1.本发明涉及检测技术领域,具体涉及一种利用电位滴定法测定氯化钾颗粒含量的方法。
背景技术:
2.氯化钾颗粒(potassium chloride granules)为氯化钾原料药与适当的辅料混合制成的淡粉色至橙色颗粒。氯化钾颗粒适用于治疗和预防伴有或不伴有代谢性碱中毒的低钾血症,对富含钾的食物进行饮食管理或减少利尿剂剂量不足的患者。氯化钾是临床常用的电解质平衡调节药,临床疗效确切,广泛用于临床各科。用于治疗和预防各种原因(进食不足、呕吐、严重腹泻、应用排钾利尿药或长期应用糖皮质刺激素和肾上腺皮质刺激素、失钾性肾病、bartter综合症等)引起的低钾血症,亦可用于心、肾性水肿以及洋地黄等强心甙中毒引起的频发性、多源性早搏或快速心率失常。
3.氯化钾为渗透性利尿药,并治低血钾症.钾离子为神经冲动传导、肌肉收缩及心脏自动机能所必需,过量或静注速度过快可引起心搏停止甚至死亡,重剧疾病长期拒食,钾离子来源枯竭、剧烈腹泻、呕吐、严重创伤,钾离子丢失过多,静注大量葡萄糖液和酸中毒时也会引起低血钾症,缺钾病畜表现精神不振、衰弱无力、极为困倦,肌肉松弛,严重时发生四肢瘫软、不能站立,有时呼吸肌麻痹,出现呼吸困难,心跳加快,心律不齐,血压下降甚至心力衰竭.胃肠蠕动减弱或停止,肚胀,引起食欲减废等症状,对上述病症可进行补钾治疗,补钾可用氯化钾加入生理盐水稀释成0.3%溶液缓慢静滴,因葡萄糖有使钾离子向细胞内转移的倾向,使血钾浓度恢复正常速度变慢.转入到血中的钾离子需15小时才能与细胞内钾离子达到平衡状态,如补钾速度过快可造成一时性高血钾症,甚至导致心脏骤停急死的严重后果,因此医生们都怕用氯化钾静注。
4.氯化钾易溶于水,通过口服容易吸收。钾是细胞内的主要阳离子,是维持细胞内渗透压的重要成分。在细胞内浓度约为150~160mmol/l,在细胞外液浓度较低,仅为3.5~5.0mmol/l。机体主要依靠细胞膜上的na
、k
及atp酶来维持细胞内外的k
、na
浓度差。正常的细胞内外钾离子浓度及浓度差与细胞的某些功能有着密切的关系,钾参与酸碱平衡的调节,糖、蛋白质的合成以及二磷酸腺苷转化为三磷酸苷需要一定量的钾参与;钾参与神经及其支配器官间、神经元间的兴奋过程,并参与神经末梢递质(乙酰胆碱)的形成;心脏内钾的含量可影响其活动,低钾时心脏兴奋性增高,临床血钾过低的患者以心律失常为主;钾是维持骨骼肌正常张力所必需的离子。钾离子不足则表现为肌无力,抽搐。
5.氯化钾的生产工艺具体有重结晶法生产氯化钾:将工业氯化钾加入盛有蒸馏水的溶解槽中进行溶解,再加人脱色剂、除砷剂、除重金属剂进行溶液提纯,经沉淀,过滤,冷却结晶,固液分离,干燥,制得食用氯化钾成品。光卤石法生产氯化钾:将岩盐光卤石粉碎,与75%的水混合,通入过热蒸汽,冷却后析出氯化钾。此粗晶体经水洗,重结晶精制而得。兑卤法:将海水析出氯化钠后的苦卤和老卤(析出氯化钾镁复盐后的母液)按一定的比例掺兑,使混合卤中硫酸镁和氯化镁的摩尔比在0.11以下,氯化镁与氯化钾的比值在11左右,在兑
卤槽中充分析出苦盐(含氯化钠90%、氯化镁2%、硫酸镁1%和氯化钾0.4%)并除去。将混合卤蒸发浓缩至128℃后放入保温沉降器,在124℃下析出高温盐(含氯化钠40%、氯化镁14%、硫酸镁13%和氯化钾1%),在85~90℃下析出低温盐(含氯化钠20%、氯化17%、硫酸镁22%和氯化钾1.3%)。分离后,滤液经冷却析出氯化钾镁复盐即人造光卤石,分离光卤石后的母液为老卤。光卤石加水分解,使氯化镁溶解,得粗氯化钾;后者经水洗、重结晶得成品。作为医药或食品用氯化钾,还需将上述产品溶于水,过滤后通入氯气至饱和。煮沸除去过量的氯,再通入氯化氢使氯化钾析出。分离后用水洗涤后再溶于水,过滤、冷却至-5℃左右得结晶,并在100~120℃下干燥得成品。
6.药用大颗粒氯化钾的生产工艺包括化盐,过滤调酸,蒸发,离心,烘干。步骤如下:以300~2000重量份的工业氯化钾为原料,经加热溶解后,加入0~11重量份的氯化钡,加热至沸,经过滤澄清后加入0~50重量份的碳酸钾,将料液加热至95~108℃,ph值为8.0~9.0,取样检测无钙、镁、钡盐。将合格的料液经过滤澄清后加入盐酸0~10重量份,调ph值至3~5,检测合格的料液进入蒸发器进行二效减压蒸发,蒸发后的结晶经离心、烘干、制粒后得到成品。氯化钾易溶于水、醚、甘油及碱类,微溶于乙醇,但不溶于无水乙醇,有吸湿性,易结块;在水中的溶解度随温度的升高而迅速地增加。
7.通常,氯化钾含量测定方法如下:一为通过火焰原子分光光度法(标准曲线法)检测钾离子测定氯化钾含量;二为普通吸附指示剂法检测氯离子测定氯化钾含量;三为电位滴定法检测氯离子测定氯化钾含量。有usp、jp、chp等法定标准收载氯化钾,usp收载氯化钾颗粒,结合法定标准收载情况有:
8.1.usp氯化钾:吸附指示剂法
9.2.jp氯化钾:吸附指示剂法
10.3.chp氯化钾:吸附指示剂法
11.4.usp氯化钾颗粒:火焰原子分光光度法
12.对于氯化钾颗粒生产、存储的过程中含量可能发生变化,需要进行严格控制,以保证产品的质量。因此,实现氯化钾颗粒含量测定,保证方法的专属性、灵敏度、精密度、准确度具有重要的现实意义。chp、usp、jp采用吸附指示剂法测定氯化钾含量,usp氯化钾颗粒采用火焰原子分光光度法测定氯化钾颗粒含量,由于氯化钾颗粒水溶液显橙色,使用吸附指示剂法测定氯化钾颗粒含量无法确定滴定终点而无法准确定量,专属性、灵敏度均较差,火焰原子分光光度法需使用原子吸收分光光度计及氯化钾标准品成本较高,不利于企业普遍采用,因此,综合考虑开发电位滴定法用于氯化钾颗粒含量测定。
技术实现要素:
13.本发明针对现有技术存在的问题,提供了一种利用电位滴定法测定氯化钾颗粒含量的方法,该方法能快速有效测定氯化钾颗粒的含量,方法灵敏度高、专属性好、精密度高、准确性好,检测结果准确可靠。
14.为实现上述目的,本发明采用的技术方案如下:
15.本发明提供了一种测定氯化钾颗粒含量的方法,包括以下步骤:
16.(1)样品溶液配制:将氯化钾颗粒研磨后加水溶解,稀释、摇匀;取氯化钾溶液第一次加入过氧化氢,第一次加热,再次加入过氧化氢,第二次加热,加入稀硝酸和水,即得;
17.(2)测定:照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定,同时做空白试验。每1ml硝酸银滴定液(0.1mol/l)相当于7.455mg的kcl。
18.步骤(1)中所述氯化钾与水的比例关系为628mg:30ml。
19.步骤(1)中所述第一次加热和第二次加热的温度均为50℃。
20.步骤(1)中所述氯化钾溶液和第一次加入的过氧化氢的体积比为5:1。
21.步骤(1)中所述氯化钾溶液和稀硝酸的体积比为1:1。
22.步骤(1)中所述第一次加入的过氧化氢与再次加入的过氧化氢的体积比为1:1。
23.步骤(1)中所述加热均为水浴加热。
24.步骤(2)中所述测定为电位滴定法。
25.步骤(2)中所述电位滴定使用硝酸银滴定液(0.1mol/l)滴定。
26.步骤(2)所述电位滴定终点的判定方法包括绘制e-v曲线法、绘制
△
e/
△
v-v曲线法或二级微商法。
27.其中,绘制e-v曲线法具体为:用加入滴定剂的体积(v)作横坐标,电动势读数(e)作纵坐标,绘制e-v曲线,曲线上的转折点即为化学剂量点。首先根据测试数据绘制e-v曲线,然后做两条余与滴定曲线相切,并与横轴夹角为45度的直线a、b,再做垂直于横轴的直线,使夹在ab间的线段被曲线交点c平分,即c点就是拐点。
28.绘制
△
e/
△
v-v曲线法具体为:
△
e/
△
v为e的变化值与相对应的加入滴定剂的体积的增量的比。曲线上存在着极值点该点对应着e-v曲线中的拐点,即为化学剂量点。以加入滴定剂的体积为为横坐标,以δe/δv为纵坐标,画出滴定曲线。曲线的最高点即为滴定终点。由最高点引横轴的垂线,交点就是消耗滴定剂的体积。
29.二级微商法具体为:以二阶微商值为纵坐标,加入滴定剂的体积为横坐标作图。
△2e/
△
v2=0所对应的体积即为滴定终点。二级微商法又称二阶微分滴定曲线,纵坐标δ2ε/δv2=0的点即为滴定终点。通过后点数据减前点数据的方法逐点计算二阶微商。具体计算公式为:
[0030][0031]
其中滴定终点的体积可由内插法求得,即取二阶微商的正、负转化处的两个点的体积值v ,v-。然后通过如下公式求得滴定终点:
[0032][0033]
在一些具体的实施方式中,本发明的一种测定氯化钾颗粒含量的方法,包括以下步骤:
[0034]
(1)样品溶液的配制:取氯化钾颗粒10袋,倒出内容物,精密称定,研细,取细粉约628mg(相当于氯化钾600mg),精密称定,置50ml量瓶中,加水30ml使溶解,用水稀释至刻度,摇匀,精密量取5ml,置滴定杯中,精密加入1ml过氧化氢,50℃水浴条件下加热15min,然后再精密加入1ml过氧化氢,50℃水浴条件下加热15min,加5ml稀硝酸和40ml水,即得。
[0035]
(2)测定:照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定,同时做空白试验。每1ml硝酸银滴定液(0.1mol/l)相当于7.455mg的kcl。
[0036]
进一步地,步骤(1)中所述稀硝酸具体配制方法为:量取22ml硝酸置盛有适量水的200ml量瓶中,有水稀释至刻度,摇匀。
[0037]
其中,空白试验具体为精密量取水5ml,置滴定杯中,精密加入1ml过氧化氢,50℃水浴条件下加热15min,然后再精密加入1ml过氧化氢,50℃水浴条件下加热15min,加5ml稀硝酸和40ml水,照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定。
[0038]
进一步地,本发明测定过程中所使用的电极为银电极。
[0039]
进一步地,本发明中的方法适用于对氯化钾颗粒中氯化钾含量的检测。本发明中的氯化钾颗粒具体为氯化钾原料药与适当的辅料混合制成的淡粉色至橙色颗粒。
[0040]
本发明所取得的技术效果是:
[0041]
1.本发明通过梯度加入过氧化氢并水浴加热等方式处理样品溶液并靠电极电位的突跃来指示滴定终点。通过梯度加入过氧化氢并水浴加热对氯化钾颗粒样品溶液处理可降低辅料对氯化钾含量测定的干扰,在滴定到达终点前后,溶液中的氯离子浓度往往连续变化n个数量级,引起电位的突跃,氯化钾的含量仍然通过消耗硝酸银滴定液(0.1mol/l)的量来计算。
[0042]
2.本发明提供的电位滴定法能有效测定氯化钾颗粒含量,且灵敏度高、专属性强、精密度及准确度好,灵敏度高对于痕量分析(ppm级)能有效检出。
附图说明
[0043]
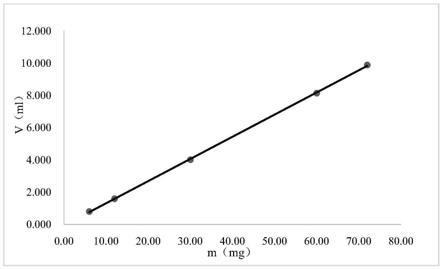
图1为氯化钾颗粒含量测定-线性关系图;
[0044]
图2为氯化钾颗粒含量测定-供试品溶液e-v图;
[0045]
图3为氯化钾颗粒含量测定-供试品溶液δe/δv-v图;
[0046]
图4为氯化钾颗粒含量测定-供试品溶液δe2/δv-v图。
具体实施方式
[0047]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0048]
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。
[0049]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本文中使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同意义。
[0050]
值得说明的是,本发明中使用的氯化钾颗粒制备过程具体为称取处方量的氯化钾、处方量的胶态二氧化硅分别进行分散处理,备用。先将约1/2称样量的氯化钾与处方量的日落黄混合,然后加入剩余的氯化钾进行预混着色。预混结束后依次加入处方量无水枸
橼酸、三氯蔗糖、桔子粉末香精、胶态二氧化硅进行总混,最后按处方装量进行包装。其余物料均为普通市售产品,因此对其来源不做具体限定。
[0051]
实施例1
[0052]
仪器:mettler toledo g10s
[0053]
电极:银电极
[0054]
一种测定氯化钾颗粒含量的方法,包括以下步骤:取氯化钾颗粒10袋,倒出内容物,精密称定,研细,取细粉约628mg(相当于氯化钾600mg),精密称定,置50ml量瓶中,加水30ml使溶解,用水稀释至刻度,摇匀,精密量取5ml,置滴定杯中,精密加入1ml过氧化氢,50℃水浴条件下加热15min,然后再精密加入1ml过氧化氢,50℃水浴条件下加热15min,加5ml稀硝酸和40ml水,照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定,同时做空白试验。每1ml硝酸银滴定液(0.1mol/l)相当于7.455mg的kcl。
[0055]
利用电位滴定法测定氯化钾颗粒含量的方法,所述的方法采用电位滴定法,将氯化钾颗粒定量配制成溶液,定量移取溶液,置滴定杯中,梯度加入过氧化氢并水浴加热,加入稀硝酸酸化用水稀释后,使用电位滴定仪测定。
[0056]
空白试验:精密量取水5ml,置滴定杯中,精密加入1ml过氧化氢,50℃水浴条件下加热15min,然后再精密加入1ml过氧化氢,50℃水浴条件下加热15min,加5ml稀硝酸和40ml水,照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定。
[0057]
对比例1
[0058]
一种测定氯化钾颗粒含量的方法,包括以下步骤:取氯化钾颗粒10袋,倒出内容物,精密称定,研细,取细粉约628mg(相当于氯化钾600mg),精密称定,置50ml量瓶中,加水30ml使溶解,用水稀释至刻度,摇匀,精密量取5ml,置滴定杯中,加5ml稀硝酸和40ml水,照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定,同时做空白试验。每1ml硝酸银滴定液(0.1mol/l)相当于7.455mg的kcl。
[0059]
其余同实施例1。
[0060]
对比例2
[0061]
一种测定氯化钾颗粒含量的方法,包括以下步骤:取氯化钾颗粒10袋,倒出内容物,精密称定,研细,取细粉约628mg(相当于氯化钾600mg),精密称定,置50ml量瓶中,加水30ml使溶解,用水稀释至刻度,摇匀,精密量取5ml,置滴定杯中,精密加入2ml过氧化氢,50℃水浴条件下加热30min,加5ml稀硝酸和40ml水,照电位滴定法,用硝酸银滴定液(0.1mol/l)滴定,同时做空白试验。每1ml硝酸银滴定液(0.1mol/l)相当于7.455mg的kcl。
[0062]
其余同实施例1。
[0063]
试验1:经过检测发现,对比例1和对比例2中测定的氯化钾含量分别为92.3%和94.6%均低于实施例1结果99.6%,说明对比例1和对比例2检测方法的辅料对氯化钾含量测定干扰较大。
[0064]
试验2-回收率、准确度试验:取空白辅料约28mg 9份,氯化钾约60mg、600mg、720mg各三份,精密称定,分别置9个不同的滴定杯中,再取空白辅料约28mg1份,精密称定,置滴定杯中,分别于实施例1、对比例1和对对比例2条件下进行实验,分别计算回收率。
[0065]
表1实施例1回收率试验结果
[0066][0067]
表2对比例1回收率试验结果
[0068][0069]
表3对比例2回收率试验结果
[0070][0071]
结果表明:对比例1和对比例2条件下氯化钾回收率均低于95%,准确度较差,实施例1条件下氯化钾回收率均在98%-101%之间,rsd小于2%,准确度良好。
[0072]
分别采用绘制e-v曲线法、绘制
△
e/
△
v-v曲线法和二级微商法进行判定实施例1测定方法对应的图分别如图2-4所示。此外,进一步验证实施例1中检测方法的精密度、专属性、线性、溶液稳定性等,具体如以下所示:
[0073]
试验3:重复性试验
[0074]
表4重复性试验结果
[0075][0076]
结果表明:6份供试品溶液含量相对标准偏差(rsd)为1.1%,≤2%;符合要求,本方法重复性较好。
[0077]
试验4:中间精密度试验
[0078]
表5中间精密度试验结果
[0079][0080][0081]
结果表明:两个试验人员6份供试品含量的相对标准偏差(rsd%)分别为1.1%、1.7%,均≤2%,满足重复性要求;两个试验人员6份供试品含量均值分别为101.6%、99.8%,差值为1.8%,≤2%,符合要求,本方法精密度较好。
[0082]
试验5:专属性试验
[0083]
表6专属试验结果
[0084][0085]
结果表明:空白溶液及空白辅料溶液对氯化钾颗粒中氯化钾含量测定几乎无干扰(<0.5%),专属性良好。
[0086]
试验6:线性试验
[0087]
表7线性试验结果
[0088][0089]
线性关系图如图1所示,结果表明:在6.00mg~72.00mg范围内,相关系数r为0.99993,y轴截距百分比为0.03%,响应因子rsd为1.34%,线性情况良好。
[0090]
试验7:溶液稳定性试验
[0091]
表8溶液稳定性试验结果
[0092]
批号消耗滴定液体积(ml)含量(%)回收率(%)空白0.027//空白辅料0.026//sample-0h7.95898.6/sample-0.5h7.97298.7100.2sample-1h7.97498.8100.2sample-1.5h7.97098.7100.2sample-2h7.98098.8100.3sample-3h7.95998.6100.0
[0093]
结果表明:氯化钾颗粒溶液在各时间点回收率(与0h含量比值)均在98.0%~102.0%以内,3h内溶液稳定性良好。
[0094]
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。