1.本发明涉及卡利拉嗪的制备方法及中间体化合物。
背景技术:
2.精神分裂症是一种严重影响人类健康的疾病,目前已影响世界约1%人口的正常生活,为患者及其家庭带来了严重的后果,是社会负担第7大的疾病。
3.抗精神病药物主要分为典型抗精神病药物和非典型抗精神病药物,目前临床一线用药以非典型抗精神病药物(如d2/5-ht2a双重拮抗剂)为主,而目前临床常用的非典型抗精神分裂症药物如利培酮、阿立哌唑、齐拉西酮、喹硫平等在治疗阳性症状的同时,对阴性症状亦有一定改善,但均具有各自特征的副作用,如椎体外系副作用(eps)概率偏高,静坐不能,失眠,焦虑,心脏毒性等,尚无一个药物在改善精神分裂症整体谱系的同时,有效降低上述副作用。
4.卡利拉嗪(cariprazine),化学名为反式-1-{4-[2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基]-环己基}-3,3-二甲基脲盐酸盐,是匈牙利gedeon richter公司和美国forest laboratories公司联合开发的d3/d2受体部分激动剂,可以用于治疗精神分裂症(注册前)、躁狂症(注册前)和重度抑郁症(iii期)。卡利拉嗪的结构式如下:
[0005][0006]
作为d3/d2受体部分激动剂,卡利拉嗪兼具优先结合d3r和da部分激动剂的特点,在超过ed50剂量100倍条件下,未见小鼠发生强直性木僵行为(僵住症为抗精神分裂症药常见副作用),锥体外系副作用(eps)低,在水迷宫实验中,显著提高东莨菪碱记忆损伤大鼠的学习认知功能。因此,卡利拉嗪在抗精神分裂症领域具有广阔的临床应用前景。
[0007]
国际专利申请wo2015056164、wo20111060363、wo2010070370、wo2010070371、wo2008142461和wo2005012266以及文献jmc2013,56(22),9199-9221,bioorganic&medicinal chemistry letters,22,(2012),3437-3440均报道了卡利拉嗪的合成方法,其流程总结如下:
[0008][0009]
其中,化合物1)经高压氢化、酯化、氨基保护得到化合物4)。化合物4)经不同的还原方法得到化合物5)或6),后续再经还原胺化、缩合,脱保护,酰基化反应得到卡利拉嗪。虽然上述方法中对于各个基团链接方式和次序以及相关官能团的形成途径存在差异,但对于核心基团“反式-1,4-二取代环己基”的形成是相同的。步骤1)硝基苯乙酸还原需要高温高压及大量的钯/铂催化剂,试剂昂贵且对设备要求高;并且还原后还需进行酯化、重结晶才能得到较纯的反式构型产物。另外,大部分文献均未提供反式构型产物的纯度数据。
[0010]
另外,synthesis(germany)2016,48(18),3120-3126公开了如下合成工艺:
[0011][0012]
其中,化合物a经wittig-horner反应、氢化还原得到化合物c(顺反构型约1:1.7),再经脱保护、酰基化得到化合物e。化合物e水解后,通过重结晶得到高纯度的反式构型化合物g,再经酯化、还原、卤化,最后与1-(2,3-二氯苯基)哌嗪缩合得到卡利拉嗪。但是,该工艺提供的制备卡利拉嗪的方法总反应步骤较长,最后与1-(2,3-二氯苯基)哌嗪缩合得到卡利拉嗪反应时间长、温度高,在长时间高温与碱性条件下卡利拉嗪容易分解。
技术实现要素:
[0013]
本发明的目的在于针对现有技术中存在的问题,提供一种新的卡利拉嗪的制备方法及中间体化合物,该制备方法流程短、产品收率高、制得的产品纯度高。
[0014]
为此,本发明第一方面提供了一种卡利拉嗪的制备方法,其包括:
[0015]
(1)以反式-(n-boc-4-氨基环己基)乙酸与1-(2,3-二氯苯基)哌嗪为原料进行反应,得到如下式i所示化合物(反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺);
[0016][0017]
(2)使式i所示化合物进行酸脱保护得到如下式ii所示化合物(反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺):
[0018][0019]
(3)使式ii所示化合物进行还原反应得到如下式iii所示化合物(反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺):
[0020][0021]
(4)使式iii所示化合物与二甲氨基甲酰卤反应得到卡利拉嗪;
[0022][0023]
优选地,步骤(1)中包括使反式-(n-boc-4-氨基环己基)乙酸任选地经活化剂活化后与1-(2,3-二氯苯基)哌嗪进行缩合反应,得到式i所示的化合物。
[0024]
根据本发明的一些优选的实施方式,步骤(1)中使反式-(n-boc-4-氨基环己基)乙酸经活化剂活化后与1-(2,3-二氯苯基)哌嗪进行缩合反应或使反式-(n-boc-4-氨基环己基)乙酸与1-(2,3-二氯苯基)哌嗪直接缩合反应得到式i所示的化合物。
[0025]
优选地,所述活化为使反式-(n-boc-4-氨基环己基)乙酸与所述活化剂反应生成酰卤、酸酐或羰基咪唑中间体化合物。
[0026]
优选地,所述直接缩合反应在缩合剂的存在下进行。
[0027]
优选地,步骤(1)中所述反应在有机溶剂的存在下进行。
[0028]
优选地,步骤(1)中所述反应在碱性条件下进行,更优选在三乙胺条件下进行。
[0029]
优选地,步骤(2)中所述酸脱保护所用酸为三氟乙酸和/或盐酸。
[0030]
优选地,步骤(3)中,所述还原反应在还原剂存在下进行,所述还原剂为硼氢化钠与三氟化硼和碘中至少一种的组合物,或所述还原剂为硼烷。
[0031]
本发明第二方面提供了一种用于合成卡利拉嗪的第一中间体化合物(反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺),其结构式如下式i所示:
[0032][0033]
本发明第三方面提供了一种用于合成卡利拉嗪的第二中间体化合物(反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺),其结构式如下式ii所示:
[0034][0035]
与现有技术相比,本发明方法采用高纯度的反式-(n-boc-4-氨基环己基)乙酸与1-(2,3-二氯苯基)哌嗪经缩合后脱保护、还原、卤化得到卡利拉嗪,大大缩短了工艺步骤,确保了最终产品的纯度,总收率明显提高。
[0036]
本发明的其他特征和优点将在下述的具体实施方式部分予以详细说明。
附图说明
[0037]
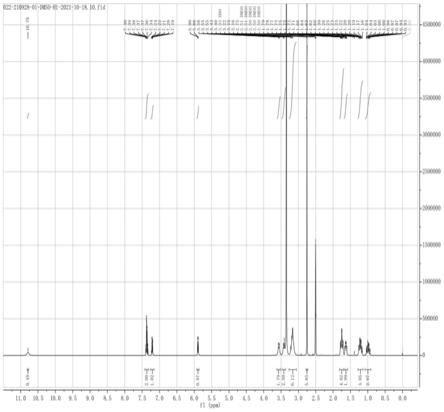
图1为实施例3中制得的中间体化合物的1h nmr图。
[0038]
图2为实施例3中制得的中间体化合物的esi-ms图。
[0039]
图3为实施例5中制得的中间体化合物的1h nmr图。
[0040]
图4为实施例5中制得的中间体化合物的esi-ms图。
[0041]
图5为实施例6中制得的中间体化合物的1h nmr图。
[0042]
图6为实施例6中制得的中间体化合物的esi-ms图。
[0043]
图7为实施例8中制得的卡利拉嗪的1h nmr图。
[0044]
图8为实施例8中制得的卡利拉嗪的
13
c nmr图。
[0045]
图9为实施例8中制得的卡利拉嗪的esi-ms图。
具体实施方式
[0046]
本发明第一方面提供了一种卡利拉嗪的制备方法,其包括:
[0047]
(1)以反式-(n-boc-4-氨基环己基)乙酸与1-(2,3-二氯苯基)哌嗪为原料进行反应,得到反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺(式1所示的化合物);
[0048]
(2)使所得反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺通过酸脱保护得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺(ii所示化合物);
[0049]
(3)使所得反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺进行还原反应得到所述反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺(iii所示化合物);
[0050]
(4)使所得反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺与二甲氨基甲酰卤进行反应得到卡利拉嗪。
[0051]
根据本发明的一些优选的实施方式,步骤(1)中包括使反式-(n-boc-4-氨基环己
基)乙酸经活化剂活化后与1-(2,3-二氯苯基)哌嗪进行缩合反应得到式1所示的化合物。
[0052]
根据本发明的一些实施方式,所述活化为使反式-(n-boc-4-氨基环己基)乙酸与所述活化剂反应生成酰卤、酸酐或羰基咪唑中间体化合物。
[0053]
根据本发明的一些优选的实施方式,所述活化剂为二氯亚砜、草酰氯、氯甲酸异丁酯或羰基二咪唑。
[0054]
根据本发明的一些优选的实施方式,步骤(1)中使反式-(n-boc-4-氨基环己基)乙酸与1-(2,3-二氯苯基)哌嗪在缩合剂存在下直接进行缩合反应得到式1所示的化合物。
[0055]
根据本发明的一些优选的实施方式,所述缩合剂选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、二环己基碳二酰亚胺和2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯中的至少一种。
[0056]
根据本发明的一些实施方式,步骤(1)中所述反应在碱性条件下进行,优选在三乙胺存在下进行。在一些优选的实施方式中,三乙胺的用量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的1-5倍。
[0057]
根据本发明的一些实施方式,步骤(1)中所述反应在有机溶剂中进行。本领域技术人员可以理解,可以通过将反应原料分散于分散溶剂中再行混合从而使充分接触。取决于具体的加料和混合方式,可以将反应原料分别各自分散于分散溶剂中再混合进行反应。所选分散溶剂取决于具体原料,选择范围较宽,以能使相应反应原料溶解分散且不影响反应为准。
[0058]
根据本发明,基于提高产物收率目的,本发明中优选将反式-(n-boc-4-氨基环己基)乙酸分散于有机溶剂如四氢呋喃和/或二氯甲烷中。
[0059]
根据本发明,基于提高产物收率目的,本发明中优选将1-(2,3-二氯苯基)哌嗪分散于二氯甲烷中。
[0060]
根据本发明的一些优选的实施方式,步骤(1)的具体操作包括以下a、b、c或d中的一种:
[0061]
a:将分散有反式-(n-boc-4-氨基环己基)乙酸的第一有机溶剂与二氯亚砜或草酰氯混合进行第一反应得到反应液,将所得反应液浓缩至干并用二氯甲烷溶解得到酰氯溶液;将1-(2,3-二氯苯基)哌嗪、三乙胺和二氯甲烷混合得到混合溶液;使所述混合溶液与所述酰氯溶液混合进行反应,后进行水淬灭,调ph至酸性,萃取,合并有机相后干燥,浓缩,打浆得到式i所示化合物;
[0062]
b:将分散有反式-(n-boc-4-氨基环己基)乙酸的第二有机溶剂与三乙胺和氯甲酸异丁酯反应得到反应液,将所得反应液浓缩至干并用二氯甲烷溶解得到混酐溶液;将1-(2,3-二氯苯基)哌嗪、三乙胺和二氯甲烷混合得到混合溶液;将所述混合溶液与所述混酐溶液混合进行反应,后进行水淬灭,调ph至酸性,萃取,合并有机相后干燥,浓缩,打浆得到式i所示化合物;
[0063]
c:将分散有反式-(n-boc-4-氨基环己基)乙酸的第三有机溶剂与三乙胺、羰基二咪唑和1-(2,3-二氯苯基)哌嗪进行混合反应,后进行水淬灭,调ph至酸性,任选的萃取步骤,分离有机相后干燥,浓缩,打浆得到式i所示化合物;
[0064]
d:将反式-(n-boc-4-氨基环己基)乙酸和1-(2,3-二氯苯基)哌嗪与第四有机溶剂混合得到混合液,并将所述混合液与缩合剂和三乙胺混合进行反应,后进行水淬灭、调ph至
酸性,任选的萃取步骤、合并有机相后干燥、浓缩、打浆得到式i所示化合物。
[0065]
根据本发明的一些优选的实施方式,具体步骤a中,所述第一有机溶剂选自四氢呋喃和二氯甲烷中的至少一种。
[0066]
根据本发明的一些优选的实施方式,具体步骤a中,将二氯亚砜或草酰氯滴加入悬浮有反式-(n-boc-4-氨基环己基)乙酸的第一有机溶剂中,优选于低温下滴加,所述低温为<15℃,优选0-15℃。
[0067]
根据本发明的一些实施方式,具体步骤a中,所述第一反应的温度为20-25℃。
[0068]
根据本发明的一些实施方式,具体步骤a中,所述第一反应的时间为2-12h。在一些实施例中,所述第一反应的时间为3-10h,优选3-5h。
[0069]
根据本发明的一些实施方式,具体步骤a中,1-(2,3-二氯苯基)哌嗪、三乙胺和二氯甲烷的混合溶液中,三乙胺的量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的1-5倍量,二氯甲烷的量为反式-(n-boc-4-氨基环己基)乙酸质量的5-10倍量。
[0070]
根据本发明的一些实施方式,具体步骤a中使所述混合溶液与所述酰氯溶液于≤15℃下混合,优选将酰氯溶液滴加入所述混合溶液中。
[0071]
根据本发明的一些实施方式,具体步骤a中,所述混合溶液与所述酰氯溶液反应的温度为20-25℃。
[0072]
根据本发明的一些实施方式,具体步骤a中,所述混合溶液与所述酰氯溶液反应的时间为2-12h,优选2-6h。
[0073]
根据本发明的一些实施方式,具体步骤a中,所述萃取所用溶剂为二氯甲烷、三氯甲烷、四氢呋喃等。
[0074]
根据本发明的一些实施方式,具体步骤a中,所述打浆通过加入乙酸乙酯进行。
[0075]
根据本发明的一些实施方式,具体步骤a中所述萃取先将反应液调节至酸性获取水相,后将所述水相用有机溶剂进行萃取。
[0076]
根据本发明的一些实施方式,具体步骤b中,所述第二有机溶剂选自四氢呋喃和二氯甲烷中的至少一种。
[0077]
根据本发明的一些实施方式,具体步骤b中,向悬浮有反式-(n-boc-4-氨基环己基)乙酸的第二有机溶剂中滴加三乙胺和氯甲酸异丁酯。
[0078]
根据本发明的一些实施方式,具体步骤b中,向悬浮有反式-(n-boc-4-氨基环己基)乙酸的第二有机溶剂中滴加三乙胺的量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的1-5倍。
[0079]
根据本发明的一些实施方式,具体步骤b中,1-(2,3-二氯苯基)哌嗪、三乙胺和二氯甲烷的混合溶液中三乙胺的用量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的1-5倍量,二氯甲烷的用量为反式-(n-boc-4-氨基环己基)乙酸质量的5-15倍量。
[0080]
根据本发明的一些实施方式,具体步骤b中,所述第二反应的温度为<-5℃,优选为-15℃~-10℃。
[0081]
根据本发明的一些实施方式,具体步骤b中,所述第二反应的时间为1-12h,例如1-4h。
[0082]
根据本发明的一些实施方式,具体步骤b中,所述混合溶液与所述混酐溶液混合进行反应的温度为20-25℃。
[0083]
根据本发明的一些实施方式,具体步骤b中,所述混合溶液与所述混酐溶液混合进行反应的时间为2-12h。
[0084]
根据本发明的一些实施方式,具体步骤b中,所述打浆通过加入乙酸乙酯进行。
[0085]
根据本发明的一些实施方式,具体步骤c中,所述第三有机溶剂选自四氢呋喃和二氯甲烷中的至少一种。
[0086]
根据本发明的一些实施方式,具体步骤c中,向悬浮有反式-(n-boc-4-氨基环己基)乙酸的第三有机溶剂中滴加三乙胺和羰基二咪唑进行第三反应。
[0087]
根据本发明的一些实施方式,具体步骤c中,加入三乙胺的量为反式-(n-boc-4-氨基环己基)乙酸摩尔质量的1-5倍量。
[0088]
根据本发明的一些实施方式,具体步骤c中,所述混合反应的温度为20-25℃。
[0089]
根据本发明的一些实施方式,具体步骤c中,所述混合反应的时间为1-16。
[0090]
根据本发明的一些优选的实施方式,具体步骤c包括将分散有反式-(n-boc-4-氨基环己基)乙酸的第三有机溶剂与三乙胺和羰基二咪唑进行第三反应得到反应液,将所述反应液与1-(2,3-二氯苯基)哌嗪混合进行反应,后进行水淬灭,调ph至酸性,任选的萃取步骤,分离有机相后干燥,浓缩,打浆得到式i所示化合物
[0091]
根据本发明的一些实施方式,具体步骤c中,所述第三反应的温度为<-5℃。
[0092]
根据本发明的一些实施方式,具体步骤c中,所述第三反应的时间为1-12h。
[0093]
根据本发明的一些实施方式,具体步骤c中,所述混合溶液与所述1-(2,3-二氯苯基)哌嗪反应的温度为20-25℃,反应的时间为1-16。
[0094]
根据本发明,具体步骤a、b、c中所述干燥可以采用本领域常用方法,例如用无水硫酸钠进行干燥。
[0095]
根据本发明,本发明中的1-(2,3-二氯苯基)哌嗪可以通过将1-(2,3-二氯苯基)哌嗪盐酸盐与碱反应后经萃取,干燥制得,例如在一些实施方式中,所述1-(2,3-二氯苯基)哌嗪通过如下方法制得:
[0096]
称取1-(2,3-二氯苯基)哌嗪盐酸盐(125.7g,0.47mol),加入1.2l水溶解,用碳酸钠调节ph至7~8后用二氯甲烷萃取(500ml*2次),无水硫酸钠干燥得到二氯甲烷萃取液。
[0097]
根据本发明的一些实施方式,具体步骤d中,所述第四有机溶剂选自四氢呋喃、二氯甲烷、n,n-二甲基甲酰胺和二甲亚砜中的至少一种。
[0098]
根据本发明的一些实施方式,具体步骤d中,所述缩合剂的用量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的0.8-3倍量,所述三乙胺的用量为反式-(n-boc-4-氨基环己基)乙酸摩尔量的1-5倍量。
[0099]
根据本发明的一些实施方式,具体步骤d中,所述反应的温度为20-25℃,
[0100]
根据本发明的一些实施方式,具体步骤d中,所述反应的时间为2-4h。
[0101]
根据本发明的一些实施方式,具体步骤d中,所述打浆通过加入乙酸乙酯进行。
[0102]
根据本发明的一些实施方式,步骤(2)中,所述酸脱保护反应包括将式i所示化合物与酸进行接触反应,优选所述酸为三氟乙酸和/或盐酸。
[0103]
根据本发明的一些实施方式,步骤(2)中,所述酸脱保护包括将式i所示化合物溶于分散溶剂中并与盐酸反应,所述分散溶剂优选为二氯甲烷和甲醇,更优选为二氯甲烷。
[0104]
根据本发明的一些优选的实施方式,步骤(2)中,所述酸脱保护通过将酸加入分散
有式i所示化合物的分散溶剂中进行,优选将所述酸在低温下加入,优选2-5℃,例如冰浴冷却至3℃加入。
[0105]
根据本发明的一些实施方式,步骤(2)中,所述酸脱保护反应包括将酸脱反应产物进行浓缩、调ph值,抽滤、重结晶得到所述式ii所示化合物。
[0106]
根据本发明的一些实施方式,步骤(2)中,所加酸的量为式i所示化合物质量的0.5-3倍量。
[0107]
根据本发明的一些实施方式,步骤(2)中,所述式i所示化合物与酸进行接触反应的温度为10-35℃,优选为10-25℃。
[0108]
根据本发明的一些优选的实施方式,步骤(2)中,将式i所示化合物分散于二氯甲烷中,并于低温下加入三氟乙酸,然后自然升温至常温进行反应。
[0109]
根据本发明的一些实施方式,步骤(2)中,所述酸脱保护的时间为1-12h。
[0110]
根据本发明的一些实施方式,步骤(3)中,所述还原反应在还原剂存在下进行,优选所述还原剂为硼氢化钠与三氟化硼乙醚的组合物、硼氢化钠与碘的组合物或硼烷。
[0111]
根据本发明的一些实施方式,步骤(3)的具体操作包括以下e、f或g中的一种:
[0112]
e:向溶有式ii所示化合物的四氢呋喃中加入硼氢化钠,接着滴加三氟化硼乙醚进行反应,后进行水淬灭,任选的萃取步骤,有机相浓缩,重结晶得到式iii所示化合物;
[0113]
f:向溶有式ii所示化合物的四氢呋喃中加入硼氢化钠,接着滴加单质碘的四氢呋喃溶液进行反应,后进行水淬灭,任选的萃取步骤,有机相浓缩,重结晶得到式iii所示化合物;
[0114]
g:使式ii所示化合物与硼烷溶液混合进行反应,后进行水淬灭,任选的萃取步骤,有机相浓缩,重结晶得到式iii所示化合物。
[0115]
根据本发明的一些实施方式,具体步骤e中,所述滴加三氟化硼乙醚进行反应的温度为10-35℃。
[0116]
根据本发明的一些实施方式,具体步骤e中,所述滴加三氟化硼乙醚进行反应的时间为1-16h。
[0117]
根据本发明的一些实施方式,具体步骤e中,硼氢化钠用量为式ii所示化合物摩尔量的1-3倍量。
[0118]
根据本发明的一些实施方式,具体步骤e中,三氟化硼乙醚的用量为式ii所示化合物摩尔量的1-3倍量。
[0119]
根据本发明的一些实施方式,具体步骤f中,所述反应的温度为10-60℃。
[0120]
根据本发明的一些实施方式,具体步骤f中,所述反应的时间为1-16h。
[0121]
根据本发明的一些实施方式,具体步骤f中,硼氢化钠用量为式ii所示化合物摩尔质量的1-3倍量。
[0122]
根据本发明的一些实施方式,具体步骤f中,碘的用量为式ii所示化合物摩尔质量的1-3倍量。
[0123]
根据本发明的一些实施方式,具体步骤g中,式ii所示化合物与硼烷溶液混合进行反应的温度为18-35℃。
[0124]
根据本发明的一些实施方式,具体步骤g中,式ii所示化合物与硼烷溶液混合进行反应的时间为1-12h。
[0125]
根据本发明的一些实施方式,步骤(4)中,所述二甲氨基甲酰卤为二甲氨基甲酰氯和/或二甲氨基甲酰溴。
[0126]
根据本发明的一些实施方式,步骤(4)包括将式iii所示化合物与二甲氨基甲酰卤、二氯甲烷和三乙胺混合进行反应,后进行水淬灭,调ph为酸性,萃取,合并有机相后干燥,浓缩,打浆得到卡利拉嗪。
[0127]
根据本发明的一些实施方式,步骤(4)包括将式iii所示化合物与三乙胺和二氯甲烷混合后与二甲氨基甲酰卤混合反应,优选加入三乙胺的量为式iii所示化合物摩尔量的1-3倍。
[0128]
根据本发明的一些实施方式,步骤(4)中在低温下将二甲氨基甲酰卤滴加入式iii所示化合物与三乙胺和二氯甲烷的混合液中,所述低温为<15℃,优选为冰浴条件下。
[0129]
根据本发明的一些实施方式,步骤(4)中还包括将式iii所示化合物与二甲氨基甲酰卤反应产物进行水淬灭,调ph、萃取,合并有机相并进行干燥、打浆的操作。
[0130]
根据本发明的一些实施方式,步骤(4)中,式iii所示化合物与二甲氨基甲酰卤反应的温度为20-35℃。
[0131]
根据本发明的一些实施方式,步骤(4)中,式iii所示化合物与二甲氨基甲酰卤反应的时间为2-48h。
[0132]
根据本发明的一些实施方式,步骤(3)、(4)中包括萃取步骤,所述萃取所用溶剂为二氯甲烷、三氯甲烷、四氢呋喃等。
[0133]
根据本发明的一些实施方式,步骤(3)、(4)中所述打浆通过加入乙酸乙酯进行。
[0134]
本发明相较现有技术的制备方法流程简单,无需高温高压条件及贵重催化剂,节约成本,且制得的中间体及卡利拉嗪收率及纯度高。
[0135]
根据本发明,“任选的”可以指含有或不含有,亦可指加入或不加入。
[0136]
为使本发明更加容易理解,下面将结合实施例来详细说明本发明,这些实施例仅用于说明本发明,而不应被视作对本发明的范围的限定。实施例中未注明具体条件者,按照常规条件或者制造商建议的条件进行。实施例中所用材料如无特殊说明,均为市售产品或可通过已知方法合成的常规产品。
[0137]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0138]
本发明中碳含量均系通过采用碳硫分析仪对成品催化剂进行测试得到。
[0139]
本发明实施例的数据通过如下方法获得:
[0140]
1、1hnmr通过:核磁共振谱用bruker avance ii 400(400mhz)记录,tms作为cdcl3或dmso溶液中的内标物。
[0141]
2、esi-ms通过:使用ab sciex qtrap 4500光谱仪收集esi质谱数据。
[0142]
实施例1
[0143]
反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的合成
[0144][0145]
将反式-(n-boc-4-氨基环己基)乙酸(100g,0.39mol)悬浮于二氯甲烷(500ml)中并于7-8℃缓慢加入二氯亚砜(69.5g,0.59mol),加毕,将体系升温至25℃搅拌4h得到反应液。将反应液减压浓缩至干,加入300ml二氯甲烷得到酰氯溶液,备用。
[0146]
称取1-(2,3-二氯苯基)哌嗪盐酸盐(125.7g,0.47mol),加入1.2l水溶解,用碳酸钠调节ph至7~8后用二氯甲烷萃取(500ml*2次),无水硫酸钠干燥得到二氯甲烷萃取液,向二氯甲烷萃取液中加入三乙胺(78.8g,0.78mol),冰水浴冷却。控温15℃滴加制备好的酰氯溶液,加毕,室温反应2h。反应完毕后,反应液加入1l水淬灭,用6mol/l盐酸溶液调至酸性,分液并将其中水相用二氯甲烷萃取一次(500ml),合并有机相并用无水硫酸钠干燥。将有机相浓缩后得到粗品,加入乙酸乙酯(200ml)打浆,过滤干燥得到白色固体157.2g,结果测得反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的收率为86.0%。
[0147]
1h nmr(400mhz,dmso-d6):δ=7.37-7.30(m,2h),7.17-7.13(m,1h),6.68-6.66(m,1h),3.62-3.60(m,4h),3.62-3.60(m,4h),3.15-3.09(m,1h),2.97-2.90(m,4h),2.25(d,j=6.4hz,2h),1.76-1.69(m,4h),1.64-1.62(m,1h),1.37(s,9h),1.19-1.09(m,2h),1.02-0.99(m,2h)。
[0148]
esi-ms:471.33[m h
]。
[0149]
实施例2
[0150]
反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的合成
[0151][0152]
将反式-(n-boc-4-氨基环己基)乙酸(100g,0.39mol)悬浮于二氯甲烷(500ml)中,加入三乙胺(78.8g,0.78mmol),反应液降温至-15℃,缓慢加入氯甲酸异丁酯(79.9g,0.59mol)。加毕,控温-15℃搅拌1h,反应液减压浓缩至干,加入300ml二氯甲烷得到混酐溶液,备用。
[0153]
称取1-(2,3-二氯苯基)哌嗪盐酸盐(125.7g,0.47mol),加入1.2l水溶解,用碳酸钠调节ph=7~8,用二氯甲烷萃取(500ml*2次),无水硫酸钠干燥,二氯甲烷滤液中加入三乙胺(78.8g,0.78mol),控温到15℃,缓慢滴入制备好的混酐溶液中。加毕,自然升温至25℃搅拌2h。反应完毕后,反应液加入1l水淬灭,用6mol/l盐酸溶液调至酸性,分离有机相。有机
相干燥浓缩后得到粗品,加入乙酸乙酯(200ml)打浆,过滤干燥得到白色固体152.7g,收率83.5%。
[0154]
1h nmr(400mhz,dmso-d6):δ=7.36-7.31(m,2h),7.18-7.13(m,1h),6.69-6.67(m,1h),3.62-3.59(m,4h),3.62-3.60(m,4h),3.15-3.09(m,1h),2.97-2.90(m,4h),2.25(d,j=6.4hz,2h),1.76-1.69(m,4h),1.64-1.61(m,1h),1.38(s,9h),1.19-1.08(m,2h),1.02-0.98(m,2h)。
[0155]
esi-ms:471.24[m h
]。
[0156]
实施例3
[0157]
反式n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的合成
[0158][0159]
称取1-(2,3-二氯苯基)哌嗪盐酸盐(115.5g,0.43mol),加入1.2l水溶解,用碳酸钠调节ph=7~8,用二氯甲烷萃取(500ml*2次),无水硫酸钠干燥,二氯甲烷滤液中加入反式-(n-boc-4-氨基环己基)乙酸(100g,0.39mol)、edci(82.4g,0.43mol),冰浴下滴加三乙胺(43.4g,0.43mol),自然升温至25℃搅拌8h。反应完毕后,反应液加入1l水淬灭,用6mol/l盐酸溶液调至酸性,分离有机相。有机相干燥浓缩后得到粗品,加入乙酸乙酯(200ml)打浆,过滤干燥得到白色固体166.7g,收率91.2%,制得的中间体化合物图谱如图1、2所示。
[0160]
1h nmr(400mhz,dmso-d6):δ=7.37-7.30(m,2h),7.17-7.13(m,1h),6.68-6.66(m,1h),3.62-3.60(m,4h),3.62-3.60(m,4h),3.15-3.09(m,1h),2.97-2.90(m,4h),2.25(d,j=6.4hz,2h),1.76-1.69(m,4h),1.64-1.62(m,1h),1.37(s,9h),1.19-1.09(m,2h),1.02-0.99(m,2h)。
[0161]
esi-ms:471.40[m h
]。
[0162]
实施例4
[0163]
反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的合成
[0164][0165]
称取100g(0.21mol)n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺加入反应瓶,加入1l甲醇,冰浴冷却至3℃,缓慢通入hcl气体搅拌5h,然后缓慢加热至35℃搅拌2h,反应完毕后减压旋蒸除去溶剂,残余物加入1l水,用碳酸钠调节ph
为7~8,搅拌1h,过滤,得到固体在50℃下鼓风干燥,得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺固体73.4g,收率93.2%。
[0166]
1h nmr(400mhz,dmso-d6):δ=8.08(s,1h),7.34-7.31(m,2h),7.16-7.13(m,1h),3.65-3.63(m,4h),2.98-2.92(m,4h),2.28-2.24(m,2h),1.98-1.96(m,2h),1.82-1.80(m,2h),1.69-1.71(m,2h),1.66-1.64(m,1h),1.41-1.33(m,2h),1.11-1.02(m,2h)。
[0167]
esi-ms:371.33[m h
]。
[0168]
实施例5
[0169]
反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺的合成
[0170][0171]
称取100g(0.21mol)n-叔丁氧羰基-4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺加入反应瓶,加入400ml二氯甲烷,冰浴冷却至3℃,缓慢加入三氟乙酸200ml,自然升温至25℃搅拌3h,反应完毕后减压旋蒸除去溶剂,残余物加入1l水,用碳酸钠调ph为7-8,析出大量固体,过滤,得到固体与50℃下鼓风干燥,得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺固体75.6g,收率96.1%,制得的中间体化合物图谱如图3、4所示。
[0172]
1h nmr(400mhz,dmso-d6):δ=8.09(s,1h),7.34-7.32(m,2h),7.16-7.12(m,1h),3.65-3.62(m,4h),2.98-2.91(m,4h),2.28-2.25(m,2h),1.98-1.95(m,2h),1.82-1.79(m,2h),1.69-1.71(m,2h),1.66-1.65(m,1h),1.41-1.31(m,2h),1.11-1.00(m,2h)。
[0173]
esi-ms:371.30[m h
]。
[0174]
实施例6
[0175]
反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺的合成
[0176][0177]
将反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺(70.0g,0.19mol),悬浮于四氢呋喃(420ml)中,冰浴下加入硼氢化钠(18.0g,0.48mol)搅拌1h,滴加三氟化硼乙醚(67.1g,0.48mol),加毕,自然升温28℃搅拌6h。tlc检测反应完全。向体系中缓慢滴加100ml水淬灭反应。淬灭完毕后,体系加入稀盐酸回流4h,减压浓缩除去大部分有机溶剂。用碳酸钠调至碱性,析出大量固体,过滤,得到固体与50℃下鼓风干燥,得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺63.9g,收率94.9%,制得的中间体化合物图谱如图5、6所示。
[0178]
1h nmr(400mhz,dmso-d6):δ=7.36-7.28(m,2h),7.14-7.12(m,1h),3.05-2.89(m,4h),2.98-2.91(m,4h),2.36-2.32(m,2h),1.93-1.90(m,2h),1.78-1.75(m,2h),1.34-1.22(m,6h),1.07-0.91(m,2h)。
[0179]
esi-ms:357.90[m h
]。
[0180]
实施例7
[0181]
反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺的合成
[0182][0183]
将反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-2-氧代-乙基}-环己胺(70.0g,0.19mol),悬浮于四氢呋喃(420ml)中,冰浴下加入硼氢化钠(18.0g,0.48mol)搅拌1h,将单质碘(144.7g,0.57mol)溶于thf(80ml)中,冰浴下滴入反应体系中,加毕,升温至60℃搅拌4h。tlc检测反应完全,反应液降至25℃。向体系中缓慢滴加100ml水淬灭反应。淬灭完毕后,体系加入稀盐酸回流3h,减压浓缩除去大部分有机溶剂。用碳酸钠调至碱性析出大量固体,过滤,得到固体与50℃下鼓风干燥,得到反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺59.5g,收率88.4%。
[0184]
1h nmr(400mhz,dmso-d6):δ=7.36-7.27(m,2h),7.14-7.13(m,1h),3.05-2.90(m,4h),2.98-2.91(m,4h),2.36-2.32(m,2h),1.93-1.90(m,2h),1.78-1.76(m,2h),1.34-1.24(m,6h),1.07-0.93(m,2h)。
[0185]
esi-ms:357.90[m h
]。
[0186]
实施例8
[0187]
卡利拉嗪的合成
[0188][0189]
称取反式n-{4-{2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基}-环己胺60.0g(0.17mol)加入到300ml二氯甲烷和三乙胺(85.9g,0.85mol)的混合溶液中。将得到的三乙胺和浓稠悬浮液在25℃的温度下搅拌1小时。冰浴降温,加入二甲氨基甲酰氯(54.8g,0.51mol)反应48小时。加入500ml水淬灭反应,浓盐酸将水相的ph调节至6~7,分液,水相用二氯甲烷萃取一次(500ml),合并有机相,无水硫酸钠干燥,有机相浓缩后得到粗品,加入乙腈(300ml)打浆,过滤干燥得到白色固体这样得到68.3g目标产物。产率为:95%。
[0190]
1h nmr(400mhz,dmso-d6):δ=7.37-7.32(m,2h),7.22-7.18(m,1h),4.12(d,j=8.0hz,1h),3.60-3.58(m,1h),3.07(s,4h),2.88(s,6h),2.63-2.61(m,4h),2.45-2.41(m,
2h),2.03-2.01(m,1h),1.79-1.76(m,3h),1.47-1.42(m,2h),1.26-0.93(m,4h)。
[0191]
13
c nmr(400mhz,dmso-d6):δ=30.39,31.39,33.13,35.00,36.35,48.21,49.71,51.53,54.39,120.30,125.78,126.52,129.15,133.22,150.04,158.04
[0192]
esi-ms:428.5[m h
]。
[0193]
应当注意的是,以上所述的实施例仅用于解释本发明,并不构成对本发明的任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发明可扩展至其他所有具有相同功能的方法和应用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。