诱导pd-l1蛋白降解的双功能分子化合物及其制备与应用
技术领域
1.本发明属于化学生物学技术领域,具体涉及诱导pd-l1蛋白降解的双功能分子化合物及其制备与应用。该化合物可以降解a375和b16 f10细胞系中的pd-l1并且可以增加t细胞杀伤肿瘤的效果。
背景技术:
2.程序性细胞死亡蛋白1配体1(pd-l1、b7-h1、cd274)是细胞表面配体b7家族的一员,该家族可调控t细胞激活和免疫应答。pd-l1在结构上与b7家族成员相似,它包含胞外igv和igc结构域和一个短细胞浆区域。pd-l1配体与pd-l1跨膜受体结合并抑制t细胞激活。研究表明,pd-l1在抗原呈递细胞、激活型t细胞、胎盘、心脏和肺等组织中表达。此外,pd-l1在黑色素瘤、卵巢癌、结肠癌、肺癌、乳腺癌和肾细胞癌等许多肿瘤类型的细胞中表达。pd-l1在癌细胞中的表达与肿瘤浸润性淋巴细胞有关,可通过释放干扰素γ介导pd-l1表达。其他研究表明pd-l1表达与病毒感染相关癌症有关。
3.蛋白质靶向嵌合体思路逐渐进入药物开发领域,其利用泛素化机制降解目标蛋白质达到对目标蛋白的抑制。传统的小分子和抗体等都是通过“占据驱动”的作用模式抑制靶蛋白的功能发挥治疗疾病的作用,这种作用模式需要抑制剂或单抗具备较高的浓度才能够占据靶点的活性位点,阻断下游信号通路的转导。而protac通过“事件驱动”机制发挥作用,不影响蛋白功能,介导靶蛋白发生降解。
4.目前用于肿瘤免疫治疗的药物多以单抗药物为主,这主要是因为pd-l1小分子抑制剂的疗效并不理想。利用protac技术可以很好地解决小分子抑制剂对pd-l1作用较差的影响,因此设计pd-l1相关的小分子降解剂可能显著提高临床免疫治疗效果。
技术实现要素:
5.本发明目的是针对现有技术补足,提供一种诱导pd-l1蛋白降解的双功能分子化合物及其制备与应用,具体为基于联苯类化合物合成的非天然pd-l1降解剂及其在肿瘤免疫治疗中的应用。通过降解细胞内pd-l1,增强t细胞对肿瘤细胞的直接杀伤作用。研究发现,以联苯类化合物bms-37为pd-l1配体的protac分子,无论选择crbn还是vhl为e3连接酶配体,均对pd-l1有一定的降解作用。本研究合成的化合物结合两种e3配体均能降解pd-l1,且这一现象可被蛋白酶体抑制剂mg132逆转,说明该类化合物是通过泛素蛋白酶体途径来发挥作用的。此外,研究结果显示该类化合物能显著增加t细胞的杀伤作用。因此,设计基于联苯类化合物的pd-l1蛋白降解剂,将为肿瘤免疫治疗提供新的治疗策略。
6.本发明的具体方案如下:
7.本发明提供了如下式i、式ii、式iii、式iv和式v所示的五种诱导pd-l1蛋白降解的双功能分子化合物,或其药学上可接受的盐、水合物或前药;
[0008][0009]
其中,b为泛素连接酶e3配体,更优选为crbn、vhl、mdm2、ciap、ubr7、rnf114、cblb、keap1中的一种。
[0010]
其中,所述的crbn优选为以下结构式中的一种:
[0011][0012]
其中:
[0013]
w选自ch2、c=o、so2、nh、n-烷基中的一种;烷基优选为c1-c4烷基;
[0014]
x选自o、s中的一种或两种;
[0015]
z选自氢、c1-c4烷基、c3-c6环烷基、卤素中的一种;
[0016]
g、g
′
各自独立表示h、c1-c4烷基、-oh、c1-c4烷基取代的5-10元杂环基,所述杂环基含有1-3个n、o或s的杂原子;
[0017]
r1选自h、d、卤素、硝基、氨基、氰基、羟基、c1-c4烷基、卤代c1-c4烷基、氘代c1-c4烷基中的一种。
[0018]
所述的vhl的结构式为:
[0019][0020]
其中:
[0021]
r2选自ch3、h中的一种。
[0022]
所述的mdm2的结构式为:
[0023][0024]
其中:
[0025]
r3为哌嗪基、哌啶基、杂环基团或以下结构的链接基团之一:
[0026][0027]
上述链接基团中,n为0-3的整数。
[0028]
其中:
[0029]
所述杂环基团为哌嗪酮基、吡咯基、吡唑基、呋喃基、噻吩基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、吡嗪基或哒嗪基中的一种。
[0030]
所述的ciap的结构为:
[0031][0032]
r4为h或boc。
[0033]
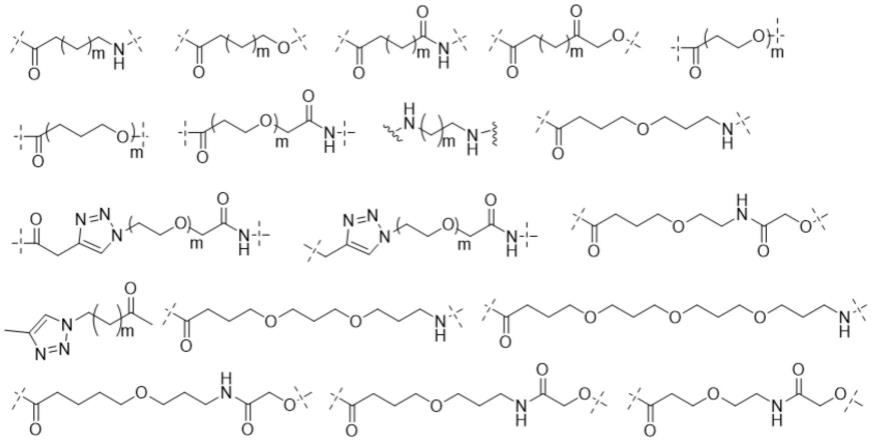
所述l优选为如下所示的任意结构之一:
[0034][0035]
其中:m选自1-10之间的整数。
[0036]
本发明优选以下式vi和式vii所示的诱导pd-l蛋白降解的双功能分子化合物或其药学上可接受的盐、水合物或前药:
[0037][0038]
上述式vi和式vii所示的诱导pd-l1蛋白降解的双功能分子化合物中,更优选为如下结构中的任意一种:
[0039][0040]
m选自1-10之间的整数。
[0041]
本发明更优选为以下式viii所示的诱导pd-l1蛋白降解的双功能分子化合物或其药学上可接受的盐、水合物或前药:
[0042][0043]
上述式viii所示的诱导pd-l1蛋白降解的双功能分子化合物中,l优选为以下结构:
[0044][0045]
m选自1-5之间的整数。
[0046]
本发明优选以下结构式所示的诱导pd-l1蛋白降解的双功能分子化合物包括,但不限于:
[0047]
[0048][0049]
根据本发明,诱导pd-l1蛋白降解的双功能分子化合物的药学上可接受的盐包括与下列酸形成的加成盐:盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸、萘二磺酸、乙酸、丙酸、乳酸、三氟乙酸、马来酸、柠檬酸、富马酸、草酸、酒石酸、苯甲酸等。盐酸、氢溴酸、硫酸、柠檬酸、酒石酸、磷酸、乳酸、丙酮酸、乙酸、三氟乙酸、马来酸、苯磺酸、琥珀酸以及类似的已知可以接受的酸成盐。
[0050]
此外,本发明还包括本发明衍生物的前药。它们自身可能具有较弱的活性甚至没有活性,但是在给药后,在生理条件下(如通过代谢、溶剂分解或另外的方式)被转化成相应的生物活性形式。
[0051]
本发明还提供了所述的诱导pd-l1蛋白降解的双功能分子化合物的制备方法,包括以下步骤:
[0052]
(一)式i所示的pd-l1蛋白降解剂的合成路线如下:
[0053]
在本合成路线中,l为m选自1-7之间的整数;
[0054][0055]
步骤1-1:化合物3的合成
[0056]
称取4-羟基-2,6-二甲氧基苯甲醛(910mg,5mmol)、三苯基膦(1.44g,5.5mmol)和(2-甲基-[1,1'-联苯]-3-基)甲醇(990mg,5mmol)加入到无水thf中,在冰浴条件下,向上述反应体系中滴加偶氮二甲酸二异丙酯(1.11g,5.5mmol)的无水thf溶液,室温下继续反应过夜,将反应混合物倒入h2o中,然后用dcm萃取混合物,有机相用盐水洗涤,经无水硫酸钠干燥,真空浓缩,得到残余物,残余物通过硅胶色谱法纯化,用乙酸乙酯/石油醚梯度洗脱,得到化合物3;
[0057]
步骤1-2:化合物6的合成
[0058]
用dcm溶解4-戊酸(200mg,2mmol),然后加入n-boc-乙二胺(320mg,2mmol)、edci(400mg,2.1mmol)、hobt(270mg,2mmol)和dipea(0.4ml)到圆底烧瓶中,混合物在室温下搅拌过夜,反应体系用dcm稀释,有机相用饱和碳酸氢钠溶液洗涤两次,无水硫酸钠干燥,减压浓缩混合物,产物通过硅胶色谱法纯化,然后用三氟乙酸脱保护,得到化合物6;
[0059]
步骤1-3:化合物7的合成
[0060]
将化合物3(3.4mmol,1.23g)和化合物6(5.1mmol,715mg)依次加入圆底烧瓶中,室温反应2小时后,加入三乙酰氧基硼氢化钠(4.1mmol,870mg)和一滴乙酸,室温搅拌过夜,用二氯甲烷稀释反应体系,用饱和碳酸氢钠溶液洗涤有机相两次,用无水硫酸钠干燥,经硅胶层析纯化,得化合物7;
[0061]
步骤1-4:式i化合物的合成
[0062]
将化合物7(0.035mmol,17mg)、b-linker、cuso4(0.044mmol,7mg)和vcna(0.105mmol,21mg)依次加入到thf和水的混合溶液(1:1),室温搅拌2小时,浓缩除去四氢呋喃,加水,二氯甲烷萃取3次,有机相用无水硫酸钠干燥,浓缩得粗品,经硅胶柱层析纯化得式i化合物;
[0063]
(二)式ii、式iii所示的pd-l1蛋白降解剂的合成路线如下:
[0064]
在本合成路线中,l为m选自1-7之间的整数;
[0065][0066]
步骤2-1:化合物9的合成;
[0067]
称取5-氯-2,4-二羟基苯甲醛(863mg,5mmol)、三苯基膦(1.44g,5.5mmol)和(2-甲基-[1,1'-联苯]-3-基)甲醇(990mg,5mmol)加入到无水thf中,在冰浴条件下,向上述反应体系中滴加偶氮二甲酸二异丙酯(1.11g,5.5mmol)的无水thf溶液,室温下继续反应过夜,将反应混合物倒入h2o中,然后用dcm萃取混合物,有机相用盐水洗涤,经无水硫酸钠干燥,真空浓缩,得到残余物,残余物通过硅胶色谱法纯化,用乙酸乙酯/石油醚梯度洗脱,得到化合物3;
[0068]
步骤2-2:化合物11的合成
[0069]
将化合物9(0.21mmol,74mg)用dmf溶解,然后依次加入3-(溴甲基)苄腈(0.25mmol,49mg),在80℃下反应30min,向反应体系中加水,用乙酸乙酯萃取,有机相用饱和盐水洗涤3次,无水硫酸钠干燥,减压蒸发得粗品,粗产物用叔丁基甲基醚纯化,过滤得到化合物11;
[0070]
步骤2-3:化合物13的合成
[0071]
将化合物11(0.13mmol,63mg)、2-氨基-2-甲基丙酸(0.54mmol,56mg)、acoh(2d)和nabh3cn(0.67mmol,42mg)在5ml dmf中的混合物置于80℃反应1h,向反应混合物中加入乙酸乙酯和水,浓缩有机萃取物,粗产物在硅胶上层析,用甲醇与二氯甲烷溶液洗脱,得到化合物13;
[0072]
步骤2-4:化合物13y的合成
[0073]
将化合物13(0.02mmol,13mg)溶于dmf中,取炔丙基胺(0.03mmol,2μl)、hatu(0.03mmol,11mg)和dipea(0.04mmol,6μl)在室温下反应2小时,产物硅胶层析石油醚乙酸乙酯体系,得到化合物13y;
[0074]
步骤2-5:式ii化合物的合成
[0075]
将化合物13y(0.035mmol,17mg)、b-linker、cuso4(0.044mmol,7mg)和vcna(0.105mmol,21mg)依次加入到thf和水的混合溶液(1:1),室温搅拌2小时,浓缩除去四氢呋喃,加水,二氯甲烷萃取3次,有机相用无水硫酸钠干燥,浓缩得粗品,经硅胶柱层析纯化得式ii化合物;
[0076]
步骤2-6:式iii化合物的合成
[0077]
化合物13(0.04mmol,22mg),edci(0.08mmol,15mg),hobt(0.08mmol,11mg),dipea(0.16mmol,28μl)和b-linker溶于dcm在室温下搅拌2h,将反应混合物用水和dcm萃取,有机层用水洗涤3次,无水硫酸钠干燥,减压蒸馏浓缩得到残渣,经硅胶层析纯化得到式iii;
[0078]
(三)式iv所示的pd-l1蛋白降解剂的合成路线如下:
[0079]
在本合成路线中,l为m选自1-7之间的整数;
[0080][0081]
步骤3-1:化合物16的合成
[0082]
在室温条件下,向2-甲基联苯-3-甲醇(1.45g,7.31mmol)的二氯甲烷溶液中分批加入戴斯-马丁试剂(3.26g,7.68mmol),将所得混合物在室温下搅拌30min,然后用碳酸氢钠溶液和硫酸钠溶液淬灭,混合物用二氯甲烷萃取,合并的萃取物用无水硫酸钠干燥并浓缩,残余物通过柱色谱法纯化,得到化合物16;
[0083]
步骤3-2:化合物18y的合成
[0084]
将3-氨基-4-羟基苯甲酸甲酯(49mg,0.29mmol)、化合物16(69mg,0.35mmol)和三氟甲磺酸锌(10mg,0.03mmol)溶于乙醇(1.5ml)回流过夜,之后将反应混合物冷却至室温,然后浓缩,将残余物溶解在二氯甲烷中,然后加入二氯二氰基苯醌(100mg,0.6mmol),将混合物在室温搅拌0.5小时,然后用乙酸乙酯稀释并用碳酸氢钠溶液、硫酸钠溶液、水和盐水洗涤,有机层用无水硫酸钠干燥并浓缩,残余物通过柱色谱法纯化,得到化合物18y;
[0085]
步骤3-3:化合物18的合成
[0086]
将化合物18y在lioh、甲醇/水的条件下进行处理,之后通过乙酸乙酯萃取和浓缩得到化合物18;
[0087]
步骤3-4:化合物19的合成,化合物19的合成同化合物13y;
[0088]
步骤3-5:式iv化合物的合成;
[0089]
将化合物19(0.035mmol,13mg)、b-linker、cuso4(0.044mmol,7mg)和vcna(0.105mmol,21mg)依次加入到thf和水的混合溶液(1:1),室温搅拌2小时,浓缩除去四氢呋喃,加水,二氯甲烷萃取3次,有机相用无水硫酸钠干燥,浓缩得粗品,经硅胶柱层析纯化得式ii化合物;
[0090][0091]
(四)式v所示的pd-l1蛋白降解剂的合成路线如下:
[0092]
在本合成路线中,l为m选自1-7之间的整数;
[0093]
步骤4-1:化合物22的合成
[0094]
将6-氯哒嗪-3-胺(5mmol,648mg)溶解在10ml二噁烷中后,依次加入nahco3(7.5mmol,630mg)和3-溴-2-氧代丙酸乙酯(5.5mmol,1.07g),混合物在100℃下反应3h,过滤反应体系,滤液用水稀释,乙酸乙酯萃取,无水硫酸钠干燥,浓缩得黑色胶体粗品,产物经硅胶柱层析纯化;
[0095]
步骤4-2:化合物24的合成
[0096]
将化合物22(10.1mmol,2.5g)、k2co3(30.5mmol,4.2g)、(r)-吡咯烷-3-基氨基甲酸叔丁酯(10.1mmol,1.9g)和40ml dmf的混合物置于110℃反应12小时,用水稀释,乙酸乙酯萃取3次,有机相用饱和盐水洗涤3次,无水硫酸钠干燥,浓缩得到粗产物,经柱层析纯化得化合物24;
[0097]
步骤4-3:化合物25的合成,化合物25的合成同化合物18;
[0098]
步骤4-4:化合物27的合成
[0099]
在20ml dmf中加入boc-d-组氨酸(2mmol,511mg)和k2co3(6mmol,829mg),然后逐滴加入苄基溴(2.2mmol,261μl),室温反应4h,将残余物通过硅胶快速色谱法纯化,得化合物27;
[0100]
步骤4-5:化合物28的合成,将化合物27用三氟乙酸脱保护基boc,得到化合物28;
[0101]
步骤4-6:化合物29的合成
[0102]
将2-甲基-[1,1'-联苯]-3-甲醇(1g,5mmol)溶于二氯甲烷中,加入三乙胺(5.5mmol,0.7ml)和三光气的二氯甲烷溶液(600mg,2mmol)在-20℃下滴加,之后转至0℃反应1.5小时,向反应体系中加入30ml正己烷,搅拌10分钟后过滤,滤饼用10ml二氯甲烷和正己烷(1:2)混合溶液洗涤3次,滤液浓缩得化合物29,因产物不稳定,未进一步纯化,直接投入下一步反应;
[0103]
步骤4-7:化合物30的合成
[0104]
将化合物24(1.86mmol,650mg)溶于thf和水的混合溶液中,加入nahco3(5.6mmol,470mg),在0℃滴加化合物29的thf(6.5ml)溶液,转移至室温并继续搅拌2小时,用水淬灭反应并用乙酸乙酯萃取3次,有机相用无水硫酸钠干燥,过滤后浓缩,残余物经硅胶柱层析纯化得到化合物30;
[0105]
步骤4-8:化合物31的合成;
[0106]
化合物28(0.04mmol,15mg),化合物30(0.04mmol,19mg),edci(0.08mmol,15mg),hobt(0.08mmol,11mg),dipea(0.16mmol,28μl)溶于dcm在室温下搅拌2h,将反应混合物用水和dcm萃取,有机层用水洗涤3次,无水硫酸钠干燥,减压蒸馏浓缩得到残渣,经硅胶层析纯化得到化合物31;
[0107]
步骤4-9:化合物31y的合成;
[0108]
化合物31(0.04mmol,32mg),丙炔胺(0.08mmol,4mg),edci(0.08mmol,15mg),hobt(0.08mmol,11mg),dipea(0.16mmol,28μl)溶于dcm在室温下搅拌2h,将反应混合物用水和dcm萃取,有机层用水洗涤3次,无水硫酸钠干燥,减压蒸馏浓缩得到残渣,经硅胶层析纯化得到化合物31;
[0109]
步骤4-10:式v化合物的合成。
[0110]
将化合物31y(0.035mmol,25.8mg)、b-linker、cuso4(0.044mmol,7mg)和vcna(0.105mmol,21mg)依次加入到thf和水的混合溶液(1:1),室温搅拌2小时,浓缩除去四氢呋喃,加水,二氯甲烷萃取3次,有机相用无水硫酸钠干燥,浓缩得粗品,经硅胶柱层析纯化得式v化合物;
[0111]
泛素连接酶e3配体与l(linker)的合成路线如下:
[0112]
(1)当泛素连接酶e3配体为crbn时,优选为th(沙利度胺衍生物),其合成方法为:
[0113][0114]
将化合物th(沙利度胺衍生物)溶于dmf中,向反应体系中加入dipea,1.0mol的linker,90℃反应2小时加入水,乙酸乙酯萃取,收集有机层,无水硫酸钠干燥,减压浓缩,经
硅胶柱层析纯化,得到化合物th-l;
[0115]
(2)当泛素连接酶e3配体为mdm2时,其合成方法为:
[0116][0117]
取1.0mol的mdm2溶于dcm中,之后在冰浴下加入1.1mol的edci、1.1mol的hobt、1.5mol的dipea,之后加入1.0mol的linker,反应5h,之后加入适量dcm稀释萃取,集有机层,无水硫酸钠干燥,减压浓缩,经硅胶柱层析纯化,得到化合物mdm2-l;
[0118]
(3)当泛素连接酶e3配体为vhl时,其合成路线同mdm2-l,区别在于将mdm2替换为vhl,相应制得vhl-l;
[0119]
(4)当泛素连接酶e3配体为ciap时,其合成路线同mdm2-l,区别在于将vhl替换为ciap,相应制得ciap-l。
[0120]
一种药物组合物,含有治疗有效量的诱导pd-l1蛋白降解的双功能分子化合物作为活性成分。
[0121]
一种药物组合物,还包括药学上可接受的载体、稀释剂、辅剂、媒介物或它们的组合。
[0122]
其中,所述的药物组合物的剂型为注射剂、片剂和胶囊剂中的任意一种。
[0123]
本发明还提供了上述的诱导pd-l1蛋白降解的双功能分子化合物及其药学上可接受的盐或上述的药物组合物在制备治疗和/或预防感染、癌症或自身免疫性疾病中药物的应用。
[0124]
所述的感染为皮肤感染、胃肠道感染、泌尿生殖系统感染、系统性感染、或由流感、丙型肝炎病毒、人类乳头状瘤病毒、巨细胞病毒、爱泼斯坦巴尔病毒、脊髓灰质炎病毒、水症带状殖彦病毒、柯萨奇病毒和人类免疫缺陷病毒中的一种或多种引起的病毒感染中的一种或多种;
[0125]
所述的癌症为骨癌、肺癌、胃癌、结肠癌、膜腺癌、乳腺癌、前列腺癌、肺癌、脑癌、卵巢癌、膀肮癌、子宫颈癌、辜丸癌、肾癌、头颈癌、淋巴癌、白血病和皮肤癌中的一种或多种。
[0126]
所述的自身免疫性疾病为类风湿性关节炎、全身性红斑狼疮、混合性结缔组织病、系统硬皮病、皮肌炎、结节性脉管炎、肾病、内分泌相关疾病、肝病、银屑病和由于感染引起的自身免疫反应中的一种或多种。
[0127]
本发明的有益效果:
[0128]
本发明以protac技术为支撑,以pd-l1抑制剂为原料,合成linker长度不同的诱导pd-l1蛋白降解的双功能分子化合物。
[0129]
本发明有效地靶向并降解pd-l1;类似于催化反应,药物起效剂量低;只提供结合活性,是事件驱动,区别于传统的占有驱动,不需直接抑制目标蛋白的功能活性;药物不需要与目标蛋白长时间和高强度的结合。本发明涉及的诱导pd-l1蛋白降解的双功能分子化
合物为pd-l1介导的肿瘤和/或其他疾病的治疗,提供了新的治疗方式。
附图说明
[0130]
图1为本发明的bms-37-c1-c5以及bms37-3c-v2、bms37-5c-v2、bms37-7c-v2对pd-l1的降解作用;
[0131]
图2为本发明的bms-37-c1-c5以及bms37-3c-v2、bms37-5c-v2、bms37-7c-v2对pd-l1降解的时间依赖性以及mg 132逆转实验;
[0132]
图3为本发明的bms-37-c1、bms-37-c3和bms37-5c-v2采用流式细胞术检测pd-l1表达的变化;
[0133]
图4为本发明的bms-37-c1以及bms-37-c3对t细胞杀伤肿瘤细胞的影响;
[0134]
图5为本发明实施例标准处理的毛细血管中通过mst测量bms-37-c1、bms-37-c3和bms37-5c-v2与pd-l1的亲和力结合曲线。
具体实施方式
[0135]
通过实施例对本发明做进一步的说明。但不应理解为本发明的范围仅限于以下实施例。
[0136]
实施例1中间化合物th-l2(m=2)的合成
[0137]
本实施例中,l为m为2;
[0138]
b为crbn,则合成路线为:
[0139][0140]
58mg化合物th(沙利度胺衍生物,可购买获得)于茄形瓶中,加入3ml dmf,搅拌下依次加入50mg叠氮基-2peg-胺和47μl dipea,90℃反应3-4小时,加入30ml水和30ml乙酸乙酯萃取,有机层用无水硫酸钠干燥,浓缩后得粗品,经硅胶柱层析纯化,石油醚-乙酸乙酯1:2至1:4梯度洗脱,得黄色油状物33.2mg,收率41%。
[0141]
th-l2,1h nmr(400mhz,cdcl3)δ9.06(br s,1h),7.49(t,j=7.8hz,1h),7.09(dd,j=
[0142]
7.2,2.0hz,1h),6.93(d,j=8.4hz,1h),6.50(t,j=5.6hz,1h),4.96
–
4.92(m,1h),3.74(t,j=5.2hz,2h),3.70
–
3.67(m,6h),3.48(q,j=5.6hz,2h),3.38(t,j=4.4hz,2h),2.80
–
2.73(m,3h),2.13
–
2.10(m,1h);
13
c nmr(100mhz,cdcl3)δ171.7,169.4,168.8,167.7,146.9,136.1,132.6,116.9,111.6,110.3,70.75,70.72,70.1,69.6,50.7,48.9,42.4,31.5,22.8。
[0143]
实施例2中间化合物m-l1(m=1)的合成
[0144]
本实施例中,b为mdm2;
[0145]
本实施例中,l为m为1;
[0146][0147]
320mg化合物m于茄形瓶中,加入3ml dcm,冰浴下依次加入65mg叠氮基-1peg-胺、132μl dipea、74mg edci、92mg hobt,反应3-4小时,加入30ml水和30ml dcm萃取,有机层用无水硫酸钠干燥,浓缩后得粗品,经硅胶柱层析纯化得白色固体237.5mg,收率:63.2%。
[0148]
m-l1,1h nmr(600mhz,dmso-d6)δ7.96(t,j=5.7hz,1h),7.56
–
7.51(m,1h),7.17
–
[0149]
7.14(m,2h),7.12(d,j=8.3hz,2h),7.07
–
7.02(m,2h),6.98(d,j=8.0hz,2h),6.62(d,j=7.3hz,2h),5.66(d,j=9.7hz,1h),5.58(d,j=9.7hz,1h),4.72(hept,j=6.0hz,1h),3.83(s,4h),3.76
–
3.67(m,2h),3.65
–
3.55(m,3h),3.43(t,j=5.9hz,2h),3.38(t,j=4.9hz,3h),3.21(q,j=5.8hz,3h),3.00(h,j=7.3hz,2h),1.27(d,j=6.0hz,3h),1.22(d,j=6.0hz,3h).hrms(esi
):m/z calculated for c
36h40
cl2n8o6[m h]
,751.2448;found,751.2443。
[0150]
实施例3中间化合物v-2l的合成
[0151]
本实施例中b为vhl;
[0152]
本实施例中,l为m为1;
[0153][0154]
215mg化合物v于茄形瓶中,加入3ml dcm,冰浴下依次加入57.5mg 3-叠氮基丙酸、132μl dipea、74mg edci、92mg hobt,反应3-4小时,加入30ml水和30ml dcm萃取,有机层用无水硫酸钠干燥,浓缩后得粗品,经硅胶柱层析纯化得白色固体279mg,收率:53%。
[0155]
v-2l,1h nmr(600mhz,dmso-d6)δ8.98(s,1h),8.57(t,j=6.0hz,1h),8.12(d,j=9.4
[0156]
hz,1h),7.47
–
7.36(m,4h),5.13(d,j=3.6hz,1h),4.58(d,j=9.4hz,1h),4.47
–
4.40(m,2h),4.39
–
4.33(m,1h),4.21(dd,j=15.8,5.5hz,1h),3.69(dd,j=10.5,4.2hz,1h),3.62(dt,j=10.8,1.8hz,1h),3.52(ddd,j=12.2,7.9,5.6hz,1h),3.47(dt,j=12.2,
6.1hz,1h),2.63
–
2.53(m,1h),2.46
–
2.45(m,1h),2.45(s,3h),2.07
–
1.99(m,1h),1.91(ddd,j=12.9,8.5,4.6hz,1h),0.95(s,9h).hrms(esi
):m/z calculated for c
25h33
n7o4s[m h]
,528.2315;found,528.2320。
[0157]
实施例4化合物3的合成
[0158][0159]
具体操作过程参见步骤1-1,淡黄色固体,收率:58%。1h nmr(400mhz,cdcl3)δ10.37(s,1h),7.42
–
7.27(m,8h),6.20(s,2h),5.15(s,2h),3.88(s,6h),2.27(s,3h).
[0160]
实施例5化合物6的合成
[0161][0162]
具体操作过程参见步骤1-2,黄色油状物。1h nmr(400mhz,cdcl3)δ6.29(s,1h),4.92(s,1h),3.40
–
3.36(m,2h),3.31
–
3.28(m,2h),2.55
–
2.51(m,2h),2.42
–
2.39(m,2h),2.01(t,j=2.4hz,1h),1.44(s,9h).
[0163]
实施例6化合物7的合成
[0164][0165]
具体操作过程参见步骤1-3,收率:48%。1h nmr(400mhz,cdcl3)δ7.48
–
7.27(m,8h),6.24(s,2h),5.08(s,2h),4.61(s,2h),3.96(s,2h),3.84(s,6h),3.47
–
3.44(m,2h),2.82(t,j=4.8hz,2h),2.51
–
2.49(m,2h),2.47
–
2.43(m,2h),2.27(s,3h),1.94(t,j=2.4hz,1h).
[0166]
实施例7化合物11的合成
[0167][0168]
具体操作过程参见步骤2-2,白色固体产物70mg,收率:71%。1h nmr(600mhz,
cdcl3)δ10.32(s,1h),7.91(s,1h),7.72(td,j=1.8,0.8hz,1h),7.68(ddt,j=7.8,6.2,1.3hz,2h),7.54(t,j=7.8hz,1h),7.46
–
7.40(m,3h),7.40
–
7.34(m,1h),7.32
–
7.27(m,4h),6.63(s,1h),5.20(d,j=9.6hz,4h),2.27(s,3h).
[0169]
实施例8化合物13的合成
[0170][0171]
具体操作过程参见步骤2-3,白色固体28mg,收率38%。1h nmr(600mhz,dmso-d6)δ8.03(s,1h),7.89(d,j=7.9hz,1h),7.83(d,j=7.8hz,1h),7.62(t,j=7.8hz,1h),7.56(s,1h),7.50
–
7.44(m,3h),7.42
–
7.36(m,1h),7.33
–
7.26(m,3h),7.21(dd,j=7.6,1.4hz,1h),7.12(s,1h),5.29(d,j=3.5hz,4h),3.87(s,2h),2.23(s,3h),1.90(s,1h),1.27(s,6h).
[0172]
实施例9化合物13y的合成
[0173][0174]
具体操作过程参见步骤2-4,白色固体,8mg,70%。1h nmr(600mhz,dmso-d6)δ8.13(s,1h),7.96(s,1h),7.84
–
7.79(m,2h),7.62(t,j=7.8hz,1h),7.49
–
7.42(m,4h),7.40
–
7.37(m,1h),7.33
–
7.30(m,2h),7.27(t,j=7.6hz,1h),7.21(dd,j=7.7,1.5hz,1h),7.06(s,1h),5.31(s,2h),5.24(s,2h),3.83(dd,j=6.0,2.6hz,2h),3.49(s,2h),3.02(t,j=2.5hz,1h),2.23(s,3h),1.99(dq,j=15.9,6.8,6.2hz,1h),1.25
–
1.22(m,3h),1.20(s,3h).
[0175]
实施例10化合物16的合成
[0176][0177]
具体操作过程参见步骤3-1,白色固体430mg,收率:86%。1h nmr(400mhz,cdcl3)δ10.39(s,1h),7.84(dd,j=7.6,1.6hz,1h),7.48-7.38(m,5h),7.29-7.27(m,2h),2.55(s,3h).
[0178]
实施例11化合物22的合成
[0179]
[0180]
具体操作过程参见步骤4-1,收率:52%。1h nmr(600mhz,dmso-d6)δ8.89(s,1h),8.30(d,j=9.6hz,1h),7.50(d,j=9.6hz,1h),4.35(q,j=7.1hz,3h),1.33(t,j=7.1hz,4h).
[0181]
实施例12化合物24的合成
[0182][0183]
具体操作过程参见步骤4-2,黑色胶体1.9g,收率44.8%。1h nmr(600mhz,dmso)δ8.35(d,j=0.7hz,1h),7.83(d,j=10.0hz,1h),7.25(d,j=6.7hz,1h),6.98(d,j=9.9hz,1h),4.27(q,j=7.1hz,2h),4.13(d,j=6.8hz,1h),3.65(dd,j=10.8,6.3hz,1h),3.57(dt,j=10.2,7.2hz,1h),3.47(ddd,j=10.3,7.9,5.7hz,1h),3.28(dd,j=10.8,4.6hz,1h),2.14(dq,j=13.6,6.9hz,1h),1.91(dq,j=12.7,6.2hz,1h),1.39(s,10h),1.30(t,j=7.1hz,3h).
[0184]
实施例13化合物27的合成
[0185][0186]
具体操作过程参见步骤4-4,收率:49%。1h nmr(600mhz,dmso-d6)δ7.33(t,j=7.7hz,1h),7.28(dd,j=8.3,6.4hz,5h),7.24
–
7.16(m,3h),6.82(s,3h),5.11(d,j=1.9hz,2h),5.03(q,j=12.5hz,2h),4.31
–
4.24(m,1h),2.84
–
2.80(m,2h),1.34(s,9h).
[0187]
实施例14化合物30的合成
[0188][0189]
具体操作过程参见步骤4-7,黄色固体512mg,收率:55%。1h nmr(600mhz,dmso-d6)δ8.35(s,1h),7.84(dd,j=10.0,0.7hz,1h),7.73(d,j=6.6hz,1h),7.44(t,j=7.5hz,2h),7.40
–
7.33(m,2h),7.28
–
7.26(m,2h),7.24(d,j=7.6hz,1h),7.16(d,j=7.6hz,1h),6.98(d,j=9.9hz,1h),5.12(s,2h),4.28(q,j=7.1hz,2h),4.23(q,j=6.0hz,1h),3.68(dd,j=10.9,6.2hz,1h),3.58(dt,j=10.3,7.3hz,1h),3.50(ddd,j=10.1,7.7,5.2hz,1h),3.37(dd,j=11.1,4.4hz,1h),2.22
–
2.17(m,1h),2.16(s,3h),2.00
–
1.91(m,1h),1.30(t,j=7.1hz,3h).
[0190]
实施例15化合物bms37-cn的合成(n=1-5,以n=1为例)
[0191]
[0192]
将化合物7(0.035mmol,17mg)、th-l1(0.035mmol,13.5mg)、cuso4(0.044mmol,7mg)和vcna(0.105mmol,21mg)依次加入到thf和水的混合溶液(1:1),室温搅拌2小时。浓缩除去四氢呋喃,加水,二氯甲烷萃取3次,有机相用无水硫酸钠干燥,浓缩得粗品,经硅胶柱层析纯化得13mg固体,收率:42.5%。1h nmr(400mhz,cdcl3)δ7.72(s,1h),7.62(s,1h),7.52
–
7.48(m,1h),7.43
–
7.41(m,3h),7.38
–
7.28(m,4h),7.12(d,j=7.2hz,1h),6.88(d,j=8.4hz,1h),6.47(t,j=5.2hz,1h),6.24(s,2h),5.10(s,2h),4.97
–
4.42(m,1h),4.54
–
4.51(m,2h),4.12(s,2h),3.88
–
3.83(m,2h),3.82(s,6h),3.67(t,j=4.8hz,2h),3.50
–
3.48(m,2h),3.43(q,j=5.2hz,2h),3.03(td,j=6.8,2.8hz,3h),2.92(t,j=4.8hz,2h),2.86
–
2.73(m,4h),2.55(t,j=7.6hz,2h),2.27(s,3h),2.09
–
2.07(m,1h).hrms m/z calcd for c
47h53
n8o
9 873.3936,found873.3930[m h
].
[0193]
实施例16化合物np19-np-th的合成(n=2-3,以n=2为例)
[0194]
具体操作及配比参考实施例15。
[0195][0196]
np19-2p-th,黄色固体,收率48%。1h nmr(600mhz,dmso-d6)δ11.09(s,1h),8.25(s,1h),7.93(s,1h),7.83(s,1h),7.80
–
7.75(m,2h),7.59(t,j=7.8hz,1h),7.54(dd,j=8.6,7.1hz,1h),7.46(q,j=7.9hz,3h),7.40
–
7.34(m,2h),7.32
–
7.29(m,2h),7.26(t,j=7.6hz,1h),7.20(dd,j=7.7,1.5hz,1h),7.07
–
6.99(m,3h),6.55(t,j=5.9hz,1h),5.26(s,2h),5.21(s,2h),5.07(dd,j=12.7,5.6hz,1h),4.48(t,j=5.2hz,2h),4.30(d,j=5.7hz,2h),3.80(t,j=5.2hz,2h),3.56(t,j=5.5hz,2h),3.48(s,2h),3.41
–
3.38(m,2h),3.17(dd,j=5.3,1.1hz,1h),2.88(ddd,j=16.8,13.6,5.4hz,1h),2.62
–
2.52(m,2h),2.22(s,3h),2.01(tdd,j=12.6,8.0,5.1hz,2h),1.46(s,1h),1.32
–
1.25(m,2h),1.23(s,3h),1.21(s,3h).hrms m/z calcd for c
55h57
cln9o
9 1022.3962,found 1022.4005[m h
].
[0197]
实施例17化合物mp-pc-nc的合成(n=4,6,以n=4为例)
[0198][0199]
化合物13(0.04mmol,22mg),edci(0.08mmol,15mg),hobt(0.08mmol,11mg),dipea(0.16mmol,28μl)和th-4l(0.04mmol,13.76mg)溶于dcm在室温下搅拌2h。混合物与水稀释和提取与dcm。结合的有机层用水洗涤,在无水硫酸钠上干燥,在减压下过滤和浓缩得到残渣。经硅胶层析纯化得黄色固体15mg,收率:43%。1h nmr(600mhz,cdcl3)δ8.17(s,1h),7.68(d,j=1.7hz,1h),7.62(ddt,j=10.8,7.8,1.4hz,2h),7.50(t,j=7.7hz,2h),7.46
–
7.40(m,4h),7.38
–
7.35(m,1h),7.31(d,j=1.5hz,1h),7.29(d,j=0.9hz,2h),7.26(d,j=1.4hz,1h),7.25(s,1h),7.04(d,j=7.1hz,1h),6.85(d,j=8.5hz,1h),6.59(s,1h),6.19
(s,1h),5.09(s,4h),4.89(dd,j=12.5,5.3hz,1h),3.61(s,2h),3.25(q,j=6.6hz,2h),3.19(q,j=6.7hz,2h),2.85(ddd,j=16.9,3.6,2.4hz,1h),2.79
–
2.66(m,2h),2.25(s,3h),2.12
–
2.07(m,1h),1.63
–
1.60(m,2h),1.55(dt,j=10.5,4.4hz,2h),1.35(s,6h).hrms m/z calcd for c
50h50
cln6o
7 881.3424,found 881.3432[m h
].
[0200]
实施例18化合物np19-2c-v2的合成
[0201]
具体操作及配比参考实施例18。
[0202][0203]
np19-2c-v2,黄色固体,收率:42%。1h nmr(600mhz,dmso-d6)δ8.98(s,1h),8.38(d,j=7.8hz,1h),7.98
–
7.91(m,2h),7.89(s,1h),7.81(dd,j=7.8,1.7hz,2h),7.62(d,j=7.7hz,1h),7.49(s,1h),7.48
–
7.45(m,2h),7.44(d,j=5.4hz,1h),7.42(s,1h),7.40
–
7.35(m,3h),7.32
–
7.29(m,2h),7.27(s,1h),7.21(d,j=1.4hz,1h),7.06(s,1h),5.35
–
5.32(m,1h),5.31(d,j=4.9hz,2h),5.24(s,2h),4.91(s,1h),4.53(d,j=9.3hz,1h),4.42(t,j=8.0hz,1h),4.27(p,j=3.3hz,1h),3.60(d,j=3.4hz,2h),3.54
–
3.49(m,2h),3.27(p,j=6.5hz,1h),3.21(p,j=6.5hz,2h),2.45(s,3h),2.35
–
2.25(m,1h),2.23(s,3h),2.05
–
1.95(m,4h),1.23(s,6h),1.18(d,j=4.5hz,3h),0.90(s,9h).hrms m/z calcd for c
59h66
cln7o7s 1052.4506,found 1052.4512[m h
]
[0204]
实施例19化合物nb系列化合物的合成(以nb-c1化合物为例)
[0205]
具体操作及配比参考实施例15。
[0206][0207]
nb-c1,黄色固体,收率:85%。1h nmr(400mhz,cdcl3)δ9.28(s,1h),8.24(d,j=1.2hz,1h),8.10(dd,j=6.8,2.4hz,1h),7.93(s,1h),7.90(dd,j=8.8,1.6hz,1h),7.58(d,j=8.4hz,1h),7.47-7.33(m,9h),7.10(d,j=6.8hz,1h),6.85(d,j=8.4hz,1h),6.47(t,j=5.2hz,1h),5.08(dd,j=12.0,5.2hz,1h),4.84-4.72(m,2h),4.62-4.58(m,2h),3.90-3.88(m,2h),3.70(t,j=5.2hz,2h),3.45-3.42(m,2h),2.88-2.76(m,3h),2.62(s,3h),2.18-2.14(m,1h).hrms m/z calcd for c
41h36
n8o7na 775.2599,found 775.2613[m na
].
[0208]
实施例20化合物pd-nc-th的合成(n=2,4,6,以n=2为例)
[0209]
具体操作及配比参考实施例18。
[0210][0211]
pd-2c-th,黄色固体,收率:53%。1h nmr(600mhz,dmso-d6)δ11.09(s,1h),8.24
–
8.18(m,2h),8.15(s,1h),7.84(d,j=9.9hz,1h),7.72(d,j=6.6hz,2h),7.58
–
7.53(m,1h),7.44(t,j=7.5hz,2h),7.40
–
7.33(m,2h),7.29
–
7.26(m,2h),7.25
–
7.21(m,2h),7.21
–
7.18(m,2h),7.16(dd,j=8.8,2.8hz,2h),7.15
–
7.11(m,2h),7.01(dd,j=7.0,1.3hz,1h),6.97(d,j=9.9hz,1h),6.91(s,1h),6.75(d,j=4.9hz,1h),5.11(d,j=15.4hz,4h),5.04(ddd,j=12.8,5.5,1.8hz,1h),4.67
–
4.62(m,1h),4.23(q,j=5.7hz,1h),3.70(dd,j=10.8,6.1hz,1h),3.59(q,j=7.8,5.4hz,1h),3.53
–
3.49(m,1h),3.37(dd,j=10.8,4.1hz,2h),3.27(t,j=6.5hz,2h),3.25
–
3.15(m,2h),2.96(s,2h),2.92
–
2.83(m,1h),2.60
–
2.54(m,1h),2.20(dt,j=12.6,6.9hz,1h),2.16(s,3h),2.04
–
1.92(m,4h).hrms m/z calcd for c
54h53n12o8 997.4104,found 997.4083[m h
].
[0212]
实施例21 western blot检测相关蛋白的改变
[0213]
1.pd-l1蛋白质表达的变化
[0214]
(1)将对数生长且融合度达到90%的细胞进行消化收集,加入新鲜培养基调整细胞浓度,然后使用细胞计数板计数。将细胞均匀接种在大皿中,培养箱中培养贴壁并开始处于对数分裂期,更换含有药物的培养基后放入培养箱继续培养48h。等待培养结束后,使用胰酶消化细胞并收集,做好标记。使用pbs清洗两遍以清除掉胰酶中多余的edta,每次5min并将细胞收集在1.5ml ep管中。清洗完毕后,根据细胞数量加入适量的ripa裂解液,裂解液提前加入磷酸酶抑制剂,全酶抑制剂以及及时加入pmsf。pmsf是一种丝氨酸蛋白酶不可逆抑制剂,可以抑制丝氨酸蛋白酶(胰蛋白酶和胰凝乳蛋白酶),也可以抑制半胱氨酸蛋白酶和乙酰胆碱酯酶,进而防止蛋白质的降解。加入裂解液后,使用涡旋振荡器混匀细胞,并在冰上进行裂解蛋白防止蛋白质降解,冰上裂解20min。裂解完成后,使用4℃离心机进行离心,转速11500rpm时间10min。等待离心结束,吸取上清液,该上清液为蛋白质;
[0215]
(2)本实验用bca法进行蛋白含量检测。在碱性条件下,蛋白将cu
2
还原为cu
,cu
与bca试剂形成紫蓝色的络合物,测定其在562nm处的吸收值,并与标准曲线对比,即可计算待测蛋白的浓度;
[0216]
(3)将定量结束的蛋白样品,根据其体积加入相应体积的6
×
loading buffer。使用前先向6
×
loading buffer中加入dtt,dtt可以将cys上的二硫键打开,使之成为线装多肽,这样便于抗体识别并结合特定的位点。使用涡旋振荡器将样品混匀并放入100℃水浴锅中煮5-10min使蛋白质充分变性。如果一个蛋白的氨基酸组成含有较多疏水性氨基酸(色氨酸w、丙氨酸a、缬氨酸v、亮氨酸l、异亮氨酸i、脯氨酸p、苯丙氨酸f等),蛋白疏水性很强,则蛋白煮了之后很容易形成聚集体对于这种富含疏水性氨基酸的蛋白,可以用37℃孵育1h,或者60℃30min代替100℃水浴锅中煮5-10min;
[0217]
(4)配制sds-page凝胶,并且将制备好的样品进行上样;
[0218]
(5)将pvdf膜放入无水甲醇中30s以激活pvdf膜。然后将膜泡进湿转液中。电转夹白色依次面放入海绵和滤纸以及pvdf膜,然后依次放入胶、滤纸和海绵,制作成三明治结构。胶和膜之间避免气泡。将三明治结构插入电转槽中,并将电转装置放入冰水混合的冰盒中,避免在转膜过程中产生过多热量导致转膜失败。使用300ma转印蛋白2h;
[0219]
(6)蛋白转印结束后,将三明治结构打开,使用立春红对膜进行染色查看转印效果并使用纯水将膜清洗干净。根据蛋白分子量将膜裁剪,然后滤纸晾干pvdf膜,再次使用甲醇进行二次激活。激活完毕后,将表面甲醇用纯水清洗干净,然后放入到5%脱脂牛奶中,使用摇床封闭2h;
[0220]
(7)将封闭完成的膜使用tbst溶液将其表面牛奶洗干净。使用5%bsa液体根据抗体说明书指明的稀释比例进行一抗稀释来配制所需抗体。然后将tbst中的pvdf膜放在湿盒上进行一抗孵育,将湿盒放入4℃冰箱过夜孵育一抗;
[0221]
(8)将湿盒取出,用tbst将膜在摇床上清洗三次,每次5min。根据一抗说明书选择不同的二抗。将清洗过的膜继续放在湿盒上并加入二抗液体并在室温封闭两个小时;
[0222]
(9)对目标条带使用凝胶成像仪进行曝光。
[0223]
如图1和图2所示,实验结果显示,化合物bms-37-c3和bms-37-c1在a375细胞系中降解效果最好。这两个化合物在0.3μm时开始抑制pd-l1蛋白质的表达水平。c2也具有降解能力但是能力差于c3。c4以及c5在a375细胞系中没有作用。同样在b16 f10细胞系中bms-37-c3与c1对pd-l1的抑制作用依旧强于其他化合物。bms-37-vhl系列化合物也对pd-l1的表达具有抑制作用。进一步使用mg132作用于细胞后,bms-37-c1以及bms-37-c3对pd-l1的抑制作用均被逆转,这也进一步说明这些化合物是通过泛素化途径降解蛋白质使pd-l1的表达量降低。
[0224]
实施例22流式细胞术检测相关蛋白的改变
[0225]
(1)将对数生长且融合度达到90%的细胞进行消化收集,加入新鲜培养基调整细胞浓度,然后使用细胞计数板计数。将细胞均匀接种在六孔板中,培养箱中贴壁并开始处于对数分裂期,更换含有药物的培养基后放入培养箱继续培养48h。
[0226]
(2)等待培养结束后,使用胰酶消化细胞并收集至1.5ml样品管中,做好标记。将样品4℃,400g,离心5min,弃去上清,加入pbs润洗一遍以清除胰酶中多余的edta和培养基中所含的血清等成分,同时将对照组一分为二,一组为阴性对照,一组为阳性对照,再次4℃,400g,离心5min,弃去上清液。
[0227]
(3)配置含5%bsa的pbs溶液,并在避光条件下将抗体与pbs以体积比为1:60(鼠源),1:40(人源)的比例混合,最终分别将igg和pd-l1抗体以30μl的体积加入样品管中。
[0228]
(4)4℃避光孵育40min,再次4℃,400g,离心5min,弃去上清液,并在样品管中加入200μl pbs溶液,上机检测。
[0229]
如图3所示,实验结果显示,在b16-f10和a375细胞中,bms-37-c1和bms-37-vhl均在0.1-1μm间降低细胞表面pd-l1的表达,但在3~10μm时出现hock效应;bms-37-c3在b16-f10和a375细胞中可浓度依赖性的降低细胞表面pd-l1的含量,效果更佳。
[0230]
实施例23化合物对t细胞杀伤肿瘤细胞的影响
[0231]
(1)将对数生长且融合度达到90%的细胞进行消化收集,加入新鲜培养基调整细
胞浓度,然后使用细胞计数板计数。将细胞均匀接种在九十六孔板中,待细胞贴壁后加入t细胞,最终使得肿瘤细胞与t细胞以1:0,1:1,1:2,1:3,1:4,1:6的比例继续共培养48h。
[0232]
(2)共培养结束后,,取出九十六孔板,缓慢吸去上清,在每孔内加入100μl pbs润洗两边,加入4%的多聚甲醛室温固定20min,再次用pbs润洗两边后用dapi染色液室温染色15~20min,pbs润洗后上机检测确定共培养最终比例。
[0233]
(3)将对数生长且融合度达到90%的细胞进行消化收集,加入新鲜培养基调整细胞浓度,然后使用细胞计数板计数。将细胞均匀接种在九十六孔板中,同时加入相应的含药物的培养基,放入培养箱中待细胞贴壁后,更换含有药物和上述比例t细胞的培养基后放入培养箱继续培养48h。
[0234]
(4)将共培养板子取出,缓慢收集上清,并在每孔内加入100μl pbs润洗两边,加入4%的多聚甲醛室温固定20min,再次用pbs润洗两边后用dapi染色液室温染色15~20min,pbs润洗后上机检测。
[0235]
如图4所示,其中,图4a为原代培养后受刺激t细胞的显微镜观察;b为t细胞与肿瘤细胞共培养比例的探讨;c为bms-37、bms-37-c1、bms-37-c3与t细胞共培养后对a375细胞的细胞毒性作用;d为bms-37、bms-37-c1、bms-37-c3和阿替唑单抗的t细胞杀伤活性统计图;实验结果显示:在探索肿瘤细胞与t细胞共培养效靶比时,肿瘤细胞与t细胞以1:3比例共培养时,t细胞可杀伤约50%的肿瘤细胞,因此后续实验中固定使用效靶比为1:3。当加入相应药物并与t细胞共培养时,pd-l1单protac bms-37-c1和bms-37-c3在0.3~1μm间可促进抗肿瘤免疫,且具有浓度依赖性,效果强于原料药bms-37和pd-l1单抗atezolizumab.
[0236]
实施例24微量热涌动实验
[0237]
用monolith nt
tm protein labeling kit red(cat#l001)对pd-l1蛋白(50nm)进行标记,样品在20mm tris-hcl(ph 7.5)和0.5(v/v)%tween-20中稀释。bms-37-c1/bms-37-c3/bms-37-5c-v2粉末溶解在5%的dmso中,浓度为200μm,倍比稀释。在室温下孵育10分钟后,将样品装入monolithtm标准处理过的毛细管中,在monolith nt.115仪器(nanotemper technologies,m
ü
nchen,germany)上孵育30分钟后,在25℃测量热泳。激光功率被设置为40%,使用30秒的开启时间。led的功率被设置为100%。通过使用ntanalysis软件(nanotemper technologies,m
ü
nchen,germany)对解离常数kd值进行拟合。
[0238]
如图5所示,实验结果表明,bms-37-c1/bms-37-c3/bms-37-5c-v2的平衡解离常数kd分别为28.70
±
4.94μm,1.61
±
0.37μm和9.10
±
2.11μm,说明合成的pd-l1 protacs能和pd-l1很好的结合。
[0239]
总的来说,本发明通过体外结合力,细胞内western blot以及流式细胞术检测pd-l1蛋白质降解水平,最终找到强有效的pd-l1降解剂bms-37-c3,该化合物在免疫治疗方面具有效果可以增强t细胞的杀伤能力,为肿瘤免疫治疗提供新的治疗方式。
[0240][0241]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。