1.本发明涉及基因变异分类技术领域,尤其涉及一种基因变异的自动分类方法、装置和电子设备。
背景技术:
2.由于基因在生命中具有非常重要的作用,当某些基因发生突变、缺失或失活时可能导致一些严重疾病的发生。
3.比如,brca1(breast cancer susceptibility gene 1,乳腺癌易感基因1)、brca2(breast cancer susceptibility gene 2,乳腺癌易感基因2)、palb2(partner and localizer of brca2,brca2定位和协作基因)属于抑癌基因,作为参与同源重组修复通路的核心基因,在卵巢癌、前列腺癌、乳腺癌、胰腺癌中都有占有重要的比例。携带上述基因的胚系致病或可能致病变异的受检者会增加罹患乳腺癌、卵巢癌、胰腺癌等相关肿瘤的风险。随着parp([poly(adp-ribose)polymerase, parp,聚腺苷二磷酸核糖聚合酶])抑制剂在卵巢癌、乳腺癌、胰腺癌及前列腺癌使用中的获批,存在brca1或brca2或palb2有害突变(致病及可能致病)的肿瘤患者可能从parp抑制剂的治疗中获益。因此快速、准确地解读出brca1、brca2、palb2变异的致病性不仅对于从事基因检测分析或临床遗传咨询的工作者来说具有重要的意义,对于受检者的遗传风险评估及肿瘤患者的用药指导也有重大价值。
4.目前,对基因变异的分类主要依据美国医学遗传学与基因组学学会(the american college of medical genetics and genomics,简称acmg)和美国分子病理学会(association for molecular pathology, 简称amp)的《序列变异解读标准和指南(2015版)》(standards and guidelines for the interpretation of sequence variants,简称acmg指南)。该指南将变异位点的致病、良性证据列为28条评判标准。首先将证据按类型分类(如人群数据、计算预测数据、功能数据等),并将证据的强度分为几类(支持,中等,强,非常强以及独立);然后使用“标准组合”的形式来评估致病性。不同组合将产生五个类别的分类:分别为“致病的(pathogenic)-5类”、“可能致病的(likely pathogenic)-4类”、“临床意义不明确的(uncertain significance)-3类”、“可能良性的(likely benign)-2类”和“良性的(benign)-1类”。其中5类和4类是靶向药物parp抑制剂及提示遗传风险的有效类别。
5.该指南虽然列出了28条证据的评判标准,但是,评判标准的描述过于模糊,在具体实践过程中,可能存在多种不确定的情况,从而无法根据评判标准得到确定的结果,导致分类结果会不够精确;此外,每条证据的评判标准都是针对所有的基因进行描述的,没有针对性,而由于每类基因的致病机制及相关表型有所差别,因此,对于不同类的基因,得到的分类结果的准确度会大不相同。
6.统计数据表明,依据acmg指南对基因变异进行分类时,临床意义未明的占比和结果冲突的情况占比都非常高,分类准确度极低。比如,在clinvar(由美国国家生物技术信息中心建立的与疾病相关的人类基因组变异的开放数据库)数据库中针对brca1、brca2、
palb2变异的临床意义未明占比分别为32.3%、39.6%、58.7%,而结果冲突的情况占比分别为: 4%、6.1%、5.9%(基于2022-02-25之前提交至clinvar的数据进行统计)。另外,采用人工的方法进行基因变异的分类,由于涉及到大量文献的查找,因此平均一个变异致病性的评价需要花费40-60分钟的时间,不仅费时,而且对解读技术人员的水平要求也非常高。
技术实现要素:
7.为了解决现有技术中存在的问题,本发明提供了如下技术方案。
8.本发明一方面提供了一种基因变异的自动分类方法,包括:获取基因变异的信息;建立所述基因变异的证据评判规则;构建所述基因变异的证据文献数据库;根据所述基因变异的信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据;根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。
9.优选地,所述基因变异的信息包括:基因名称、变异位点、变异类型以及变异在疾病数据库的收录情况。
10.优选地,所述基因变异的信息在测定序列后,经过生物信息学分析获得。
11.优选地,所述建立所述基因变异的证据评判规则包括:根据相关指南和标准中描述的针对所述基因变异的证据评判规则,对《acmg遗传变异分类标准与指南》中的证据评判规则进行内容补充,和/或,证据级别的升降修改,形成所述基因变异的证据评判规则。
12.优选地,所述构建所述基因变异的证据文献数据库包括:搜索所述基因变异并将通过人工评价确定为所述基因变异的证据的文献、及其对应的证据及强度进行保存,形成证据文献数据库。
13.优选地,所述根据所述基因变异的信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据,包括:根据所述基因变异的类型,确定待匹配的证据;在公共数据库和所述文献数据库中检索所述待匹配的证据,判断检索到的证据是否满足所述基因变异的证据评判规则,如果满足则匹配得到所述基因变异的证据并输出。
14.优选地,所述根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类包括:基于《acmg遗传变异分类标准与指南》规定的变异分类标准,根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。
15.本发明第二方面还提供了一种基因变异的自动分类装置,包括:变异信息获取模块,用于获取基因变异的信息;证据评判规则建立模块,用于建立所述基因变异的证据评判规则;文献数据库构建模块,用于构建所述基因变异的证据文献数据库;证据匹配模块,用于根据所述基因变异的信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据;
变异分类模块,用于根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。
16.第三方面提供了一种存储器,存储有多条指令,所述指令用于实现如上述的方法。
17.本发明还提供了一种电子设备,包括处理器和与所述处理器连接的存储器,所述存储器存储有多条指令,所述指令可被所述处理器加载并执行,以使所述处理器能够执行如上述的方法。
18.本发明的有益效果是:本发明根据基因的特异性,建立了针对基因变异的证据评判规则和证据文献数据库,再基于该证据评判规则和证据文献数据库,结合公共数据库,对基因变异进行证据的自动化匹配以及分类输出。由于重新建立的证据评判规则更具实操性以及变异的自动化判读分类的实现,使得临床工作者可以更快捷更方便地获取受检者的变异分类结果,相较人工分类,节省了操作时间;同时降低了临床意义未明的比例,从而避免由于检出临床意义未明变异给受检者带来的心理压力,也减少了临床工作者的解释成本。本发明的方法可以广泛应用于相关基因对应疾病的精准诊疗,比如基因brca1、brca2、palb2变异对应的相关肿瘤如卵巢癌、乳腺癌、胰腺癌、前列腺癌等的遗传风险评估,以及肿瘤患者parp抑制剂用药指导。
附图说明
19.图1为本发明所述基因变异的自动分类方法流程示意图;图2为本发明所述基因变异的自动分类装置功能结构示意图。
具体实施方式
20.为了更好的理解上述技术方案,下面将结合说明书附图以及具体的实施方式对上述技术方案做详细的说明。
21.本发明提供的方法可以在如下的终端环境中实施,该终端可以包括一个或多个如下部件:处理器、存储器和显示屏。其中,存储器中存储有至少一条指令,所述指令由处理器加载并执行以实现下述实施例所述的方法。
22.处理器可以包括一个或者多个处理核心。处理器利用各种接口和线路连接整个终端内的各个部分,通过运行或执行存储在存储器内的指令、程序、代码集或指令集,以及调用存储在存储器内的数据,执行终端的各种功能和处理数据。
23.存储器可以包括随机存储器(random access memory,ram),也可以包括只读存储器(read-only memory,rom)。存储器可用于存储指令、程序、代码、代码集或指令。
24.显示屏用于显示各个应用程序的用户界面。
25.除此之外,本领域技术人员可以理解,上述终端的结构并不构成对终端的限定,终端可以包括更多或更少的部件,或者组合某些部件,或者不同的部件布置。比如,终端中还包括射频电路、输入单元、传感器、音频电路、电源等部件,在此不再赘述。
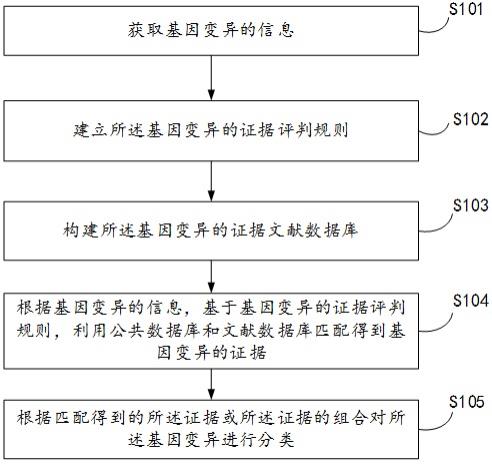
26.如图1所示,本发明实施例提供了一种基因变异的自动分类方法,包括:s101,获取基因变异的相关信息;s102,建立所述基因变异的证据评判规则;s103,构建所述基因变异的证据文献数据库;
s104,根据所述基因变异的相关信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据;s105,根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。
27.其中,在步骤s101中,所述基因变异的相关信息包括:基因名称、变异位点、变异类型以及变异在疾病数据库的收录情况。
28.在本发明实施例中,所述基因变异的相关信息,作为一个示例,比如对于抑癌基因brca1、brca2、palb2变异的相关信息可以按照如下方法获得:对申请进行包括brca1、brca2、palb2等基因变异检测及识别的受检者,首先获取符合要求的样本,比如:甲醛固定石蜡包埋(ffpe,formalin-fixed and paraffin-embedded)组织、新鲜组织、血浆/血液等。以ngs(next-generation sequencing,二代测序)检测基因为例,主要包括实验操作和生物信息学分析两部分。实验操作部分包括样本准备、文库制备、编码(barcoding)、目标区域富集、测序等;生物信息学分析部分包括定位(mapping)、比对、变异识别、变异注释、变异解读及报告等。ngs数据的生物信息分析可分为两个主要步骤:一是对测序数据进行质控分析及过滤。二是对通过质控的序列进行变异位点鉴定分析并注释,在ngs检测panel中,主要检测snv和indel两种突变类型,参数调整后可分析cnv,完成变异鉴定后,下一步进行变异的注释。生物信息学分析过程具体可以为:1、数据预处理1)数据分析:使用fastp(v0.19.4)软件去除实验及测序环节引入的接头序列及低质量碱基序列获得高质量测序数据,简称为clean data(清洁数据)。2)数据质控:数据质量需满足q30≥80%,否则判定为质控不通过,需要重新实验。
29.2、数据比对1)数据分析:使用bwa(0.7.17)序列比对软件将clean data比对至hg19(grch37)参考基因组上,生成记录比对结果的bam格式文件。然后使用samtools(v1.9)和genomeanalysistk(v4.1.0)工具对bam文件进行排序、去重及碱基质量校正,获得最终bam文件,并基于此文件进行后续分析。2)数据质控:参考基因组比对率需≥90%,对照样本目标区域平均测序深度需≥100
×
,肿瘤样本目标区域平均测序深度需≥500
×
,目标区域内深度大于平均深度
×
0.2的位点比例需≥90%,肿瘤和对照样本配对一致性需≥90%。以上任一条件不满足均判定质控不通过,需要重新实验。
30.3、突变分析1)点突变(snv)和插入缺失(indel)分析:使用genomeanalysistk(v4.1.0)的变异鉴定模块对样本中的点突变、插入缺失突变进行分析。2)拷贝数(cnv)分析:使用拷贝数变异分析模块(包含cnvkit(v 0.9.6)软件和自主开发过滤模块)对样本中的拷贝数变异进行分析。
31.4、突变注释使用基于annovar(v2018.04.16)(生物信息学软件工具)搭建的变异注释模块对点突变、插入缺失、及拷贝数变异进行注释,注释使用的数据库及工具包括clinvar、lovd (leiden open variation database,莱顿开放变异数据库)、hgmd (the human gene mutation database,人类基因突变数据库)、gnomad (the genome aggregation database,基因组聚合数据库)、revel(rare exome variant ensemble learner,罕见的外
显子组变异集合学习者)等。
32.执行步骤s102,建立所述基因变异的证据评判规则包括:根据相关指南和标准中描述的针对所述基因变异的证据评判规则,对《acmg遗传变异分类标准与指南》(acmg于2015年发布)中的证据评判规则进行内容补充,和/或,证据级别的升降修改,形成所述基因变异的证据评判规则。
33.其中,《acmg遗传变异分类标准与指南》将基因变异分类的证据列为了28条,并对每条证据的评判规则都进行了描述。比如,其中的一条证据ba1,其评判规则为:esp数据库(exome sequencing project,外显子组测序项目)、千人数据库(1000 genome project)、exac数据库(exome aggregation consortium,外显子组整合数据量库)中等位基因频率》5%。但是,该评判规则的描述过于概括,没有针对性,没有体现基因的特异性,是对所有的基因变异的证据评判规则,因此,在实际的证据评判中,就可能出现较大的评判误差。本发明实施例中,为了避免或减小评判误差,针对基因的特异性,对证据评判规则进行了重新建立。在实际建立过程中,主要是以《acmg遗传变异分类标准与指南》中的证据评判规则为基础,根据相关指南和标准中记载的有关基因变异的证据评判规则,对《acmg遗传变异分类标准与指南》中的证据评判规则进行内容补充,和/或,证据级别的升降修改。
34.由于相关指南和标准中记载的有关基因变异的证据评判规则更加具有针对性,内容更加细化,因此,通过内容补充、对证据进行升级或降级的修改可以使得证据的评判规则更加详细,更加具有实操性,能够更好的指导证据匹配,使得匹配到的证据更加正确。其中,相关指南和标准包括:欧洲分子基因诊断质量联盟(emqn)的遗传性乳腺癌/卵巢癌分子遗传分析最佳实践指南(2008版)(来源:emqn官方网站)、enigma-brca1/2基因变异分类标准【enigma(evidence-based network for the interpretation of germline mutant alleles,胚系突变等位基因解读实证联盟) 专家组制定的brca1/2基因变异分类标准(version 2.5.1 29 june 2017)】(来源: enigma官方网站)、《brca数据解读中国专家共识(2021版)》(来源:中华病理学杂志官方网站)、基于acgs(association for clinical genomic science, 临床基因组学会)变异分类最佳实践指南的canvig-uk(the cancer variant interpretation group uk,英国癌症变异解读组)癌症易感基因共识(v1.2 03/03/20)【canvig-uk consensus specification for cancer susceptibility genes of acgs best practice guidelines for variant classification (v1.2 03/03/20)】(来源:canvig-uk官方网站)。
35.作为一个实施例,比如,证据bs1作为支持将变异分类为可能良性或良性的强证据,在《acmg遗传变异分类标准与指南》评判规则为:一般情况下,某一等位基因在对照人群的频率大于疾病预期人群,可认为是罕见孟德尔疾病良性变异的强证据(bs1);但是该指南中并没有明确针对某些基因的疾病预期人群频率设置的定义方法,比如,针对brca1、brca2及palb2基因的疾病预期人群频率设置的定义方法。
36.根据本发明提供的方法,建立证据bs1的评判规则的过程可以为:按照canvig-uk consensus specification for cancer susceptibility genes of acgs best practice guidelines for variant classification (v1.2 03/03/20)的推荐,参考whiffinetal 2017(pmid:28518168),通过在线应用allele frequency app(等位频率计算应用,a shiny app for allele frequency calculations copyright
ꢀ©ꢀ
2016 james ware)计算相关基
因的最大预期变异频率(maximum credible variant frequency)。以brca1为例,采用如下保守性参数对bs1进行计算:常染色体显性遗传;疾病(乳腺癌)流行率设置为1:10;等位异质性设置为10%(变异在所有brca1致病变异等位中占比10%);遗传异质性设置为1%(1%的乳腺癌由brca1胚系致病或可能致病突变导致);brca1胚系致病或可能致病突变导致乳腺癌的外显率为72%(pmid:28632866),计算出最大预期变异频率为6.94e-05,对该参数进行保守性处理,得到最大预期变异频率为0.01%(0.0001)。根据上述方法计算brca2及palb2的最大预期变异频率,与brca1基本一致。根据本发明提供的方法建立的评判规则为:在gnomad v2.1.1 (non-cancer)数据库中,当质控合格的某一变异对应的popmax filtering af(最大人群等位基因过滤频率)(95% confidence)大于0.01%(0.0001)时,则满足bs1,(需排除满足bs1的已知致病变异)。
37.本发明建立的证据bs1的评判规则,与《acmg遗传变异分类标准与指南》中描述的bs1的评判规则相比,细化了针对基因特异性,比如brca1、brca2、palb2的基因特异性的bs1的标准,使得按照该标准计算最大预期频率更具有操作性,与此同时,大量的实践数据表明,与intervar(intervar是一种生物信息学软件工具,用于通过acmg/amp 2015指南对遗传变异进行临床解释)半自动化解读bs1设置的阈值1%相比,本发明的bs1规则更为严谨,能识别出更多的支持良性分类的变异。
38.作为一个实施例,比如,对于证据pm1,在《acmg遗传变异分类标准与指南》中,pm1的级别为中等强度致病证据。根据本发明提供的方法,对pm1的级别进行调整的方法为:pm1作为支持将变异分类为可能致病或致病的中等强度证据,在《acmg遗传变异分类标准与指南》评判规则为:位于热点突变区域,和/或位于已知无良性变异的关键功能域(如酶的活性位点)。比如针对brca1、brca2、palb2等抑癌基因,与癌基因不同,无热点突变位置,但在其蛋白序列中存在保守的结构域或模体,且在这些区域内存在重要临床意义的氨基酸残基,而其他非保守区域暂未发现致病或可能致病的氨基酸替代或框内插入/缺失变异。据此,本发明制定brca1、brca2、palb2基因特异性的pm1规则时,区分了重要结构域(pm1_sup)及重要临床意义的氨基酸残基(pm1_mod)。最终,将pm1的级别进行调整为:以brca1基因为例,brca1的pm1_sup为:发生在ring domains(1-101aa)或brct(1642-1863aa)的错义变异;pm1_mod定义为涉及到ring:18,22,37,39,41,44,47,61,64,71;brct:1685,1688,1697,1699,1706,1708,1715,1736,1738,1739,1748,1764,1766,1770,1775,1786,1837,1838,1839,1853的残基的错义变异。
39.本发明中规定的pm1级别,与《acmg遗传变异分类标准与指南》中描述的pm1级别相比,不仅对brca1、brca2、palb2的基因特异性的pm1的标准进行了细化,还增加了支持强度的级别设置,使得该标准具有可操作性,同时区分重要功能域及重要的氨基酸残基的致病强度,实现对发生在重要结构域的错义变异进行致病性强度分层,使得变异的分类更为精准。
40.执行步骤s103,构建所述基因变异的证据文献数据库包括:搜索所述基因变异相关的文献,将通过人工评价确定为所述基因变异证据的文献、及其对应的证据及强度进行保存,形成证据文献数据库。
41.在28条证据中,有些证据需要通过查找文献并根据文献进行评判。在现有技术中,对于一条证据的评判,可能需要从不同的数据库中查找大量的文献,从而需要花费大量的
时间和精力。技术人员在根据文献对证据进行评判时,需要具有较高的技术水平并对每条证据的评判规则非常熟悉,因此,对技术人员具有较高的能力要求,而由于人员的不确定性,会带来证据结果的不确定性。本发明为了解决这个问题,构建了基因变异的证据文献数据库。从而根据该文献数据库,实现自动化匹配证据。
42.作为一个实施例,比如,对于证据ps3/bs3,可以按照如下方法构建文献数据库:首先搜索基因相关的功能实验的文献,然后,依据本发明提供的关于证据ps3/bs3的评判规则进行人工评价,最终确定数万个变异的功能实验结果对应的证据及证据强度,并将其保存在文献数据库中。根据该方法建立的文献数据库包括如下内容:变异所在的染色体(chr),变异发生的区间(start)(end),变异所在位置参考序列(ref)及改变后的序列(alt),变异所在的基因(gene),参考转录本(transcript),核苷酸变化(hgvsc),蛋白变化(hgvsp),ps3及bs3的定级(ps3_bs3_final_one),定级所参考的来源(source)。在实际应用过程中,可以直接根据文献数据库匹配得到基因变异发生的物理位置,及对应的ps3或bs3相关级别的功能实验证据。可见,对于需要查找文献的证据,通过建立文献数据库,可以直接匹配得到证据,而无需在使用过程中再行查找文献,也无需人工评价,从而不仅节约了大量的时间和精力,而且降低了对人员的技术要求,可以提高准确度和工作效率,降低成本。
43.执行步骤s104,根据所述基因变异的相关信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据,包括:根据所述基因变异的类型,确定待匹配的证据;在公共数据库和所述文献数据库中检索所述待匹配的证据,判断检索到的证据是否满足所述基因变异的证据评判规则,如果满足则匹配得到所述基因变异的证据并输出。
44.其中,由于不同的变异类型,待匹配的证据不同,因此,本发明中,首先确定待匹配的证据,从而将需要评判的证据数量减少,范围缩小,极大地减小工作量,缩短评判和匹配的时间。
45.作为一个实施例,比如,对于brca2基因变异nm_000059.3(brca2):c.4677del(p.phe1559leufster9),属于插入/缺失变异,预测的蛋白后果是移码,因此该变异类型属于移码变异。根据该变异类型确定其需要评判的证据至少包括:pvs1、pm2、ps4、pp5。在公共数据库和所述文献数据库中检索这些证据,根据所述基因变异的证据评判规则,对检索到的证据进行评判,得到基因变异的证据并输出为:pp5_expert pm2_mod ps4_sup pvs1_vst。
46.执行步骤s105,根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类包括:基于《acmg遗传变异分类标准与指南》规定的变异分类标准,根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。比如,作为一个实施例,如下的证据组合:pp5_expert pm2_mod ps4_sup pvs1_vst的基因变异,可划分为5类。
47.实施例一本发明实施例以brca2基因变异nm_000059.3(brca2):c.4677del(p.phe1559leufster9)为例,说明采用本发明的方法对其进行解读并划分类别的过程。
48.该变异属于插入/缺失变异,预测的蛋白后果是移码,因此该变异类型属于移码变异。根据该变异类型确定其需要评判的证据至少包括:pvs1、pm2、ps4、pp5。在公共数据库和所述文献数据库中依次检索证据pvs1、pm2、ps4、pp5。经过检索发现:
对于pvs1,根据本发明建立的证据评判规则,满足“当一个疾病的致病机制为功能丧失(lof)时,无功能变异(无义突变、移码突变、经典
±
1或2的剪接突变、起始密码子变异、单个或多个外显子缺失),注:1.该基因的lof是否是导致该疾病的明确致病机制(如gfap、myh7)。2. 3'端末端的功能缺失变异需谨慎解读。3.需注意外显子选择性缺失是否影响到蛋白质的完整性。4.考虑一个基因存在多种转录本的情况。”。同时结合文献:abou tayoun an, pesaran t, distefano mt, et al. recommendations for interpreting the loss of function pvs1 acmg/amp variant criterion. hum mutat. 2018;39(11):1517-1524. doi:10.1002/humu.23626(pmid:30192042)中的pvs1决策树,该变异位于倒数第二个外显子最后50 bp之外,预测会发生nmd(nonsense-mediated mrna decay,无义介导的mrna降解)。因此判断该变异满足pvs1_vst的证据。
49.对于pm2,检索到该变异在gnomad中人群频率缺失,满足本发明的评判规则:“pm2_mod:在gnomad v2.1.1cancer-free female controls (of any/all ethnicities)(gnomad v2.1.1无癌女性对照人群下的所有亚群)中未观测到该变异的发生,可使用pm2_mod。”对于ps4,检索到该变异在clinvar中被6家单位提交,提示满足ps4_sup。即ps4:(case counting,病例计数):该变异在满足在对照人群中极低或缺失的情况下(满足pm2或pm2_sup),在≥5个不相关的hboc家系中观测到该变异(ps4_sup);在≥10个不相关的hboc家系中观测到该变异(ps4_mod)。变异可来源公共疾病数据库、文献数据库等。公共疾病数据库:变异在hgmd, lovd, clinvar共出现的次数。
50.对于pp5,检索到该变异在clinvar中被专家组认证为致病变异,按照本发明建立的评判规则“获批clingen(clinical genome resource,临床基因组资源中心)认证的专家组(clinvar上获3星)、enigma、lovd,输出对应分类,证据标签同时输出pp5 _expert”。
51.因此,采用本发明提供的方法得到该变异的证据组合为:pp5_expert pm2_mod ps4_sup pvs1_vst,根据该证据组合可将该变异分类为5类。
52.对比例一采用现有技术中的解读分类方法对brca2基因变异nm_000059.3(brca2):c.4677del(p.phe1559leufster9)进行解读并划分类别的过程为:第一步,该变异为移码突变,可评估该变异是否会发生无义介导的mrna降解(nmd),从而评估pvs1的适用性。即将变异输入到mutalyzer(检测基因变异命名是否符合hgvs规则的在线工具,详见lefter m et al. (2021). mutalyzer 2: next generation hgvs nomenclature checker. bioinformatics, btab051),提示brca2基因有27个外显子,该变异发生在11号外显子,即不位于倒数第二个外显子最后50bp内,预测该变异会导致nmd,根据pvs1决策树(pmid:30192042),该情况可以匹配pvs1的证据。第二步,查看该变异在普通人群中的人群频率,以评估pm2的适用性,登录gnomad数据库(目前收录范围较广的基因组变异数据库之一,由麻省剑桥博德研究所提供),输入该变异的物理位置,发现该变异在gnomad数据库中未被观测到,提示人群频率缺失,可使用pm2_mod;第三步,搜索疾病数据库clinvar,该变异在clinvar中被专家组认证为三星致病变异,可匹配pp5的证据;第四步,查阅与该变异相关的文献报道,以评估ps4的适用性,该变异在两个不相关的乳腺癌-卵巢癌高风险个体中检出(pmid:20104584,30322717),按照《acmg遗传变异分类标准与指南》的规则,不满足ps4
(casecontrolstudy,病例对照研究,or>5)的标签。至此,通过上述过程,可获得该变异满足pvs1、pm2、pp5,翻阅acmg的组合规则,该变异的分类符合:1pvs合并≥1pm合并≥1pp,可分类为5类。
53.尽管该实施例采用现有技术与本发明的分类方法,结果一致,但本发明在时间耗费及标签使用的准确性上更有优势。时间耗费方面:本发明的分类方法只涉及输入变异及获取结果的过程,预估在1分钟内就可获得该变异的分类结果及证据标签;对比之下,现有的技术方法需要对每一条证据进行在线查阅,并人工进行相应证据的匹配,全程预估20-30分钟。标签使用方面:本发明对ps4的标签进行了升级及降级,并结合clinvar数据库进行该标签的运用,使得该变异可自动匹配ps4_sup的标签,而现有技术受限于查阅到的文献数量及ps4在《acmg遗传变异分类标准与指南》中的不具操作性,进而放弃使用。(ps4:变异出现在患病群体中的频率显著高于对照群体。注:1、可选择使用相对风险值或者or值来评估,建议位点or大于5.0且置信区间不包括1.0的可列入此项。2、极罕见的变异在病例对照研究可能无统计学意义,原先在多个具有相同表型的患者中观察到该变异且在对照中未观察到可作为中等水平证据。)实施例二本发明实施例以brca1基因变异brca1:nm_007294.3(brca1):c.80 4a》t为例,说明采用本发明的方法对其进行解读并划分类别的过程。该变异属于内含子变异,不能排除对mrna的剪切产生影响。根据该变异类型确定其需要评判的证据至少包括:pp3、pm2、ps3。在公共数据库和所述文献数据库中依次检索证据pp3、pm2、ps3。经过检索发现:对于pp3,根据本发明建立的证据评判规则,满足“当内含子变异满足spliceai,(anyδscore≥0.2)”,则可判断该变异符合pp3的证据。对于pm2,检索到该变异在gnomad中人群频率缺失,满足本发明的评判规则:“pm2_mod:在gnomad v2.1.1cancer-free female controls (of any/all ethnicities)中未观测到该变异的发生,可使用pm2_mod。”同时该变异被本发明中文献数据库收录,提示满足ps3_str,该标签的使用的背后逻辑如下:该变异在文献pmid:20104584中被报道,该研究利用crispr-cas9基因编辑技术获得brca1变异体,并将变异体整合进人类单倍体细胞系,通过检测变异频率反应细胞的存活率以对brca1关键功能域单核苷酸变异来指导变异的分类。该文献用到的方法中纳入了较多数量的阳性对照及阴性对照,且有技术重复,确定为验证良好的功能实验,对应的致病性odd path(odds of pathogenicity,致病性的几率)为21.7,良性的odd path为0.007,按照clingen推荐的定级标准(pmid:31892348),定级为ps3_str,bs3_str。该变异在数据库中提示为loss of function(功能丧失),对应ps3_str。
54.因此,采用本发明提供的方法得到该变异的证据组合为:pp3 pm2_mod ps3_str,根据该证据组合可将该变异分类为4类。
55.为验证本发明的自动化结果的正确性,对具有变异解读经验及背景的人员进行本发明规则的培训,采用本发明的规则对该变异进行人工分类,过程为:第一步,通过软件预测评估该变异是否影响剪切,登录spliceai(一种可以预测变异剪切影响的在线工具,由麻省剑桥博德研究所提供),输入变异,查看δscore的分数,显示最大值为0.63,提示影响剪切,根据本发明的规则定义,符合pp3。第二步,查看该变异在普通人群中的人群频率,以评估pm2的适用性,登录gnomad数据库,输入该变异的物理位置,发现该变异在gnomad数据库
中未被观测到,提示人群频率缺失,可使用pm2_mod;第三步,查阅与该变异相关的文献报道,以评估ps4或ps3的适用性;在pubmed及google中仅搜索到一篇与该变异相关的文献(pmid:20104584),按照pmid:31892348,可定级为ps3_str。至此,通过上述过程,可获得该变异满足pp3 pm2_mod ps3_str,翻阅acmg的组合规则,该变异的分类符合:1ps合并≥1-2pm,可分类为4类,该分类结果与上述自动化结果一致。
56.对比例二clinvar数据库作为重要的疾病数据库,收录了不同检测机构对变异分类的信息,也反应了当下不同机构对变异分类的理解水平。而brca1:nm_007294.3(brca1):c.80 4a》t变异在clinvar中被两家提供分类标准的单位提交,其中一家(ambry genetics-检测机构a)分类为致病,一家分类为意义未明(invitae-检测机构b),总体评估为致病性冲突;且该变异在breast cancer information core (bic,乳腺癌信息中心)中提示意义未明。bic数据库中变异的致病性评价未经过acmg指南评价,检测机构a和b都是基于acmg指南制定的分类规则,尽管如此,却仍然针对同一变异给予了不同的分类结果。且不同于检测机构a或b的结果。
57.在该实施例二中,采用本发明获取的结果更为直接、准确和高效。如直接参考clinvar中提交者针对该变异的分类,即临床意义未明或致病,都不够准确。通过对比检测机构a和检测公司b提供的证据描述,发现检测公司b并未采用a公司所述的功能实验证据(pmid:20104584),分析原因可能检测公司b提交该证据时,上述文献并未发表,导致分类结果存在差异。尽管a公司采用了功能实验的证据,但并未对该变异匹配具体证据标签,在无其他临床证据支持的情况下,将该变异分类为致病并不合理。原因在于该变异属于非经典剪切切点内含子变异,不支持pvs1的使用,而该变异支持致病性证据组合只能是如下情况:(i)≥2个强(ps1-ps4)或(ii)1个强(ps1)和(a)≥3个中等(pm1-pm6)或(b)2个中等(pm1-pm6)和≥2个支持(pp1-pp5)或(c)1个中等(pm1-pm6)和≥4个支持(pp1-pp5)。
58.基于人工搜索到的a公司提供的证据描述,并不能满足该变异支持致病。而本发明中针对该变异的分类不仅考虑了功能实验证据的使用,还参考相关标准进行了ps3的定级,从而确保ps3_str的正确使用。除pm2_mod及pp3之外,通过具有解读资质的人员对该变异进行相关文献的搜索,并未发现其余支持该变异致病的证据或良性的证据,且在本发明规则指导下,人工分类及自动化分类结果一致,充分说明了本发明在准确性及时间成本方面的优势。
59.实施例三本发明实施例以palb2基因变异nm_024675.4(palb2):c.1054g》c(p.glu352gln)为例,说明采用本发明的方法对其进行解读并划分类别的过程。
60.该变异属于错义变异,暂不能确定变异的致病性影响,根据该变异类型确定其需要评判的证据至少包括:bs1、bp4。
61.在公共数据库和所述文献数据库中依次检索证据bs1、bp4。经过检索发现:对于bs1,根据本发明建立的证据评判规则,满足“在gnomad v2.1.1(non-cancer)
数据库中,当质控合格的某一变异对应的popmax filtering af(95% confidence)大于0.01%(0.0001)时,则满足bs1。”对于bp4,根据本发明建立的证据评判规则,需同时满足蛋白功能及保守性无影响和无剪切影响,输出bp4。即revel(《0.4)且spliceai(allδscores《0.2)。
62.因此,采用本发明提供的方法得到该变异的证据组合为:bs1 bp4,根据该证据组合可将该变异分类为2类。
63.为验证本发明的自动化结果的正确性,对具有变异解读经验及背景的人员进行本发明规则的培训,采用本发明的规则对该变异进行人工分类,过程为:第一步,通过revel软件获取该变异分数为0.152,提示不影响蛋白功能;同时通过软件预测评估该变异的是否影响剪切,采用spliceai在线预测工具,输入变异,查看δscore的分数,显示最大值为0,提示不影响剪切,因此,同时根据本发明的规则定义,符合bp4。第二步,查看该变异在普通人群中的人群频率,以评估bs1或ba1的适用性,登录gnomad数据库,输入该变异的物理位置,发现该变异gnomad(non-cancer)数据库中popmax filtering af为0.0004777,大于0.01%,提示符合bs1;第三步,查阅与该变异相关的文献报道,暂未发现相关文献可匹配致病性证据标签或良性证据标签。至此,通过上述过程,可获得该变异满足bs1 bp4,翻阅acmg的组合规则,该变异的分类符合:1bs合并1bp,可分类为2类。
64.在本发明建立的规则指导下,人工分类与机器分类结果一致,且极大比例降低了时间耗费。
65.对比例三clinvar数据库作为重要的疾病数据库,收录了不同检测机构对变异分类的信息,也反应了当下不同机构对变异分类的理解水平。而palb2基因变异nm_024675.4(palb2):c.1054g》c(p.glu352gln)在clinvar中被5家单位提交,其中2家分类为可能良性,3家分类为意义未明,总体评估为致病性冲突。
66.在该实施例三中,采用本发明的方法获取的结果更为直接、准确和高效。该变异所在的氨基酸位置并不保守,提示不大可能影响蛋白的功能,且该变异并不位于一致性剪切区域,经spliceai预测不影响剪切,提示bp4使用的合理性,且该变异在gnomad(non-cancer)数据库中的东亚人群中,存在15个等位计数(等位总数=18394),对应人群频率为0.0008155,超过疾病的预期发生率,提示bs1使用的合理性。且在本发明规则指导下,人工分类及自动化分类结果一致,充分说明了本发明在准确性及时间成本方面的优势。
67.随着二代测序技术在临床应用中的进展,越来越多的受检者被推荐进行基因检测,相应的针对brca基因、palb2基因的变异也逐渐被检出,采用现有技术进行变异的解读非常耗时,尤其是当变异的数量非常巨大的情况下,耗时的缺陷越发明显,不能满足应用需求。采用本发明提供的方法,对于大量的需要解读的brca1/2及palb2变异,可批量处理。在执行过程中,只需要在计算机程序中输入变异,即可根据本发明提供的方法逻辑,输出变异的类别,而得到每个变异的分类结果只需数秒钟;而且,基于enigma专家组认证的7371个位点,采用本发明提供的方法,在良性及可能良性变异的分类上,一致性达到99.2%;在致病及可能致病变异的分类上,一致性达到99.9%,因此,采用本发明的方法进行变异解读分类具有非常高的准确度。
68.如图2所示,本发明的另一方面还包括和前述方法流程完全对应一致的功能模块架构,即本发明实施例还提供了一种基因变异的自动分类装置,包括:变异信息获取模块301,用于获取基因变异的相关信息;其中,所述基因变异的相关信息包括:基因名称、变异位点、变异类型以及变异在疾病数据库的收录情况。
69.所述基因变异的相关信息在测定序列后,经过生物信息学分析获得。
70.证据评判规则建立模块302,用于建立所述基因变异的证据评判规则,包括:根据相关指南和标准中描述的针对所述基因变异的证据评判规则,对《acmg遗传变异分类标准与指南》中的证据评判规则进行内容补充,和/或,证据级别的升降修改,形成所述基因变异的证据评判规则。
71.文献数据库构建模块303,用于构建所述基因变异的证据文献数据库,包括:搜索所述基因变异相关的文献,将通过人工评价确定为所述基因变异的证据的文献、及其对应的证据及强度进行保存,形成证据文献数据库。
72.证据匹配模块304,用于根据所述基因变异的相关信息,基于所述基因变异的证据评判规则,利用公共数据库和所述文献数据库匹配得到所述基因变异的证据,包括:根据所述基因变异的类型,确定待匹配的证据;在公共数据库和所述文献数据库中检索所述待匹配的证据,判断检索到的证据是否满足所述基因变异的证据评判规则,如果满足则匹配得到所述基因变异的证据并输出。
73.变异分类模块305,用于根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类,包括:基于《acmg遗传变异分类标准与指南》规定的变异分类标准,根据匹配得到的所述证据或所述证据的组合对所述基因变异进行分类。
74.该装置可通过上述实施例一提供的基因变异的自动分类方法实现,具体的实现方法可参见实施例一中的描述,在此不再赘述。
75.本发明还提供了一种存储器,存储有多条指令,所述指令用于实现如实施例一所述的方法。
76.本发明还提供了一种电子设备,包括处理器和与所述处理器连接的存储器,所述存储器存储有多条指令,所述指令可被所述处理器加载并执行,以使所述处理器能够执行如实施例一所述的方法。
77.尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
