达数据的非肌层浸润型膀胱癌(nmibc)患者向肌层浸润型膀胱癌(mibc)恶性进展及进展速度快慢预测的分子标志物的筛选和鉴定。
背景技术:
2.尿路上皮非肌层浸润型膀胱癌(non-muscle-invasive bladder cancer,nmibc)是最常见的膀胱癌类型,50%-70%的nmibc患者出现疾病复发,高达20%的nmibc患者进展为肌肉浸润型膀胱癌(muscle-invasive bladder cancer,mibc)。nmibc进展为mibc与肿瘤分期和分级相关。因此,尽管nmibc患者5年生存率良好(》90%),但大多数患者必须接受终生膀胱镜检查和多种治疗干预,而nmibc患者一旦进展为mibc,其预后风险和治疗手段将发生很大变化。对于nmibc患者而言,肿瘤细胞转移可能性较少,可以行经尿道膀胱电切或激光剜除术。由于nmibc容易复发,即使肿瘤恶性程度较低,术后也需要密切随访,包括膀胱内灌注化疗和定期膀胱镜检查。即使nmibc恶性程度较低,也有一部分患者约20%会出现疾病进展,转变成mibc。定期随访和疾病监控有利于及时发现nmibc的复发和进展。而对于mibc患者而言,由于肿瘤已经侵犯膀胱肌层,肿瘤细胞通过血管和淋巴向周围及远处转移的可能性大大增加,原则上对于mibc患者需要膀胱全切和盆腔淋巴结清扫,同时行尿流改道手术。如果nmibc进展成mibc,不仅膀胱全切除将对患者生活质量带来极大麻烦,而且肿瘤转移风险也会大大增加。因此在nmibc患者预后和动态监控中,开发nmibc患者向mibc恶性进展的分子预测标志物具有重大的临床应用意义,可以实时监控nmibc患者是否有向mibc发生恶性进展的潜能,以及nmibc发生恶性进展的速度。而目前临床和医学研究中很少针对nmibc患者向mibc恶性进展开发有效的分子预测标志物。
3.关于nmibc患者向mibc恶性进展的研究中,以往研究有发现少量基因与nmibc向mibc恶性进展有关,但基本上很少有研究专门针对nmibc患者向mibc恶性进展的预后监控阶段开发无创分子预测标志物,包括“nmibc患者是否向mibc恶性进展”和“nmibc患者向mibc进展快慢”的肿瘤病变阶段。以往研究中,一般认为nmibc的高危患者即pt1高级别肿瘤患者很可能会进展为mibc。对于高风险即pt1肿瘤患者,以往研究发现p53的积累与nmibc患者膀胱内复发、进展为mibc相关,但也有研究表明p53不能预测nmibc患者的临床进展结果。另外,有研究者针对929名pt1尿路上皮膀胱癌患者的26项研究进行荟萃分析发现,p53可以预测nmibc患者是否进展为mibc。除了p53,以往研究实验报告了在nmibc患者中p21的缺失与膀胱内复发、mibc进展以及总体生存率os降低有关,不过其它研究并没有验证出p21与nmibc预后的关联性。
技术实现要素:
4.本发明要解决的技术问题之一是提供一种非肌层浸润型膀胱癌预后预测分子标志物的筛选方法,它可以筛选出与nmibc恶性进展为mibc高度相关的分子预测标志物,实现
对nmibc患者的无创预后预测。
5.为解决上述技术问题,本发明的非肌层浸润型膀胱癌预后预测分子标志物的筛选方法,主要包括以下步骤:
6.获取nmibc患者样本的转录组数据和临床信息,根据nmibc进展情况分组;
7.将转录组基因表达谱数据转化为原始reads count表达谱数据;
8.筛选不同分组之间的差异表达基因;
9.对所述差异表达基因的reads count表达谱数据进行标准化;
10.基于所述差异表达基因的标准化表达值对所述nmibc患者样本进行聚类分析,获得差异基因群;
11.以所述差异基因群的标准化表达值作为分子特征,利用机器学习分类算法,筛选nmibc预后预测分子标志物。
12.上述临床信息包括nmibc是否进展为mibc、nmibc在多长时间内发生了向mibc的恶性进展,可以根据不同的预测目的,选择不同的临床信息。
13.上述转录组基因表达谱为fpkm基因表达谱,将fpkm基因表达谱数据转化为reads count表达谱数据,可以采用如下方法:
14.获取每个样本的测序reads数量信息;
15.计算每个基因编码区域的长度;
16.将fpkm基因表达谱数据转换为reads count表达谱数据,转换公式为:
[0017][0018]
其中,i为样本编号,j为基因编号,r
ij
为样本i基因j的reads count数值,f
ij
为样本i基因j的fpkm数值,lj为基因j的编码区域长度,ti为样本i的测序reads数量。
[0019]
上述差异表达基因的筛选,可以采用如下方法:
[0020]
设计分组矩阵;
[0021]
进行不同分组的差异表达分析;
[0022]
过滤掉差异表达基因中低丰度且变化小的基因,获得不同分组的差异表达基因分析结果;
[0023]
根据校正后的p值《0.05、倍数变化log2转换值的绝对值》1、倍数变化log2转换值的标准误差《1三个阈值筛选差异表达基因。
[0024]
上述差异表达基因的reads count表达谱数据的标准化,可以采用如下方法:
[0025]
对样本的原始reads count表达值进行基因编码区域长度的标准化,计算公式为:
[0026][0027]
其中,i为样本编号,j为基因编号,n
ij
为样本i基因j的编码区域标准化表达值,r
ij
为样本i基因j的原始reads count表达值,lj为基因j的编码区域长度;
[0028]
对样本的编码区域标准化表达值进行tpm标准化,计算公式为:
[0029]
[0030]
其中,i为样本编号,j为基因编号,tpm
ij
为样本i基因j的tpm标准化表达值,n
ij
为样本i基因j的编码区域标准化表达值,total genes in sample i为样本i中所有基因总数。
[0031]
上述聚类分析方法优选为:利用pheatmap工具的pheatmap函数,基于差异表达基因的标准化表达值,对样本进行层次聚类分析,挑选在分组中具有明显聚类特征的差异基因群。
[0032]
上述利用机器学习分类算法筛选nmibc预后预测分子标志物的方法,优选为:利用随机森林的机器学习分类算法,构建随机森林模型,并评估差异基因群的每个基因特征在模型构建中的重要性,挑选重要基因特征,作为nmibc预后预测分子标志物。
[0033]
本发明要解决的技术问题之二是提供两组用上述分子标志物筛选方法筛选的nmibc预后预测分子标志物。其中,一组分子标志物包括表1所示的385个分子标志物,用于预测nmibc患者是否向mibc恶性进展;另一组分子标志物包括表2所示的32个分子标志物,用于预测nmibc患者向mibc进展的快慢。
[0034]
本发明要解决的技术问题之三是提供基于上述分子标志物的非肌层浸润型膀胱癌预后预测模型的构建方法。该方法用上述nmibc预后预测分子标志物对不同分组的nmibc患者进行umap降维映射,构建分类器(优选为利用randomforest工具的randomforest函数,构建随机森林分类器)作为最佳预后预测模型。
[0035]
本发明要解决的技术问题之四是提供上述分子标志物的检测试剂以及包含该检测试剂的试剂盒,以及所述试剂、试剂盒在非肌层浸润型膀胱癌预后预测中的应用。
[0036]
本发明通过nmibc患者转录组高通量测序,结合差异分析、聚类模式识别和机器学习特征选择等多种方法,筛选出与nmibc恶性进展为mibc高度相关的分子预测标志物,根据分子标志物基因表达数据,对nmibc患者是否向mibc恶性进展以及nmibc患者向mibc进展快慢的肿瘤病变阶段进行分子预测分析,从而为nmibc恶性进展的动态监控和预后提供了一种快速、高效、无创的分子检测方法。
附图说明
[0037]
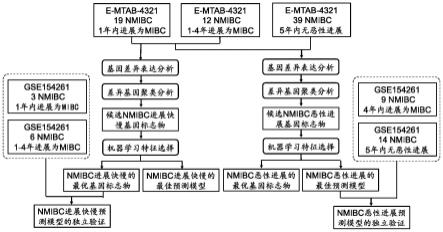
图1为本发明基于转录组基因表达筛选预测nmibc向mibc恶性进展及进展速度快慢的分子标志物及构建预测模型的方法流程示意图。
[0038]
图2为本发明基于预测nmibc患者向mibc恶性进展的分子标志物对2组nmibc患者进行umap降维映射。
[0039]
图3为本发明基于预测nmibc患者向mibc进展速度快慢的分子标志物对2组nmibc患者进行umap降维映射。
[0040]
图4为本发明的nmibc患者向mibc恶性进展预测模型的测试数据roc曲线。
[0041]
图5为本发明的nmibc患者向mibc进展速度快慢预测模型的测试数据roc曲线。
具体实施方式
[0042]
为对本发明的技术内容、特点与功效有更具体的了解,以下结合附图及具体实施例,对本发明的技术方案进行详细的说明。
[0043]
实施例1基于转录组基因表达筛选nmibc预后预测分子标志物
[0044]
1.获取nmibc患者转录组数据和临床信息
[0045]
从arrayexpress数据库获取具有详细临床随访信息的nmibc患者转录组数据作为
发现队列——e-mtab-4321(https://www.ebi.ac.uk/arrayexpress/experiments/e-mtab-4321/)。该发现队列的临床随访信息包括nmibc是否进展为mibc,以及在多长时间内(以月份为单位)发生了向mibc的恶性进展。
[0046]
根据临床随访信息,将发现队列中的nmibc患者(总计70例患者样本)分为3组:a组——nmibc患者在1年内进展为mibc,共19例;b组——nmibc患者在1-4年进展为mibc,共12例;c组——nmibc患者在5年内无恶性进展,共39例。
[0047]
2.转录组数据预处理
[0048]
针对步骤1中的发现队列e-mtab-4321的转录组数据进行预处理。e-mtab-4321提供了fpkm处理后的基因表达谱,由于转录组差异表达分析需要原始reads count表达谱数据,而非fpkm处理后的基因表达谱,因此需要对fpkm基因表达谱数据进行转换,得到转录组原始reads count表达谱数据。具体转换步骤如下:
[0049]
(2.1)获取每个样本测序reads数量信息:从ega数据库中获取e-mtab-4321数据集每个样本测序序列reads数量信息(https://ega-archive.org/datasets/egad00001001939/files)。
[0050]
以一个样本egaf00000977420为例,在ega数据库页面(https://ega-archive.org/datasets/egad00001001939/files)中点击egaf00000977420样本的qc report链接(https://filesportal.ega-archive.org/egaf00000977420),获得total reads——20529121,作为样本测序序列reads数量。样本egaf00000977420的名称信息(u0560)可以从bam header链接中的
‑‑
rg-sample参数获得。
[0051]
(2.2)计算每个基因编码区域的长度:从ensembl数据库(http://ftp.ensembl.org/pub/release-104/gff3/homo_sapiens/)中下载人类基因编码区域注释文件homo_sapiens.grch38.100.gtf,对于每个基因,计算每个转录本的外显子区域长度之和作为转录本长度,挑选最长转录本的长度作为基因编码区域的长度。
[0052]
(2.3)对于样本i基因j,由fpkm数值向reads count数值转换的计算公式如下:
[0053][0054]
其中,r
ij
表示样本i基因j的reads count数值,f
ij
表示样本i基因j的fpkm数值,lj表示基因j的编码区域长度,ti表示样本i的测序reads数量。
[0055]
3.筛选差异表达基因
[0056]
结合deseq2和htsfilter工具,筛选出a b组与c组的差异表达基因,作为nmibc患者是否发生向mibc恶性进展的差异表达基因,同时筛选出a组与b组的差异表达基因,作为nmibc患者向mibc进展快慢的差异表达基因。具体步骤如下:
[0057]
(3.1)分别针对a b组vs c组,以及a组vs b组,利用deseq2工具的deseqdatasetfrommatrix函数设计2个分组矩阵;
[0058]
(3.2)利用deseq2工具的deseq函数进行a b组vs c组,以及a组vs b组的差异表达分析;
[0059]
(3.3)利用htsfilter工具的htsfilter函数对差异表达基因中低丰度且变化很小的基因进行过滤,获得过滤后a b组vs c组(过滤条件设置为:基因表达最大值《62.966),以及a组vs b组(过滤条件设置为:基因表达最大值《68.464)的差异表达基因分析结果;
[0060]
(3.4)将以下3个条件作为筛选差异表达基因的阈值:校正后的p值(padj)《0.05;倍数变化log2转换值(log2foldchange)的绝对值》1;倍数变化log2转换值的标准误差(lfcse)《1。在a b组vs c组比较中筛选获得798个差异表达基因,在a组vs b组比较中筛选获得227个差异表达基因。
[0061]
4.转录组reads count基因表达谱数据标准化
[0062]
对转录组reads count基因表达谱数据进行tpm标准化,具体步骤如下:
[0063]
(4.1)对样本i基因j的原始reads count表达值进行基因编码区域长度的标准化:
[0064][0065]
其中,n
ij
表示样本i基因j的编码区域标准化表达值,r
ij
表示样本i基因j的原始read count表达值,lj表示基因j的编码区域长度,该长度由步骤2.2获得。
[0066]
(4.2)对样本i基因j的编码区域标准化表达值进行tpm标准化:
[0067][0068]
其中,tpm
ij
表示样本i基因j的tpm标准化表达值,n
ij
表示样本i基因j的编码区域标准化表达值,total genes in sample i表示样本i中所有基因总数。
[0069]
5.差异表达基因聚类分析
[0070]
利用pheatmap工具的pheatmap函数分别基于a b组vs c组,以及a组vs b组差异表达基因的tpm表达谱数据对样本进行层次聚类分析。函数的标准化参数scale设置为行归一化scale=row,分别挑选在a b组(即向mibc恶性进展的nmibc患者)以及a组(快速向mibc恶性进展的nmibc患者)中具有上调等明显聚类特征的差异基因群,分别获得2个差异基因群,其中,a b组vs c组的差异基因群包含741个差异基因,a组vs b组的差异基因群包含150个差异基因。
[0071]
6.nmibc恶性进展及进展快慢分子标志物筛选
[0072]
分别以a b组vs c组,以及a组vs b组差异基因群的tpm标准化表达值作为分子特征,利用随机森林的机器学习分类算法,即利用randomforest工具的randomforest函数,进行随机森林模型构建,并评估差异基因群的每个基因特征(即tpm标准化表达值)在模型构建中的重要性,函数中设置importance=true,分别挑选a b组vs c组,以及a组vs b组中重要性排名前~50%或~20%的基因特征,分别获得a b组vs c组的385个(表1和表2)和a组vs b组的32个(表3和表4)重要基因特征,这些重要基因特征分别作为nmibc患者向mibc恶性进展预测模型和nmibc患者向mibc进展速度快慢预测模型的分子标志物,用于下一步模型优化。
[0073]
表1 nmibc患者向mibc恶性进展预测模型的385个分子标志物
[0074]
[0075]
[0076]
[0077]
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085][0086]
表2 nmibc患者向mibc恶性进展的385个分子标志物功能注释
[0087]
[0088]
[0089]
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097][0098]
表3 nmibc患者向mibc进展快慢预测模型的32个分子标志物
[0099]
[0100][0101]
表4 nmibc患者向mibc进展快慢的32个分子标志物功能注释
[0102]
[0103]
[0104][0105]
7.nmibc预后预测模型构建
[0106]
以a b组vs c组,以及a组vs b组的重要基因特征作为分子标志物,利用randomforest工具的randomforest函数,分别重新构建随机森林分类器作为最佳预测模型,用于预测nmibc患者是否向mibc恶性进展,以及nmibc患者向mibc进展快慢。
[0107]
图2展示了基于nmibc患者向mibc恶性进展预测的分子标志物对a b组和c组的nmibc患者进行umap降维映射。图3展示了基于nmibc患者向mibc进展速度快慢预测的分子标志物对a组和b组的nmibc患者进行umap降维映射。
[0108]
8.预后预测模型验证
[0109]
从geo数据库中下载gse154261数据集作为独立验证队列(总计23例患者样本),验证队列的临床信息与发现队列相似,包括nmibc是否进展为mibc,以及在多长时间内(以月份为单位)发生了向mibc的恶性进展。根据临床随访信息,将nmibc患者分为3组:a’组——nmibc患者在1年内进展为mibc,共3例;b’组——nmibc患者在1-4年进展为mibc,共6例;c’组——nmibc患者在5年内无恶性进展,共14例。
[0110]
验证队列gse154261提供了原始reads count基因表达谱,按照步骤4对验证队列的转录组reads count基因表达谱数据进行tpm标准化。
[0111]
利用randomforest工具的randomforest函数,基于a’ b’和c’组验证数据的tpm标准化表达谱,对nmibc患者是否向mibc恶性进展的385个基因标志物和预测模型进行独立验证,基于a’组和b’组验证数据的tpm标准化表达谱,对nmibc患者向mibc进展快慢的32个基因标志物和预测模型进行独立验证,分别计算预测模型的灵敏度、特异性和auc。如图4所示,nmibc患者是否向mibc恶性进展的预测模型的灵敏度=100%,特异性=71.43%,auc=80.16%。如图5所示,nmibc患者向mibc进展快慢的预测模型的灵敏度=100%,特异性=100%,auc=100%。
[0112]
上述实施例仅为本发明的可行或较佳实施例而已,是用来说明本发明的,并非用以限制本发明申请专利的范围,因此,凡依本发明申请专利范围所作的均等变化与修饰,均应属于本发明专利涵盖的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。