1.本发明涉及兽用药物技术领域,尤其涉及一种复方羟氯扎胺混悬液及其制备方法。
背景技术:
2.羟氯扎胺又名氯羟柳胺、羟氯柳苯胺等,化学名2,3,5,-三氯-n-(3,5-二氯-2-羟基苯基)-6-羟基苯甲酰胺,是一种水杨酸苯胺类驱虫药,是一种氧化磷酸化解偶联剂,主要用于治疗牛羊等动物各种肝片吸虫(fasciola)、蛔虫和绦虫感染。它可以降低虫体内三磷酸腺甙(atp)的合成量,从而导致虫体因能量代谢紊乱而终于死亡。其作用机理主要是抑制寄生虫体内的氧化磷酸化过程,减少atp的产生,并降低糖原含量,使琥珀酸积累,从而影响虫体的能量代谢过程,使虫体死亡。
3.羟氯扎胺属于难溶性物质,目前,羟氯扎胺混悬液在制备时辅料种类较多,成本高,使用时容易产生气泡,且再分散性能差。因此,发展成本低,所用原料种类少,且再分散性优异的羟氯扎胺混悬液成为本领域亟需。
技术实现要素:
4.有鉴于此,本发明提供了一种复方羟氯扎胺混悬液及其制备方法,解决了现有的羟氯扎胺混悬液在制备时辅料种类较多,成本高,使用时容易产生气泡,且再分散性能差的问题。
5.为了达到上述目的,本发明采用如下技术方案:
6.本发明提供一种复方羟氯扎胺混悬液,每100l复方羟氯扎胺混悬液中,包括以下组分:
[0007][0008][0009]
本发明还提供所述复方羟氯扎胺混悬液的制备方法,包括如下步骤:
[0010]
(1)将羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄进行细化;
[0011]
(2)将细化后的黄原胶与40~60vt%的水混合后进行溶胀,得黄原胶混合液;加入细化后的亚硫酸氢钠、柠檬黄、盐酸左旋咪唑原料药,得第一溶液;
[0012]
(3)将羟氯扎胺原料药和丙二醇混合,得第二溶液;
[0013]
(4)将第一溶液、第二溶液、苯甲酸和剩余水混合,得复方羟氯扎胺混悬液。
[0014]
作为优选,所述步骤(1)中,羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄的粒径目数独立的为≥80目。
[0015]
作为优选,所述步骤(2)中,混合时间为20~24h;搅拌溶胀的速度为200~300r/min,时间为20~40min。
[0016]
作为优选,所述步骤(4)中,第一溶液、第二溶液、苯甲酸和剩余水混合方式为:将第一溶液和第二溶液混合,后加入苯甲酸和剩余水。
[0017]
作为优选,所述第一溶液和第二溶液混合方式为:将第二溶液在搅拌下加入至第一溶液中;所述搅拌速度为100~200r/min。
[0018]
本发明还提供了所述复方羟氯扎胺混悬液的另外一种制备方法,包括如下步骤:
[0019]
(1)将羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄细化;
[0020]
(2)40~60vt%的丙二醇与水混合,得丙二醇溶液,加入细化后的黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄进行分散,过筛后得第一混合液;
[0021]
(3)取剩余丙二醇与水混合,得丙二醇溶液,加入细化后的羟氯扎胺原料药、盐酸左旋咪唑原料药,得第二混合液;
[0022]
(4)将第一混合液和第二混合液混合,得复方羟氯扎胺混悬液。
[0023]
作为优选,所述步骤(1)中,细化后的羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄的粒径独立的为≤1μm。
[0024]
作为优选,所述步骤(2)中,过筛所用筛为80~100目筛。
[0025]
作为优选,所述步骤(2)和步骤(3)中,丙二醇溶液的浓度独立的为40~60%。
[0026]
经由上述的技术方案可知,与现有技术相比,本发明有益效果如下:
[0027]
本发明所得复方羟氯扎胺混悬液的主成分含量在95~105%,单杂含量小于0.5%,总杂含量小于1%,纯度高,在高温和光照条件下无显著变化,稳定性强,且具有优异的再分散性;
[0028]
本发明所述复方羟氯扎胺混悬液的制备成本低,所用原料种类少,制备工艺简单,适于推广应用。
附图说明
[0029]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
[0030]
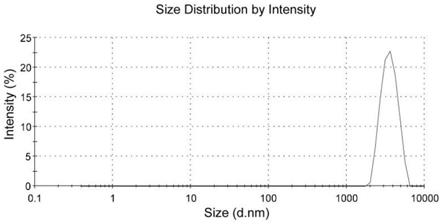
图1为本发明实施例1所得样品1的粒径分布图;
[0031]
图2为本发明实施例2所得样品2的粒径分布图;
[0032]
图3为本发明实施例3所得样品3的粒径分布图;
[0033]
图4为本发明实施例1所得样品1的含量测定色谱图;
[0034]
图5为本发明实施例2所得样品2的含量测定色谱图;
[0035]
图6为本发明实施例3所得样品3的含量测定色谱图;
[0036]
图7为本发明实施例1所得样品空白辅料的有关物质测定色谱图;
[0037]
图8为本发明实施例1所得样品1的有关物质测定色谱图;
[0038]
图9为本发明实施例2所得样品空白辅料的有关物质测定色谱图;
[0039]
图10为本发明实施例2所得样品2的有关物质测定色谱图;
[0040]
图11为本发明实施例3所得样品空白辅料的有关物质测定色谱图;
[0041]
图12为本发明实施例3所得样品3的有关物质测定色谱图。
具体实施方式
[0042]
本发明提供一种复方羟氯扎胺混悬液,每100l复方羟氯扎胺混悬液中,包括以下组分:
[0043][0044]
本发明还提供所述复方羟氯扎胺混悬液的制备方法,包括如下步骤:
[0045]
(1)将羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄进行细化;
[0046]
(2)将细化后的黄原胶与40~60vt%的水混合后进行溶胀,得黄原胶混合液;加入细化后的亚硫酸氢钠、柠檬黄、盐酸左旋咪唑原料药,得第一溶液;
[0047]
(3)将羟氯扎胺原料药和丙二醇混合,得第二溶液;
[0048]
(4)将第一溶液、第二溶液、苯甲酸和剩余水混合,得复方羟氯扎胺混悬液。
[0049]
在本发明中,所述步骤(1)中,羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄的粒径目数独立的优选为≥80目,进一步优选为≥100目。
[0050]
在本发明中,所述步骤(2)中,所述细化后的黄原胶溶胀时优选为采用45~55vt%的水进行,进一步优选为采用50vt%的水进行;混合时间优选为20~24h,进一步优选为21.5~23h;搅拌溶胀的速度优选为200~300r/min,进一步优选为220~280r/min;搅拌溶胀的时间优选为20~40min,进一步优选为25~35min。
[0051]
在本发明中,所述步骤(4)中,第一溶液、第二溶液、苯甲酸和剩余水混合方式为:将第一溶液和第二溶液混合,后加入苯甲酸和剩余水。
[0052]
在本发明中,所述第一溶液和第二溶液混合方式为:将第二溶液在搅拌下加入至第一溶液中;所述搅拌速度优选为100~200r/min,进一步优选为125~170r/min。
[0053]
本发明还提供了所述复方羟氯扎胺混悬液的另一种制备方法,包括如下步骤:
[0054]
(1)将羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄细化;
[0055]
(2)40~60vt%的丙二醇与水混合,得丙二醇溶液,加入细化后的黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄进行分散,过筛后得第一混合液;
[0056]
(3)取剩余丙二醇与水混合,得丙二醇溶液,加入细化后的羟氯扎胺原料药、盐酸左旋咪唑原料药,得第二混合液;
[0057]
(4)将第一混合液和第二混合液混合,得复方羟氯扎胺混悬液。
[0058]
在本发明中,所述步骤(1)中,细化后的羟氯扎胺原料药、盐酸左旋咪唑原料药、黄原胶、亚硫酸氢钠、苯甲酸、柠檬黄的粒径独立的优选为≤1μm,进一步优选为≤0.8μm。
[0059]
在本发明中,所述步骤(2)中,所述丙二醇溶液优选为45~55vt%的丙二醇与水混合,进一步优选为50vt%的丙二醇与水混合;过筛所用筛优选为80~100目筛,进一步优选为80目。
[0060]
在本发明中,所述步骤(2)和步骤(3)中,丙二醇溶液的浓度独立的优选为40~60%,进一步优选为45~55%。
[0061]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0062]
实施例1
[0063]
有效成分及辅料投料量如下:
[0064][0065]
制备工艺:
[0066]
用球磨机将原料药及各辅料分别研磨后过80目筛备用。提前22h将黄原胶浸泡于处方量一半的蒸馏水中使其初步溶胀,再以250r/min转速搅拌30min使其充分溶胀,后将亚硫酸氢钠、柠檬黄、盐酸左旋咪唑依次缓慢加入至容器中,继续搅拌至充分分散,得第一溶液;取处方量丙二醇加入至容器中,再加入羟氯扎胺,搅拌,使其充分润湿,得第二溶液,调整电动搅拌机的转速为150r/min后,将第二溶液在搅拌下缓慢加入至第一溶液中,搅拌均匀后加入苯甲酸,蒸馏水定容后搅拌即得样品1。
[0067]
样品空白辅料的制备:同所述样品1的制备,区别在于不添加羟氯扎胺原料药和盐酸左旋咪唑原料药。
[0068]
实施例2
[0069]
有效成分及辅料投料量如下:
[0070][0071][0072]
制备工艺:
[0073]
用球磨机将原料药及各辅料分别研磨后过100目筛备用。提前24h将黄原胶浸泡于处方量一半的蒸馏水中使其初步溶胀,再以280r/min转速搅拌30min使其充分溶胀,后将亚硫酸氢钠、柠檬黄、盐酸左旋咪唑依次缓慢加入至容器中,继续搅拌至充分分散,得第一溶液;取处方量丙二醇加入至容器中,再加入羟氯扎胺,搅拌,使其充分润湿,得第二溶液,调整电动搅拌机的转速为165r/min后,将第二溶液在搅拌下缓慢加入至第一溶液中,搅拌均匀后加入苯甲酸,蒸馏水定容后搅拌即得样品2。
[0074]
样品空白辅料的制备:同所述样品2的制备,区别在于不添加羟氯扎胺原料药和盐酸左旋咪唑原料药。
[0075]
实施例3
[0076]
有效成分及辅料投料量如下:
[0077][0078]
制备工艺:
[0079]
将原料药及各辅料进行气流超微粉碎至粒径小于1μm,备用取处方量丙二醇于容器中,加水稀释为50%丙二醇水溶液,将50%丙二醇水溶液等分为两份,一份加入处方量各辅料搅拌均匀,过胶体磨后过80目筛得第一溶液,另一份加入羟氯扎胺及盐酸左旋咪唑搅拌均匀得第二溶液,将第一溶液和第二溶液混合均匀后定容并搅拌均匀即得样品3。
[0080]
样品空白辅料的制备:同所述样品3的制备,区别在于不添加羟氯扎胺原料药和盐酸左旋咪唑原料药。
[0081]
对实施例1~3所得三个样品进行质量评价,结果如下:
[0082]
1、外观及ph
[0083]
制备的样品1~3均为亮黄色均匀、粘性、无丁达尔效应的混悬液,静止较长时间可能出现分层现象,上清液黄色透明,下层淡黄色的沉降物,但轻轻振摇后能重新恢复均匀体系。测得样品1~3的ph分别为4.43、4.37、4.35,平均ph为4.38。
[0084]
2、沉降体积比和再分散次数
[0085]
如表1所示,样品1~3的沉降体积比均大于0.9,再分散性良好。
[0086]
表1沉降体积比和再分散性测定结果
[0087]
样品沉降体积比再分散次数10.99221130.991
[0088]
3、粒径测定结果
[0089]
测得样品1~3的粒径大小及分布如图1~3所示,平均粒径大小分别为3514nm、3421nm、3238nm,均小于10μm。本发明所得复方羟氯扎胺混悬液的粒径分布均匀。
[0090]
4、含量测定
[0091]
按照高效液相色谱法进行测定。
[0092]
色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂(zorbax sb-c18,150
×
4.6mm,5μm或等效色谱柱);以0.1%磷酸-7mm磷酸氢二钠水溶液为流动相a,甲醇为流动相b,按下表2进行梯度洗脱;流速为每分钟0.7ml;检测波长为230mn;柱温为35℃;理论板数按羟氯扎胺峰与盐酸左旋咪唑计算均不低于2500。
[0093]
表2 hplc洗脱梯度
[0094]
时间(min)a相(%)b相(%)0703081090131090157030187030
[0095]
测定法:精密称定羟氯扎胺对照品60mg,盐酸左旋咪唑对照品30mg,置于100ml量瓶中,用80%甲醇水超声溶解后稀释至刻度,摇匀,再精密量取1ml,置于10ml量瓶中,用80%甲醇水稀释至刻度,作为对照品溶液;取本品适量(约相当于羟氯扎胺60mg,盐酸左旋咪唑30mg),精密称定,置于100ml容量瓶中,加80%甲醇水溶解并稀释至刻度,充分混匀,离心,精密量取上清液1ml,置于10ml量瓶中,用80%甲醇水稀释至刻度,摇匀,作为供试品溶液;精密量取对照品溶液和供试品溶液各5μl,分别注入液相色谱仪,记录色谱图。按外标法以峰面积计算,即得。
[0096]
样品1~3的含量检测结果如表3所示,对应图谱为图4~6,表明本发明所得复方羟氯扎胺混悬液的主成分含量均在95%~105%,符合要求。
[0097]
表3样品含量测定结果
[0098][0099][0100]
5、有关物质
[0101]
按照高效液相色谱法进行测定。
[0102]
色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂(poroshell ec-c18,150
×
4.6mm,2.7μm或等效色谱柱);以0.5%磷酸二氢铵水溶液(1m naoh溶液调节ph至6.6)为流动相a,甲醇为流动相b,按下表4进行梯度洗脱;流速为每分钟0.7ml;检测波长为230mn;柱温为35℃;理论板数按羟氯扎胺峰与盐酸左旋咪唑计算均不低于2500。
[0103]
表4 hplc洗脱梯度
[0104]
时间(min)a相(%)b相(%)0505012505013307022307022.15050605050
[0105]
测定法:取本发明所得复方羟氯扎胺混悬液(复方羟氯扎胺混悬液的用量相当于羟氯扎胺60mg,盐酸左旋咪唑30mg),精密称定,置于100ml量瓶中,加20ml纯水使其分散,再加入甲醇超声溶解并充分混匀,用甲醇稀释至刻度,摇匀,离心,取上清液作为供试品溶液;精密量取供试品溶液1ml,置于100ml量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。精密量取对照溶液和供试品溶液各10μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3倍。按不加校正因子的主成分自身对照法以峰面积计算,单个杂质不得过0.5%,杂质总量不得过1.0%,供试品溶液色谱图中小于对照溶液主峰面积0.05倍的色谱峰忽略不计。
[0106]
样品1~3的有关物质检测结果如表5所示,对应图谱为图7~9,表明本发明所得复方羟氯扎胺混悬液的单杂含量均小于0.5%,总杂含量均小于1.0%,符合要求。
[0107]
表5有关物质检测结果
[0108][0109]
6、稳定性
[0110]
表6高温试验
[0111][0112][0113]
表7光照试验
[0114][0115][0116]
对样品1~3进行影响因素试验,试验结果显示该复方羟氯扎胺混悬液各指标在高温和光照条件下无显著变化,表明本发明所得复方羟氯扎胺混悬液性状稳定。
[0117]
对实施例1~3所得三个样品进行了临床试验,结果如下:
[0118]
选择国外市售同类产品(主要成分羟氯扎按(30mg/ml)和盐酸左旋咪唑(15mg/ml))作为药物对照组,临床试验如下:
[0119]
1、实验动物筛选
[0120]
选取临床症状为腹泻、消瘦、消化不良、精神倦怠、对外界刺激反应迟钝的羊,进一步进行粪便虫卵检测,确定线虫以及肝片吸虫感染阳性病例。
[0121]
2、实验方法
[0122]
经阳性病例筛选,筛选出125头感染肝片吸虫以及线虫的羊进入临床试验,试验羊随机分为5组,分别为本发明所得复方羟氯扎胺混悬液低、中、高剂量组,药物对照组和空白对照组,试验动物数分别为20、45、20、20、20头。试验组按照羟氯扎按计,给药量分别为7.5mg/kg.bw、15mg/kg.bw、30mg/kg.bw;药物对照组为国外同类制剂,按照羟氯扎按计,给
药量为15mg/kg.bw,空白组不进行药物治疗。给药方式为单次口服给药。如表1所示。
[0123]
表8试验药物分组和剂量
[0124][0125][0126]
3、结果判定
[0127]
在给药后的第3d,7d,14d,21d,28d和56d,根据粪便虫卵数的改变,以虫卵减少率(fecr)和虫卵转阴率(cpcr)来评价羟氯扎按、盐酸左旋咪唑混悬液的治疗效果;使用的公式如下:
[0128]
fecr%=(用药前epg-用药后epg)/用药前epg
×
100%
[0129]
cpcr%=虫卵转阴动物数/实验动物数
×
100%
[0130]
试验组的虫卵减少率在90%以上,与空白对照组虫卵减少率有显著差异(p《0.05),可视为药物有效。
[0131]
4、结果分析
[0132]
表9试验组羊不同阶段平均每克粪便虫卵数(epg值)及给药56d后统计结果
[0133]
[0134][0135]
注:**表示与空白对照组比较组间差异极显著p《0.01
[0136]
从上述结果中可见,与对照药物相比,本发明所述复方羟氯扎胺混悬液对线虫和吸虫的治疗效果明显优于国外同类复方左旋咪唑混悬口服液。本发明所得复方混悬液的中剂量组以及高剂量组对治疗感染线虫和吸虫的羊的治愈率达到95%以上,明显优于国外同类制剂(主要成分羟氯扎按即氯羟柳胺(30mg/ml)和盐酸左旋咪唑(15mg/ml))的92.13%。
[0137]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
