一种
β-烟酰胺单核苷酸及其相关物质的检测方法和应用
技术领域
1.本发明属于核磁定量分析检测技术领域,具体涉及一种β-烟酰胺单核苷酸及其相关物质的检测方法和应用。
背景技术:
2.β-烟酰胺单核苷酸(nicotinamide mononucleotide,nmn),是烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,nad,辅酶i)的生物合成前体。nad是维生素pp(烟酸和烟酰胺)的衍生物,是一种在糖酵解、糖异生、三羧酸循环即呼吸链中,发挥重要作用的辅酶,广泛参与新陈代谢、衰老、细胞死亡、dna修复和基因表达等多种生命活动。
3.nmn相关物质包括了nmn的生物合成和分解代谢物质,在本发明指烟酸(nicotinic acid,na)、烟酰胺(nicotinamide,nam)、β-烟酰胺核糖(β-nicotinamide riboside,nr)、β-烟酰胺腺嘌呤二核苷酸(β-nicotinamide adenine dinucleotide,nad)或简称辅酶i、还原型烟酰胺腺嘌呤二核苷酸或简称还原型辅酶i(nadh)。
4.研究表明,nad在衰老和长寿种均有重要作用na、nam、nmn、nr均可以通过不同的代谢途径补充nad,nadh作为nad的还原态,以一定比例与nad共存,作为各类氧化还原反应的底物维持着机体氧化还原稳态。外源性补充na、nam、nmn、nr、nad和nadh,均可能提高体内nad的水平,从而产生一定的功效性。
5.为了进一步的研究nad补充剂的效果,控制相关食源性补充剂的食品安全和质量,na、nam、nmn、nr、nad和nadh在补充剂中含量的检测技术,越来越受到检验检测认证服务行业的关注。现阶段,食源性nad补充剂检测的关注点主要集中在nmn这一种物质上,并没有关注相关物质的检测,使得nad补充剂相关产品的市场监管和质量把控成为一大难题。
技术实现要素:
6.因此,本发明要解决的技术问题在于克服现有技术中的不能实现nmn及其相关物质同时检测的缺陷,从而提供一种β-烟酰胺单核苷酸及其相关物质的检测方法和应用。
7.为此,本发明提供如下技术方案:
8.本发明提供一种β-烟酰胺单核苷酸及其相关物质的检测方法,包括如下步骤:
9.s1,配制tsp的重水溶液,作为内标溶液;
10.s2,利用内标溶液分别配制不同浓度的na,nam,nmn,nr,nad,nadh标准工作溶液;
11.s3,利用内标溶液将待测样品溶解,离心,过滤,得待测样品溶液;
12.s4,设置核磁测试的操作参数,分别取一定浓度的na,nam,nmn,nr,nad,nadh标准工作溶液进行氢谱检测,确定各待测组分的特征峰;
13.s5,分别取不同浓度的na,nam,nmn,nr,nad,nadh标准工作溶液进行氢谱检测,经过数据处理,绘制标准曲线;
14.s6,取待测样品溶液进行氢谱检测,解析氢谱信号峰并进归属,根据上述标准曲线和确定的特征峰得到待测样品溶液中各待测组分na,nam,nmn,nr,nad,nadh的浓度,经计算
得待测样品中各待测组分na,nam,nmn,nr,nad,nadh的含量。
15.可选的,步骤s4中确定各待测组分的特征峰为na,9.12
±
0.05ppm;nam,7.58
±
0.05ppm;nmn,9.47
±
0.05ppm;nr,9.62
±
0.05ppm;nad,9.40
±
0.05ppm;nadh,6.95
±
0.05ppm。受检测溶液ph的影响,信号峰会略有漂移,一般在
±
0.05ppm内。
16.可选的,步骤s4中核磁测试的操作参数包括采集次数,弛豫时间,脉冲宽度,采样温度,脉冲角度,谱宽。
17.可选的,所述采集次数为大于等于8次;可选的,所述采集次数维8-32次;
18.和/或,所述弛豫时间为大于等于60s;可选的,所述弛豫时间为60-70s。
19.可选的,核磁测试的其它操作参数可根据仪器型号或选定的脉冲序列进行设置,典型非限定性的,选定核磁共振氢谱(proton)的脉冲序列,测定参数为:
20.采样温度设置为298k,谱宽(spectral width)16ppm,脉冲宽度(observe pulse)9.6μs,脉冲角度(observe degrees)90度。优选的,采集次数为8次,弛豫时间为60s。
21.可选的,步骤s2中各标准工作溶液的浓度为0.1-100mmol/l;
22.可选的,不同浓度的标准工作溶液的浓度为0.1mmol/l,0.2mmol/l,1mmol/l,2mmol/l,10mmol/l,20mmol/l,100mmol/l或50mmol/l。
23.可选的,步骤s1中,内标溶液中tsp的浓度为0.1mmol/l以上;优选的,内标溶液中tsp的浓度为0.1-10mmol/l。
24.可选的,步骤s3中采用0.22或0.45μm的滤膜过滤;优选的,采用0.22μm的滤膜过滤。
25.可选的,步骤s4中一定浓度的标准工作溶液的浓度为0.1-100mmol/l。
26.可选的,步骤s5中的标准曲线以标准工作溶液中待测组分的浓度为横坐标,以待测组分的特征峰峰面积与tsp峰面积的比值为纵坐标。
27.可选的,按照如下公式计算待测组分的含量:
[0028][0029]
x-试样中na、nam、nmn、nr、nad或nadh的含量,单位为毫克每百克(mg/100g);
[0030]
ω-由标准曲线得到的待测样品溶液中na、nam、nmn、nr、nad或nadh的浓度,单位为毫摩尔每升(mmol/l或mm);
[0031]
v-溶解试样的内标溶液总体积,单位为毫升(ml);
[0032]
m-试样的称样量,单位为毫克(mg);
[0033]
m-待测组分的相对分子质量,单位为克每摩尔(g/mol)。
[0034]
可选的,所述待测样品为保健食品,膳食营养补充剂,食品添加剂,保健食品原料或化妆品。
[0035]
典型非限定性的,待测样品溶液的制备过程中可根据待测组分的含量确定取样量,例如:
[0036]
保健食品、膳食营养补充剂:取20粒样品,如胶囊制剂则去掉胶囊壳,研细后,根据待测样品中na、nam、nmn、nr、nad、nadh的浓度,精密称取10mg(精确到0.01mg),于2ml ep离心管中,加入1ml tsp重水溶液,涡旋或超声使其充分溶解,离心后取上清液0.22μm过膜后,转移至5mm核磁管中待测。
[0037]
食品添加剂、保健食品原料(nmn原料):精密称量20mg样品,于2ml ep离心管中,加入1ml tsp重水溶液,涡旋或超声使其充分溶解,4500r/min离心,取上清液0.22μm过膜后,转移至5mm核磁管中待测。
[0038]
本发明技术方案,具有如下优点:
[0039]
本发明提供的β-烟酰胺单核苷酸及其相关物质的检测方法,包括以下步骤,s1,配制tsp的重水溶液,作为内标溶液;s2,利用内标溶液分别配制不同浓度的na,nam,nmn,nr,nad,nadh标准工作溶液;s3,利用内标溶液将待测样品溶解,离心,过滤,得待测样品溶液;s4,设置核磁测试的操作参数,分别取一定浓度的na,nam,nmn,nr,nad,nadh标准工作溶液进行氢谱检测,确定各待测组分的特征峰;s5,分别取不同浓度的na,nam,nmn,nr,nad,nadh标准工作溶液进行氢谱检测,经过数据处理,绘制标准曲线;s6,取待测样品溶液进行氢谱检测,解析氢谱信号峰并进归属,根据上述标准曲线和确定的特征峰得到待测样品溶液中各待测组分na,nam,nmn,nr,nad,nadh的浓度,经计算得待测样品中各待测组分na,nam,nmn,nr,nad,nadh的含量。本发明提供的检测方法,可“一锅法”进行检测,无需分离,可同时对6种相关联的化合物进行定量,检测用时短、效率高。另外,不同的化合物的核磁谱图,均是成组地出现,单一出现的峰,可判断为非待测化合物,因此,不会出现假阳性。为后续nad补充剂相关产品的生产研发、质量把控、安全检测、市场监管等提供有力的技术支持。
[0040]
本发明提供的β-烟酰胺单核苷酸及其相关物质的检测方法,通过对核磁测试的操作参数的限定,特别是弛豫时间和采集次数的限定,能够进一步降低信噪比,提升结果准确性。
附图说明
[0041]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0042]
图1是本发明实施例中nmn的特征峰与内标峰的峰面积比值与弛豫时间的变化趋势图;
[0043]
图2是本发明实施例中nmn的特征峰与内标峰的峰面积比值与采集次数的变化趋势图;
[0044]
图3是本发明实施例中nmn的核磁氢谱;
[0045]
图4是本发明实施例中nad的核磁氢谱;
[0046]
图5是本发明实施例中nadh的核磁氢谱;
[0047]
图6是本发明实施例中nr的核磁氢谱;
[0048]
图7是本发明实施例中na的核磁氢谱;
[0049]
图8是本发明实施例中nam的核磁氢谱;
[0050]
图9是本发明实施例中tsp的核磁氢谱;
[0051]
图10是本发明实施例中mla的核磁氢谱;
[0052]
图11是本发明实施例中sca的核磁氢谱;
[0053]
图12是本发明实施例中10mm的标准工作溶液核磁氢谱;
[0054]
图13是本发明实施例中获取的标准工作曲线;
[0055]
图14是本发明实施例中样品1的核磁氢谱;
[0056]
图15是本发明实施例中样品2的核磁氢谱;
[0057]
图16是本发明实施例中样品3的核磁氢谱;
[0058]
图17是本发明实施例中样品4的核磁氢谱;
[0059]
图18是本发明实施例中样品5的核磁氢谱;
[0060]
图19是本发明实施例中样品6的核磁氢谱。
具体实施方式
[0061]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0062]
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0063]
实施例
[0064]
本实施例提供一种β-烟酰胺单核苷酸及其相关物质的检测方法,包括以下步骤:
[0065]
(一)、检测原理为:使用2,2,3,3-氘代三甲基硅烷丙酸钠(tsp)的重水溶液作为内标溶液,将待测物质充分溶解,采用核磁共振波谱(nmr)对试样的重水溶液进行检测,以tsp为化学位移零点(δ=0.00ppm),及标准品、待测组分特征峰积分参照物,外标法定量。
[0066]
(二)、适用范围为:适用于利用核磁共振波谱法(nmr),定量测定食品添加剂、保健食品原料、保健食品、膳食营养补充剂中,烟酸(na)、烟酰胺(nam)、β-烟酰胺核糖(nr)、β-烟酰胺单核苷酸(nmn)、β-烟酰胺腺嘌呤二核苷酸(nad)和还原型烟酰胺腺嘌呤二核苷酸(nadh)的含量。
[0067]
(三)、试剂和材料
[0068]
(1)重水(d2o):氘代度99.9%,品牌:剑桥同位素实验室(cambridge isotope laboratories,inc.)。
[0069]
(2)2,2,3,3-氘代三甲基硅烷丙酸钠(tsp):98atom%d,品牌:剑桥同位素实验室(cambridge isotope laboratories,inc.)。
[0070]
(3)β-烟酰胺单核苷酸(nmn)标准品:β-烟酰胺单核苷酸,cas no.:1094-61-7,99%,品牌:麦克林(macklin),产品编号:b886299。
[0071]
(4)β-烟酰胺腺嘌呤二核苷酸(nad)标准品:β-烟酰胺腺嘌呤二核苷酸,cas no.:53-84-9,》98.0%,品牌:麦克林(macklin),产品编号:n1511。
[0072]
(5)β-烟酰胺核糖(nr)标准品:烟酰胺核糖氯化物(nrc),cas no.:23111-00-4,≥98%,品牌:麦克林(macklin),产品编号:n889451。
[0073]
(6)烟酰胺(nam)标准品:烟酰胺,cas no.:98-92-0,≥99.5%,品牌:麦克林(macklin),产品编号:n814696。
[0074]
(7)烟酸(na)标准品:烟酸,cas no.:59-67-6,99%,品牌:麦克林(macklin),产品
编号:n814565。
[0075]
(8)还原型辅酶i(nadh)标准品:还原型辅酶i二钠盐,cas no.:606-68-8,98%,品牌:麦克林(macklin),产品标准:n814671。
[0076]
(四)、仪器设备
[0077]
(1)核磁共振波谱仪(agilent technologies 600mhz dd2),配有onenmr5mm宽带二合一液体探头;7510自动进样器。
[0078]
(2)分析天平(mettler toledo xs104):感量0.01mg。
[0079]
(3)涡旋混合器(as one trio tm-1n)。
[0080]
(4)可调节移液器(eppendorf):20~100μl;100~1000μl。
[0081]
(5)台式高速微量小型离心机(大龙d2012plus):2ml
×
12,500r/min-15000r/min。
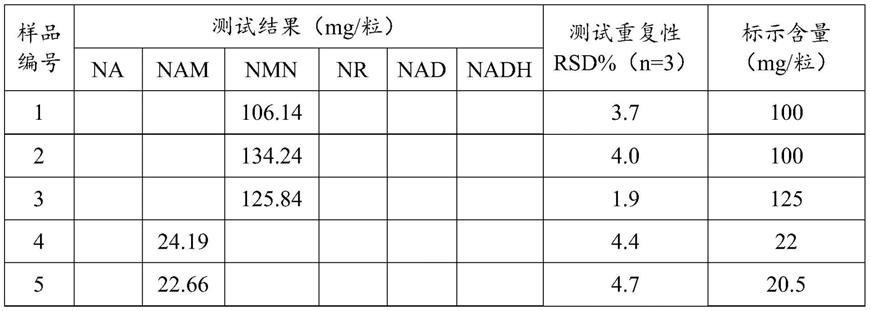
[0082]
(6)超声仪(昆山舒美kq5200de):200w;10l;40khz通用型。
[0083]
(7)核磁管(norell):5mm。
[0084]
(五)分析步骤
[0085]
1.tsp重水溶液(内标溶液)的配置
[0086]
使用棕色容量瓶,准确称取68.91mg tsp固体粉末,用重水溶解并定容至100ml,获得4mmol/l的tsp重水溶液,室温避光保存。
[0087]
2.标准中间溶液的配置
[0088]
上机测试前,分别称取12.21mg nam、29.07mg nrc、12.31mg na、33.42mg nmn、33.17mg nad、70.94mg nadh于2ml ep离心管中,加入1ml 4mmol/l tsp重水溶液,涡旋充分混溶,获得以下浓度的标准中间溶液:
[0089]
nam:100mmol/l;
[0090]
nr:100mmol/l;
[0091]
na:100mmol/l;
[0092]
nmn:100mmol/l;
[0093]
nad:50mmol/l;
[0094]
nadh:100mmol/l。
[0095]
3.标准工作溶液的配置
[0096]
采用逐级稀释的方法,在2ml ep离心管中配置以下浓度的na、nam、nmn、nr、nadh、nad标准工作溶液,配置的试液使用涡旋混合均匀,转移至5mm核磁管中待测。
[0097]
20mmol/l:200μl 100mmol/l标准中间溶液 800μl tsp重水溶液;
[0098]
10mmol/l:500μl 20mmol/l标准工作溶液 500μl tsp重水溶液;
[0099]
2mmol/l:200μl 10mmol/l标准工作溶液 800μl tsp重水溶液;
[0100]
1mmol/l:500μl 2mmol/l标准工作溶液 500μl tsp重水溶液;
[0101]
0.2mmol/l:200μl 1mmol/l标准工作溶液 800μl tsp重水溶液;
[0102]
0.1mmol/l:500μl 0.2mmol/l标准工作溶液 500μl tsp重水溶液;
[0103]
其中nad的20mmol/l:400μl 50mmol/l标准中间溶液 600μl tsp重水溶液。
[0104]
4.待测样品溶液的制备
[0105]
保健食品或膳食营养补充剂:取20粒样品,如胶囊制剂则去掉胶囊壳,研细后,根据试样中na、nam、nmn、nr、nad、nadh的浓度,精密称取10mg(精确到0.01mg),于2ml ep离心
管中,加入1ml tsp重水溶液,涡旋或超声使其充分溶解,本实施例中采用涡旋,离心后取上清液0.22μm过膜后,转移至5mm核磁管中待测。
[0106]
食品添加剂或保健食品原料(nmn原料):精密称量20mg样品,于2ml ep离心管中,加入1ml tsp重水溶液,涡旋或超声使其充分溶解,本实施例中采用超声,离心后取上清液0.22μm过膜后,转移至5mm核磁管中待测。
[0107]
5.仪器参数
[0108]
选定核磁共振氢谱(proton)的脉冲序列,测定参数为:
[0109]
采样温度设置为298k,谱宽(spectral width)16ppm,脉冲宽度(observe pulse)9.6μs,脉冲角度(observe degrees)90度。
[0110]
(1)弛豫时间参数的设定
[0111]
nmr检测需要足够长的弛豫时间(relaxation delay,d1),使受核磁脉冲激发的质子完全恢复到基态后,再进行下一次的核磁脉冲激发。本发明以10s为弛豫时间参数设定间隔,考察了30s至70s弛豫时间参数设定条件下,nmn的特征峰与内标峰的峰面积比值变化趋势。如图1所示,弛豫时间为60s以上时,峰面积比值与弛豫时间的一阶导数无限趋近于0。为了提高测试效率,选取60s为本发明弛豫时间的仪器参数设定值。
[0112]
(2)扫描采集次数参数的设定
[0113]
nmr的扫描采集次数(scans requested),与检测结果的信噪比密切相关,扫描采集次数越多,信噪比越低,检测用时越长。本发明考察了扫描采集次数设定分别为2、4、8、16、32条件下,nmn的特征峰与内标峰的峰面积比值变化趋势,如图2所示,扫描采集次数设定为8以上时,峰面积比值与弛豫时间的一阶导数无限趋近于0。为了提高测试效率,选取8为本发明扫描采集次数的仪器参数设定值。
[0114]
6.数据处理
[0115]
使用mestrenova软件处理采集的核磁共振波信号,采用核磁仪的软件vnmrj自动调节相位和基线校正,以内标物tsp信号峰为化学位移的零点,对tsp和各待测组分特征峰进行积分。
[0116]
7.定性分析
[0117]
使用合适浓度的标准中间溶液(浓度没有特别要求,只要保证在仪器检出限以上即可,例如,可以是0.1-100mmol/l,本实施例中为10mmol/l),分别对待测组分nmn、nad、nadh、nr、na、nam进行氢谱检测,采集其氢谱信号,对谱图分别进行分析,归纳氢信号峰的化学位移、裂分及比例,解析氢谱信号峰并进行归属。
[0118]
(1)nmn的核磁氢谱如图3所示,1h nmr(600mhz,d2o)δ9.47(s,1h),9.30(d,j=6.3hz,1h),8.99(dt,j=8.2,1.5hz,1h),8.31(dd,j=8.1,6.3hz,1h),6.23(d,j=5.4hz,1h),4.65(t,j=2.6hz,1h),4.57(t,j=5.2hz,1h),4.45(dd,j=5.1,2.6hz,1h),4.31(ddd,j=11.9,4.4,2.4hz,1h),4.16(ddd,j=11.9,5.1,2.1hz,1h)。
[0119]
(2)nad的核磁氢谱如图4所示,1h nmr(600mhz,d2o)δ9.40(s,1h),9.29(d,j=6.3hz,1h),8.95(dt,j=7.9,1.6hz,1h),8.59(s,1h),8.39(s,1h),8.31(dd,j=8.0,6.3hz,1h),6.19(d,j=5.5hz,1h),6.13(d,j=5.4hz,1h),4.74(t,j=5.3hz,1h),4.62(t,j=2.6hz,1h),4.58(t,j=5.3hz,1h),4.53(t,j=4.5hz,1h),4.49(dd,j=5.1,2.7hz,1h),4.44-4.38(m,2h),4.32-4.26(m,2h),4.26-4.20(m,1h)。
[0120]
(3)nadh的核磁氢谱如图5所示,1h nmr(600mhz,d2o)δ8.46(s,1h),8.16(s,1h),6.95(s,1h),6.11(d,j=5.4hz,1h),6.00(d,j=7.9hz,1h),4.79-4.75(m,2h),4.71(t,j=5.2hz,1h),4.53(t,j=4.6hz,1h),4.40(d,j=3.8hz,1h),4.32
–
4.26(m,1h),4.25(d,j=5.6hz,2h),4.23-4.18(m,1h),4.13-4.08(m,3h),2.78(d,j=18.0hz,1h),2.69(dd,j=18.1,3.2hz,1h)。
[0121]
(4)nr的核磁氢谱如图6所示,1h nmr(600mhz,d2o)δ9.62(s,1h),9.28(d,j=6.3hz,1h),8.99(d,j=8.0hz,1h),8.29(dd,j=8.0,6.3hz,1h),6.26(d,j=4.5hz,1h),4.53(t,j=4.8hz,1h),4.49(q,j=3.6hz,1h),4.37(t,j=4.7hz,1h),4.06(dd,j=12.9,2.9hz,1h),3.91(dd,j=12.9,3.6hz,1h)。
[0122]
(5)na的核磁氢谱如图7所示,1h nmr(600mhz,d2o)δ9.12(s,1h),8.90(d,j=8.1,1h),8.84(d,j=5.8hz,1h),7.95(dd,j=8.1,5.8hz,1h)。
[0123]
(6)nam的核磁氢谱如图8所示,1h nmr(600mhz,d2o)δ8.91(s,1h),8.70(d,j=5.0hz,1h),8.23(d,j=8.1hz,1h),7.58(dd,j=8.0,5.1hz,1h)。
[0124]
8.定量分析
[0125]
(1)内标化合物
[0126]
选择合适的内标化合物,采集内标化合物的核磁共振氢谱信号。内标化合物选择的原则为稳定,信号简单,与目标化合物的氢谱信号峰无重叠。本技术方案中,对比了对2,2,3,3-氘代三甲基硅烷丙酸钠(tsp)、马来酸(mla)和丁二酸(sca)的核磁氢谱图,选用tsp为化学位移定位内标。图9为tsp的核磁氢谱,1h nmr(600mhz,d2o)δ0.00(s,9h),从图中可以看出,其所出现的信号峰不会出现与待测化学组分信号峰重叠的情况,tsp既可以用做化学位移定位内标,又可用于计算含量用内标。
[0127]
图10为马来酸(mla)的核磁氢谱,1h nmr(600mhz,d2o)δ6.39(s,2h),从图中可以看出,nmn、nad、nadh在6.0-6.5ppm区间有信号峰,易混淆;图11为丁二酸(sca)的核磁氢谱,1h nmr(600mhz,d2o)δ2.69(s,4h),从图中可以看出,nadh有2.69的信号峰。因此未采用这两种化合物作为内标
[0128]
(2)定量特征峰
[0129]
选择特征峰应考虑峰型的完整性,峰与峰之间留有足够的化学位移差值,不可与其他目标物的信号峰重合,不受杂质的干扰,根据以上原则选出nmn、nad、nadh、nr、na、nam的特征峰的化学位移δ分别为9.47ppm、9.40ppm、6.95ppm、9.62ppm、9.12ppm、7.58ppm。
[0130]
图12是10mm的标准工作溶液核磁氢谱,图中各待测化合物定量特征峰:1.nr,9.62ppm;2.nmn,9.47ppm;3.nad,9.40ppm;4.na,9.12ppm;5.nam,7.58ppm;6.nadh,6.95ppm。
[0131]
(3)标准工作曲线
[0132]
在仪器参数设定下,分别对na、nam、nmn、nr、nadh、nad标准工作溶液进行氢谱检测,经过数据处理后,以标准工作溶液中化合物的毫摩尔每升浓度为横坐标,化合物的特征峰峰面积与tsp峰面积的比值为纵坐标,绘制各待测化合物的制标准工作曲线,各标准曲线的截距设置为0.0。
[0133]
(4)试样的测定
[0134]
在相同仪器条件下,将经过前处理的试样进样,进行nmr分析。以标准品的氢信号
峰的化学位移、裂分及比例定性,定量特征峰与tsp内标峰面积的比值外标法定量,根据线性回归方程计算出试样溶液中待测组分的浓度。
[0135]
如试样溶液中待测组分的浓度超出相应的线性范围,则适当增加tsp重水溶液的添加体积后,重新测定。
[0136]
(5)分析结果的表述
[0137]
试样中待测组分(na、nam、nmn、nr、nad或nadh)的含量,按以下公式计算:
[0138][0139]
x-试样中na、nam、nmn、nr、nad或nadh的含量,单位为毫克每百克(mg/100g);
[0140]
ω-由标准曲线得到的试样tsp重水溶液中,na、nam、nmn、nr、nad或nadh的浓度,单位为毫摩尔每升(mmol/l或mm);
[0141]
v-溶解试样的tsp重水溶液总体积,单位为毫升(ml);
[0142]
100-将结果单位由毫克每克换算为毫克每百克样品中含量的换算系数;
[0143]
m-试样的称样量,单位为毫克(mg);
[0144]
m-待测组分的相对分子质量,单位为克每摩尔(g/mol)。其中,nmn 334.22g/mol、nad 663.43g/mol、nr 255.25g/mol、nam 122.12g/mol、na 123.11g/mol、nadh 709.6g/mol。
[0145]
计算结果保留小数点后两位。
[0146]
(6)精密度
[0147]
在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的10%。
[0148]
(六)实样测试
[0149]
1.标准工作曲线
[0150]
按照本案“(五)分析步骤”,配置na、nam、nmn、nr、nad、nadh的标准工作溶液,设定仪器参数,进行nmr的测定,制作定量用标准工作曲线,如图13所示:
[0151]
nmn在0.1mm至100mm浓度范围内,呈现良好的线性关系,线性回归方程为y=0.0358x(r2=1.0000)。
[0152]
nad在0.1mm至50mm范围内,呈现良好的线性关系,线性回归方程为y=0.0330x(r2=1.0000)。
[0153]
nadh在0.2mm至100mm范围内,呈现良好的线性关系,线性回归方程为y=0.0339x(r2=0.9999)。
[0154]
nr在0.2mm至100mm范围内,呈现良好的线性关系,线性回归方程为y=0.0335x(r2=1.0000)。
[0155]
na在0.1mm至100mm范围内,呈现良好的线性关系,线性回归方程为y=0.0361x(r2=1.0000)。
[0156]
nam在0.2mm至100mm范围内,呈现良好的线性关系,线性回归方程为y=0.0361x(r2=0.9999)。
[0157]
2.测定结果
[0158]
(1)本发明测试了3款市售nmn胶囊/片剂(样品1~3),2款含nam的复合维生素保健
食品(样品4~5)。按照本发明“(五)分析步骤”对样品进行前处理,按与标准工作曲线测试相同的仪器条件,对进样试液进行nmr检测,核磁氢谱如图14-18所示。检测结果表示单位,换算为测试样品的营养标签数值单位,mg/粒。
[0159]
表1.实际样品测试结果
[0160][0161]
(2)本发明配置了同时含有六种物质的待测样品的混合样品(样品6),按照本发明“(五)分析步骤”对样品进行前处理,按与标准工作曲线测试相同的仪器条件,对进样试液进行nmr检测,其配制含量和检测含量见表2,核磁氢谱如图19所示。
[0162]
表2样品6的配制含量和测试结果
[0163]
样品6配制含量(mmol/l)测试含量(mmol/l)rsd(%)na14.2114.520.22nam12.4512.800.43nr6.096.023.67nmn7.487.322.82nad3.893.963.13nadh3.093.064.59
[0164]
本发明选择的内标化合物为2,2,3,3-氘代三甲基硅烷丙酸钠(tsp),其常用做水相体系化学位移参考试剂使用,本发明首次提出用该化合物作为内标化合物,其优点为通常情况下其信号峰位置不与其他化合物重叠。因此该内标物的选择可谓“一石二鸟”,不仅是化学位移参考试剂,还可以作为检测内标使用。
[0165]
本发明中弛豫时间的选择是保证结果检测准确的关键,这是因为,在核磁共振波谱检测过程中,每次脉冲激发,需待上次激发全部恢复到基态后,再进行下次的激发,否则会造成信号峰的比例偏差,故d1的时间设定为检测结果准确的关键,在本发明中需大于60s。
[0166]
本发明中扫描次数的增加,可降低信噪比,本发明的扫描次数需大于8。
[0167]
本发明中6种待测化合物均水溶性较好,因此选择重水为氘代试剂,其相比其他氘代有机溶剂,价格便宜,且绿色环保。
[0168]
本发明中目标化合物特征峰的选取,需尽可能选取峰形简单,裂分少的信号峰,且与其他目标物以及内标物,无重叠。因此选出nmn、nad、nadh、nr、na、nam的特征峰化学位移δ分别为9.47ppm,9.40ppm,6.95ppm,9.62ppm,9.12ppm,7.58ppm。
[0169]
本发明提供的检测方法可用于保健食品、膳食补充剂、保健食品原料中β-烟酰胺
单核苷酸及其代谢相关物质6种化合物(nmn、nad、nadh、nr、na、nam)的同时测定。
[0170]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。