本发明属于表观遗传数据分析领域,特别是涉及一种基于甲基化的肿瘤数据分析系统。
背景技术
DNA甲基化代表着基本的表观遗传修饰,在细胞周期、细胞增殖、细胞凋亡、DNA复制、染色质结构和基因转录等生物过程中扮演着重要作用。在许多疾病中,如癌症、退行性疾病以及衰老等都表现出异常的甲基化模式影响疾病表型的变化。因此,以基因DNA甲基化水平的变化作为切入点探究疾病的分子分型或病理机制已成为表观遗传学中的研究热点。但目前针对于DNA甲基化数据的分析方法主要集中于假设检验为主的传统统计学分析方法和基于机器学习思想的特征选择算法识别差异甲基化位点或差异甲基化区域,未考虑在甲基化水平上,基因与基因之间的协同或拮抗作用。以基因表达数据作为计算基础应用机器学习算法或统计学方法构建有向基因调控关系已成为基因表达网络研究的主要趋势。而各种针对于时间序列的分析方法被广泛应用于时序基因共表达网络分析,且WGCNA算法被广泛应用于表型数据的关联分析,但WGCNA仍存在一定局限性,WGCNA算法作为一种层次聚类算法,未考虑拓扑学对基因的影响将多数基因归于灰色模块造成某些潜在关键基因的丢失。
技术实现要素:
鉴于DNA甲基化水平上,基于加权基因调控网络数据分析的空白,为解决上述技术问题,本发明提供了如下方案:一种基于甲基化的肿瘤数据分析系统,包括:
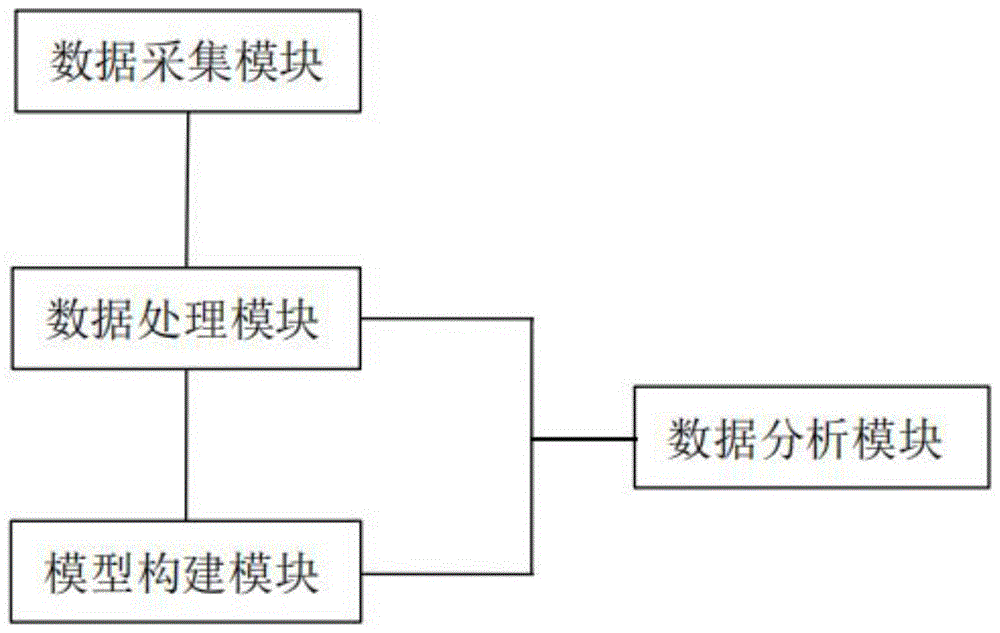
数据采集模块,用于采集肿瘤原始数据;
数据处理模块,与所述数据采集模块连接,用于对所述肿瘤原始数据进行预处理,获得目标肿瘤数据;
模型构建模块,与所述数据处理模块连接,用于构建甲基化加权基因调控网络,识别所述甲基化加权基因调控网络的基因模块;
数据分析模块,分别与所述数据处理模块和所述模型构建模块连接,用于基于所述基因模块和所述目标肿瘤数据进行分析,获得分析结果。
优选地,所述肿瘤原始数据至少包括甲基化数据、临床样本数据。
优选地,所述数据处理模块包括第一处理单元、第二处理单元;
所述第一处理单元用于清除低表达甲基化位点;
所述第二处理单元用于识别差异甲基化基因。
优选地,所述第一处理单元通过针对癌组织和癌旁组织甲基化位点的β值,去除低表达甲基化位点;
所述第二处理单元通过对同一甲基化位点在癌组织和癌旁组织的β值进行分析,当所述甲基化位点的β值变化达到预设阈值时,所述第二处理单元识别所述甲基化位点对应的基因为差异甲基化基因。
优选地,所述模型构建模块包括基因提取单元、特征降维单元、权重计算单元、网络构建单元;
所述基因提取单元用于提取基因调控网络只包含差异甲基化基因的差异基因调控网络;
所述特征降维单元用于基于所述差异基因调控网络的差异甲基化基因值,对所述差异基因调控网络的节点基因的甲基化位点值进行特征降维;
所述权重计算单元用于计算所述节点基因间主成分的相关性,获得网络边权重;
所述网络构建单元用于基于所述网络边权重构建所述甲基化加权基因调控网络。
优选地,所述数据分析模块包括第一分析单元、第二分析单元、第三分析单元;
所述第一分析单元用于分析所述基因模块与临床特征信息的相关性;
所述第二分析单元用于所述基因模块的GO富集分析;
所述第三分析单元用于识别和验证关键甲基化位点。
优选地,所述第一分析单元通过识别差异甲基化位点;对所述差异甲基化位点进行特征降维,获得模块特征基因;计算所述模块特征基因与临床特征信息的相关性,基于所述相关性构建关联矩阵分析所述基因模块与临床信息的相关性。
优选地,所述第二分析单元通过费舍尔精确检验,对与临床特征相关的基因模块得到的GO富集分析结果进行检验。
优选地,所述第三分析单元包括位点鉴定单元、生存分析单元;
所述位点鉴定单元用于鉴定关键甲基化基因模块中的关键甲基化位点;
所述生存分析单元用于对所述关键甲基化位点进行生存分析。
本发明公开了以下技术效果:
本发明结合生物信息学与DNA甲基化芯片分析技术,提出了一种基于甲基化的肿瘤数据分析系统,来确定在肿瘤的发展中起到关键作用的生物过程以及与影响预后的关键DNA甲基化位点,从基因网络角度弥补了DNA甲基化水平常见的窗函数分析方法,通过对基因调控网络的加权,从染色体的空间水平分析影响外部临床特征的关键甲基化修饰基因模块,为肿瘤甲基化水平机制的研究提供新的理论支撑。利用本发明的分析方法处理后的数据,不仅可以用于关键甲基化基因模块识别,还可以应用于其他的数据分析任务。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例的系统结构示意图;
图2为本发明实施例的甲基化基因模块与临床信息相关性图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
如图1所示,本发明提供了一种基于甲基化的肿瘤数据分析系统,包括:
数据采集模块,用于采集肿瘤原始数据;
数据处理模块,与数据采集模块连接,用于对肿瘤原始数据进行预处理,获得目标肿瘤数据;
模型构建模块,与数据处理模块连接,用于构建甲基化加权基因调控网络,识别甲基化加权基因调控网络的基因模块;
数据分析模块,分别与数据处理模块和模型构建模块连接,用于基于基因模块和目标肿瘤数据进行分析,获得分析结果。
肿瘤原始数据至少包括甲基化数据、临床样本数据。
数据处理模块包括第一处理单元、第二处理单元;
第一处理单元用于清除低表达甲基化位点;
第二处理单元用于识别差异甲基化基因。
第一处理单元通过针对癌组织和癌旁组织甲基化位点的β值,去除低表达甲基化位点;
第二处理单元通过对同一甲基化位点在癌组织和癌旁组织的β值进行分析,当甲基化位点的β值变化达到预设阈值时,第二处理单元识别甲基化位点对应的基因为差异甲基化基因。
模型构建模块包括基因提取单元、特征降维单元、权重计算单元、网络构建单元;
基因提取单元用于提取基因调控网络只包含差异甲基化基因的差异基因调控网络;
特征降维单元用于基于差异基因调控网络的差异甲基化基因值,对差异基因调控网络的节点基因的甲基化位点值进行特征降维;
权重计算单元用于计算节点基因间主成分的相关性,获得网络边权重;
网络构建单元用于基于网络边权重构建甲基化加权基因调控网络。
数据分析模块包括第一分析单元、第二分析单元、第三分析单元;
第一分析单元用于分析基因模块与临床特征信息的相关性;
第二分析单元用于基因模块的GO富集分析;
第三分析单元用于识别和验证关键甲基化位点。
第一分析单元通过识别差异甲基化位点;对差异甲基化位点进行特征降维,获得模块特征基因;计算模块特征基因与临床特征信息的相关性,基于相关性构建关联矩阵分析基因模块与临床信息的相关性。
第二分析单元通过费舍尔精确检验,对与临床特征相关的基因模块得到的GO富集分析结果进行检验。
第三分析单元包括位点鉴定单元、生存分析单元;
位点鉴定单元用于鉴定关键甲基化基因模块中的关键甲基化位点;
生存分析单元用于对关键甲基化位点进行生存分析。
实施例一
进一步地,本发明基于甲基化的肿瘤数据分析系统的分析过程包括:
步骤一、获取原始数据集,原始数据集包括肿瘤的甲基化数据和临床随访数据。
步骤二、对原始数据进行预处理,获得385475个甲基化位点;
步骤三、筛选差异甲基化基因
筛选出在FC>0.235且p<0.05下的所有差异甲基化位点,并定义差异甲基化位点对应的基因为差异甲基化基因。
步骤四、构建甲基化加权基因调控网络
通过PCA算法对背景网络中每个基因所包含的所有位点进行特征降维并通过CCA算法计算两两基因之间所有主成分的相关性作为网络边的权重,构建加权基因调控网络。
步骤五、识别加权基因调控网络的基因模块
具体通过multilevel算法实现对加权基因调控网络的基因模块识别。
步骤六、分析基因模块和临床信息的关联,主要包括以下几个方面:
①识别具有显著差异的甲基化位点,即通过champ算法识别具有显著差异的甲基化位点(p<0.05);
②特征降维,即通过PCA算法对每一基因模块的差异甲基化位点进行特征降维,并定义第一主成分为模块特征基因(ME);
③构建关联矩阵,通过spearman相关性算法计算ME与临床特征之间相关性,从而构建关联矩阵;
步骤六、识别和验证关键甲基化位点,主要包括以下几个方面:
①鉴定关键甲基化位点,即通过MM算法和GS算法鉴定m4和m10两个关键甲基化基因模块中的关键甲基化位点;
②生存分析,即通过KM算法实现对关键甲基化位点进行生存分析。
实施例2直肠腺癌的生物信息学分析
基于本发明的分析方法对直肠腺癌进行生物信息学分析,具体包括以下步骤:
(1)原始数据集获取和预处理
具体包括以下步骤:
①原始数据集来源为TCGA数据库中直肠腺癌的甲基化数据和临床样本数据,包括103个组织样本,其中,6个癌旁组织样本和97个癌组织样本。
②基于原始数据集,筛选甲基化样本与临床样本的交集样本,通过stats R包(v3.6.1)中的函数hclust()进行层次聚类,去除离群样本,获得84个癌组织样本。
③去除所有样本中β值小于0的甲基化位点,获得385475个甲基化位点。
差异基因筛选流程
通过champ算法,以|log2FC|>0.235&p<0.05为阈值,筛选得到5983个差异甲基化基因(19064个差异甲基化位点)。
加权基因调控网络的构建和基因模块的鉴定
具体包括以下步骤:
①通过Cui等构建的信号调控网络作为基础,构建以差异甲基化基因信号调控网络作为背景网络
②以所有基因DNA甲基化数据为背景数据,通过CCA算法计算基因之间甲基化的相关性作为网络边的权重,得到一个包含4719个基因的大网和若干个基因数小于10个小网,定义大网为加权基因调控网络。
③通过mutilevel算法对加权基因调控网络进行模块划分得到20个基因模块(modularity=0.59)
④去除基因数小于100的基因模块,获得16个基因模块。
(4)基因模块与临床信息关联分析
如图2所示,具体包括以下步骤:
①以符合统计学意义的甲基化位点的癌组织甲基化数据(p<0.05)为背景数据,通过PCA算法计算每一模块的第一主成分,定义为该模块的特征基因(ME)
②应用Spearman相关性分析,计算临床特征Event、age、M、N、T与ME之间的相关性如图2所示,T是指原发肿瘤的状况。N是指局部淋巴结受累。M指远距离转移,通过图2可以看出:与Event相关性最高的模块为m2;与age相关性最高的模块为m2;与M相关性最高的模块为m10;与N相关性最高的模块为m4;与T相关性最高的模块为m1。
(5)基因模块的富集分析
通过GO富集分析探究模块的生物学意义,发现age、Event特征密切相关的模块主要参与细胞代谢、细胞极性以及细胞周期调控等生物过程;M特征密切相关的模块主要参与成纤维细胞生长因子的趋化和免疫细胞的活化调控等生物过程;N特征密切相关的模块主要参与以微管和鞭毛为主的细胞骨架形成的调控、突触发生的调节以及细胞有丝分裂的调控;T特征密切相关的模块主要参与细胞外基质的调控、微丝形成的调控以及免疫细胞的迁移等生物过程。
(6)关键甲基化位点识别
本发明通过GS/MM算法筛选模块m4、m5中关键甲基化位点,其中,模块m4中关键甲基化位点为MAP4(cg04441191)、KSR2(cg05658717)、GRIN2A(cg09622330)、YWHAG(cg10698404)、SPAG9(cg17047993)、AZI1(cg17917959)、CLASP1(cg22055790)、CEP135(cg24504843)、CEP250(cg24531267)、PIN1(cg26231243)。模块m10中关键甲基化位点为HGFAC(cg02658564)、DLG2(cg08432013,cg26449294)、PDCD1(cg09319815,cg10526431)、EPHB6(cg14842771)、PIK3CD(cg15499275)、FGF12(cg16376000)、PPP2R2C(cg22325673)、PDGFD(cg23852348)。
(7)关键甲基化位点的生存分析
对关键甲基化位点进行生存分析发现MAP4(cg04441191)、KSR2(cg05658717)、GRIN2A(cg09622330)、YWHAG(cg10698404)、SPAG9(cg17047993)、CEP135(cg24504843)、CEP250(cg24531267)的DNA甲基化情况与肿瘤的预后生存密切相关。
本发明结合生物信息学与基因芯片技术,从测序得到的海量基因样本数据、临床个体样本数据中挖掘其中潜在联系,构建DNA甲基化数据网络针对基因与基因之间的内在调控关系的表达模型,确定在肿瘤的发展中起到关键作用的生物过程,以及与肿瘤预后密切相关的基因甲基化位点,进而从DNA甲基化层面上解释肿瘤的病理学调控机制,为肿瘤甲基化水平机制的研究提供新的方向。
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
本文用于企业家、创业者技术爱好者查询,结果仅供参考。