电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法及产品和应用
技术领域
1.本发明涉及电化学合成领域,具体涉及一种电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法、及根据该方法制备得到的包含4-氨基-3,6-二氯吡啶甲酸的产品和应用。
背景技术:
2.4-氨基-3,6-二氯吡啶甲酸,商品名为胺草定、氯氨吡啶酸、二氯氨基吡啶酸,是一种吡啶羧酸类除草剂,它能迅速进入植物体内,从而导致植物生长中断并迅速死亡,主要用于牧场、种植园和非农作物区的杂草控制。另外,4-氨基-3,6-二氯吡啶甲酸还是氟氯吡啶酯和氯氟吡啶酯合成的关键中间体。氟氯吡啶酯和氯氟吡啶酯是陶氏益农公司开发的新型芳基吡啶甲酸酯类除草剂,是激素类除草剂中的新品种,具有药量更低,杀草谱更广的特点。
3.4-氨基-3,6-二氯吡啶甲酸(即氯氨吡啶酸)的合成比较常见的方法是电化学合成,以4-氨基-3,5,6-三氯吡啶甲酸为原料电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法。但仍然存在一些问题问题:(1)产品收率偏低且纯度不高、颜色发红;(2)碱消耗大、产生废盐量大;(3)电解后后期槽压升高的问题。
技术实现要素:
4.发明目的
5.为克服上述不足,本发明的目的在于提供一种电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法、及根据该方法制备得到的包含4-氨基-3,6-二氯吡啶甲酸的产品和应用;本发明不仅可以提高4-氨基-3,6-二氯吡啶甲酸产品的合成收率、品质,还避免了“碱消耗大、产生废盐量大”、“电解电压不稳”、“后期槽压高”的问题。
6.解决方案
7.为实现本发明目的,本发明采用的技术方案如下:
8.第一方面,本发明提供了一种电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法,包括如下步骤:采用阳离子膜将电解池分为阳极区和阴极区,以酸性电解液为阳极液,以含有4-氨基-3,5,6-三氯吡啶甲酸(如式i)或其盐的碱性电解液为阴极液,进行电解反应,电解结束后从阴极液中分离出4-氨基-3,6-二氯吡啶甲酸(如式ii),其中,所述酸性电解液为酸溶液或含有金属盐的酸性溶液。
9.优选的,所述酸性电解液为酸溶液。
10.11.本发明的阳离子膜使阳离子液中的h
穿过至阴极液中。
12.本发明在电解过程中,阴极上主要发生还原脱氯反应(如反应式(1))和析氢反应(如反应式(2),主要发生在电解后期);阳极上主要发生析氧反应(如反应式(3))或者析氯反应(如反应式(4))。
[0013][0014]
2h2o 2e-→
h2 2oh-ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(2)
[0015]
2h2o 2e-→
1/2o2 2h
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(3)
[0016]
2cl- 2
e-→
cl2ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4)
[0017]
进一步地,当所述酸性电解液为含有金属盐的酸性溶液时,金属离子的浓度不大于氢离子浓度的5倍,可选地不大于2倍,可选地,不大于1倍,可选地,不大于0.5倍;可选地,所述金属离子为碱金属离子;可选地,氢离子的浓度大于0.01m。
[0018]
进一步地,所述阳极液中含有0.1~5m h2so4或者0.1~5m hcl或者0.1~5m h3po4或者0.1~5m的混酸,可选地含有0.5~5m h2so4或0.5~5m hcl,优选地含有0.5~2.0m h2so4。
[0019]
采用的阳极材料不是关键因素,只要电解过程中不会被腐蚀的导电材料都可以作为阳极材料,例如硫酸h2so4水溶液为阳极液时,可以用钛基氧化铱涂层、钛基二氧化铅涂层、钛镀铂等作为阳极材料(这个条件下发生析氧反应);hcl水溶液作为阳极液时,可以用钛基氧化钌涂层、石墨等作为阳极材料(这个条件下发生析氯反应)。
[0020]
进一步地,所述阴极液的ph为8~14。
[0021]
进一步地,所述阴极液中含0.1~1m 4-氨基-3,5,6-三氯吡啶甲酸或者4-氨基-3,5,6-三氯吡啶甲酸盐,优选含有0.3~0.6m 4-氨基-3,5,6-三氯吡啶甲酸或者4-氨基-3,5,6-三氯吡啶甲酸盐。
[0022]
加入的4-氨基-3,5,6-三氯吡啶甲酸或者4-氨基-3,5,6-三氯吡啶甲酸盐可以是粉末也可以是浆料。
[0023]
为了控制阴极液的ph和增加阴极液的导电性,所述阴极液中还含有碳酸盐,可选地,碳酸盐选自碳酸钠、碳酸钾和碳酸锂中的一种或几种。
[0024]
进一步地,所述阴极液中含有0.1~1.2m naoh,可选地含有0.6~1.2m naoh。
[0025]
进一步地,所述阴极液在电解过程中进行搅拌,在实验室条件下通常采用搅拌磁子。
[0026]
电解过程中电流从阳极依次通过阳极液、阳离子膜和阴极液最后到达阴极,电解过程的电流密度为1~10a/dm2,可选地为2~5a/dm2。
[0027]
进一步地,电解过程的温度范围为10~90℃,优选为30~60℃。
[0028]
进一步地,阴极电极的形状可以为片状、网状和泡沫状,优选为网状银。
[0029]
进一步地,所述阴极电极为活化银电极,例如可以用电化学氧化还原法进行活化,也可以用纯化学的氧化还原法进行活化,可选地活化银的电化学氧化还原法包括:在含有
氯离子或溴离子的水溶液液中,先以银为阳极进行氧化至有氧气析出,然后以银为阴极进行还原至有氢气析出;可选地氧化还原过程的电流密度为0.1~5a/dm2,优选0.5~2a/dm2;温度为0~50℃,优选20~40℃。
[0030]
在电化学氧化还原反应前,将新鲜的银或者使用过的银在含5~35wt%的盐酸水溶液中浸泡5~300min,浸泡温度为20~50℃。
[0031]
进一步地,所述阳极电极的涂层采用钛基氧化铱涂层、钛基二氧化铅涂层和钛镀铂中的一种或几种的混合涂层,可选地为钛基氧化铱涂层。
[0032]
进一步地,所述阳离子膜为耐酸碱耐腐蚀的阳离子膜,可选地为均相阳离子膜。
[0033]
阳极反应为析氧反应时,阳离子膜只要耐受酸碱即可;阳极反应为析氯反应时,阳离子膜不仅要耐酸碱还要耐氯气及其生成的次氯酸的氧化腐蚀,例如杜邦的nafion 324阳离子膜就符合这个条件,包括氯碱中常用的全氟磺酸和全氟羧酸复合膜也符合这个条件。
[0034]
进一步地,所述阳极液和阴极液均用去离子水配制。
[0035]
进一步地,从阴极液中分离出4-氨基-3,6-二氯吡啶甲酸的方法可以采用常规方法,也可以采用如下方法:用盐酸将阴极液ph调到0.5~1.5,得到白色固体;对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末。进一步可选地,采用如下方法:在85~90℃下用浓盐酸将阴极液ph调到0.5~1.5,冷却得到白色固体,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末。
[0036]
进一步地,阴极和阳极之间的电压变化范围可以在0.2~1.2v,优选地在0.2~0.9v;
[0037]
和/或,阴极和阳极之间的电压可以为4.1-5.6v范围。
[0038]
进一步地,提供一种所述的方法获得的4-氨基-3,6-二氯吡啶甲酸产品。
[0039]
进一步可选地,所述4-氨基-3,6-二氯吡啶甲酸产品形态为白色固体或白色固体粉末。
[0040]
还一方面,提供一种所述的4-氨基-3,6-二氯吡啶甲酸产品的应用,所述4-氨基-3,6-二氯吡啶甲酸产品作为除草剂。通过本发明所述的方法获得的4-氨基-3,6-二氯吡啶甲酸产品可以与至少一种农业上可接受的助剂或载体混合,直接被施用于植被或其部位或在植被出土前施用于土壤中,来杀灭或控制不需要植被,优选地,不需要植被是指阔叶草。
[0041]
有益效果
[0042]
本发明采用特定的酸性电解液,在电解过程中不仅可以提高4-氨基-3,6-二氯吡啶甲酸产品的合成收率、品质,还避免了“碱消耗大、产生废盐量大”、“电解电压不稳”、“后期槽压高”的问题,且电解过程中电压变化范围小、电压在4.1-5.6v之间,并降低能耗,解决了现有技术中因电压难以稳定控制的问题,使产品的品质更优,产品收率在95%以上,纯度能达到96%以上,甚至能达到98.5%。
附图说明
[0043]
一个或多个实施例通过与之对应的附图中的图片进行示例性说明,这些示例性说明并不构成对实施例的限定。在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。
[0044]
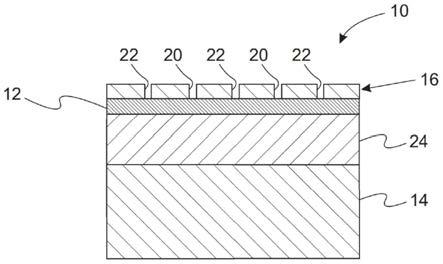
图1本发明的设有隔膜的h型电解槽的一个实施例的示意图;
[0045]
图2本发明的实施例1、对比例1的电解液及水洗产品的hplc图,其中,a为实施例1电解后的阴极电解液,b为对比例1电解后的阴极电解液,c为实施例1的水洗产品,d为对比例1的水洗产品;hplc图中的谱峰从左到后(标记出的竖线)分别对应于未知副产物1、4-氨基-3,6-二氯吡啶甲酸(目标产品)、未知副产物2和4-氨基-3,5,6-三氯吡啶甲酸(原料)。
具体实施方式
[0046]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0047]
另外,为了更好的说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在一些实施例中,对于本领域技术人员熟知的原料、元件、方法、手段等未作详细描述,以便于凸显本发明的主旨。
[0048]
除非另有其它明确表示,否则在整个说明书和权利要求书中,术语“包括”或其变换如“包含”或“包括有”等等将被理解为包括所陈述的元件或组成部分,而并未排除其它元件或其它组成部分。
[0049]
银电极的活化
[0050]
以nafion 324阳离子膜为隔膜的h型电解槽中(如图1所示),银网(纯度为99.99wt%,尺寸为0.3cm
×
4.0cm
×
6.0cm)为工作电极(在进行下述氧化还原反应之前,将银网浸泡在静止的10wt%盐酸水溶液中20min);相同面积的316不锈钢为对电极;银/氯化银为参比电极。工作电极室为0.5m nacl水溶液,对电极室为1.0m氢氧化钠水溶液。控制工作电极室的水溶液的温度为20~25℃,首先对银电极施加0.5a/dm2的阳极氧化电流直至电极电位到达 0.4vs.vs.ag/agcl;然后对银电极施加0.5a/dm2的阴极还原电流直至电极电位到达-0.4vs.ag/agcl。重复上述步骤2次后,取出银电极,置于去离子中备用。
[0051]
本发明的高效液相色谱测定条件为:c18对称柱(250mm length_4.6mm i.d.,5mm particle size)为分离柱;含有30mm磷酸的乙腈/甲醇/水(体积比1:3:6)混合溶液为流动相;流速为:1ml/min;检测波长为230nm;waters 2996 pda为检测器。
[0052]
实施例1:
[0053]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0054]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含0.5m h2so4的水溶液为阳极液,100ml含0.3m 4-氨基-3,5,6-三氯吡啶甲酸 0.6m naoh的水溶液为阴极液。在30-35℃下,搅拌阴极液,通入2a/dm2的电流,反应6小时后停止电解。
[0055]
电解过程中,观察到阴极和阳极之间的电压稳定在4.4-5.1v范围内。
[0056]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.2%,4-氨基-3,6-二氯吡啶甲酸收率为96.5%;以酚酞为指示剂酸碱滴定阳极液中的h2so4浓度为0.5m,阴极液中naoh的浓度为0.02m。
[0057]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为98.5%。
[0058]
实施例2
[0059]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0060]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含2.0m h2so4的水溶液为阳极液,100ml含0.6m 4-氨基-3,5,6-三氯吡啶甲酸 1.2m naoh的水溶液为阴极液。在55-60℃下,搅拌阴极液,通入5a/dm2的电流,反应2小时后将电流密度调为2a/dm2再电解5小时,停止电解。
[0061]
电解过程中,观察到阴极和阳极之间的电压稳定在4.6-5.5v范围内。
[0062]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.4%,4-氨基-3,6-二氯吡啶甲酸收率为97.2%;以酚酞为指示剂酸碱滴定阳极液中h2so4的浓度为2.0m,阴极液中naoh的浓度为0.01m。
[0063]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为98.5%。
[0064]
实施例3
[0065]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0066]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含5.0m h2so4水溶液为阳极液,100ml含1.0m 4-氨基-3,5,6-三氯吡啶甲酸 1.0m k2co3的水溶液为阴极液(ph~8)。在85-90℃下,搅拌阴极液,通入10a/dm2的电流,反应5小时后停止电解。
[0067]
电解过程中,观察到阴极和阳极之间的电压稳定在4.5-4.7v范围内。
[0068]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.8%,4-氨基-3,6-二氯吡啶甲酸收率为95.2%;分别以酚酞和甲基橙为指示剂酸碱滴定阳极液中h2so4的浓度为5.0m,阴极液中k2co3的浓度为0m。
[0069]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为96.7%。
[0070]
实施例4
[0071]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0072]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化钌涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含1.0m hcl的水溶液为阳极液,100ml含
0.6m 4-氨基-3,5,6-三氯吡啶甲酸 1.2m naoh的水溶液为阴极液。在55-60℃下,搅拌阴极液,通入5a/dm2的电流,反应2小时后将电流密度调为2a/dm2再电解5小时,停止电解。
[0073]
电解过程中,观察到阴极和阳极之间的电压稳定在4.8-5.6v范围内。
[0074]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.9%,4-氨基-3,6-二氯吡啶甲酸收率为96.1%;以酚酞为指示剂酸碱滴定阳极液中hcl的浓度为0.4m,阴极液中naoh的浓度为0.01m。
[0075]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为97.2%。
[0076]
实施例5
[0077]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0078]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以实施例1方法制备的活化银网电极为阴极,相同面积的钛镀铂片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含1.0m h3po4的水溶液为阳极液,100ml含0.6m 4-氨基-3,5,6-三氯吡啶甲酸 1.2m naoh的水溶液为阴极液。在55-60℃下,搅拌阴极液,通入5a/dm2的电流,反应2小时后将电流密度调为2a/dm2再电解5小时,停止电解。
[0079]
电解过程中,观察到阴极和阳极之间的电压稳定在6.8-7.9v范围内。
[0080]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为97.2%,4-氨基-3,6-二氯吡啶甲酸收率为95.2%;以酚酞为指示剂酸碱滴定阳极液中h3po4的浓度为1.0m,阴极液中naoh的浓度为0.01m。
[0081]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为96.2%。
[0082]
实施例6
[0083]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0084]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含0.005m h2so4 0.05m na2so4的水溶液为阳极液,100ml含0.1m 4-氨基-3,5,6-三氯吡啶甲酸 1.1m naoh的水溶液为阴极液(ph~14)。在10-15℃下,搅拌阴极液,通入1a/dm2的电流,反应5小时后停止电解。
[0085]
电解过程中,观察到阴极和阳极之间的电压稳定在6.6-8.6v范围内。
[0086]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.1%,4-氨基-3,6-二氯吡啶甲酸收率为91.8%;以酚酞为指示剂酸碱滴定阳极液中h2so4的浓度为0.005m,阴极液中naoh的浓度为0.9m。
[0087]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为
94.1%。
[0088]
本实施例中,阳极液中,na
浓度是h
浓度的10倍,收率和纯度均下降明显。
[0089]
实施例7
[0090]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0091]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以上述方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含0.1m h2so4 2.0m na2so4水溶液为阳极液,100ml含0.3m 4-氨基-3,5,6-三氯吡啶甲酸 0.6m naoh的水溶液为阴极液。在30-35℃下,搅拌阴极液,通入2a/dm2的电流,反应6小时后停止电解。
[0092]
电解过程中,观察到阴极和阳极之间的电压稳定在4.2-4.8v范围内。
[0093]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.4%,4-氨基-3,6-二氯吡啶甲酸收率为91.2%;以酚酞为指示剂酸碱滴定阳极液中h2so4的浓度为1.1m,阴极液中naoh的浓度为1.1m。
[0094]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为93.8%。
[0095]
本实施例7与实施例1相比,仅在于实施例7中的阳极液的na
浓度是h
浓度的20倍,虽然均为酸性阳极液,但在金属盐离子(na
)浓度大于h
浓度10倍以上时,产品的收率和纯度均下降,用于中和阴极液中naoh的盐酸需要的更多,从而导致产生的废盐nacl也更多。
[0096]
实施例8
[0097]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0098]
与实施例1的电解条件相比,区别仅在于采用0.5m h2so4 2.5m na2so4的水溶液为阳极液,其余电解条件相同。
[0099]
电解过程中,观察到阴极和阳极之间的电压稳定在4.5-5.4v范围内。
[0100]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.8%,4-氨基-3,6-二氯吡啶甲酸收率为95.1%;以酚酞为指示剂酸碱滴定阳极液中h2so4的浓度为0.8m,阴极液中naoh的浓度为0.5m。
[0101]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为97.6%。
[0102]
实施例9
[0103]
以酸性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0104]
与实施例1的电解条件相比,区别仅在于采用0.5m h2so4 1m na2so4的水溶液为阳极液,其余电解条件相同。
[0105]
电解过程中,观察到阴极和阳极之间的电压稳定在4.5-5.3v范围内。
[0106]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为98.9%,4-氨基-3,6-二氯吡啶甲酸收率为96.1%;以酚酞为指示剂酸碱滴定阳极液中
h2so4的浓度为0.48m,阴极液中naoh的浓度为0.022m。
[0107]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为97.3%。
[0108]
对比例1
[0109]
以碱性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0110]
与实施例1的电解条件相比,区别仅在于采用0.6m naoh的水溶液为阳极液,其余电解条件相同。
[0111]
电解过程中,观察到阴极和阳极之间的电压稳定在6.1-8.9v范围内。
[0112]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.5%,4-氨基-3,6-二氯吡啶甲酸收率为91.8%;以酚酞为指示剂酸碱滴定阳极液中naoh的浓度为0.07m,阴极液中naoh的浓度为1.1m。
[0113]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为93.6%。
[0114]
同等电解条件下,实施例1与对比例1的区别仅在于阳极液不同,对比例1使用碱性水溶液作为阳极液在电解过程中,电解电压更高(6.1-8.9v)且不稳定(电压变化范围高达2.7v),产品收率(91.8%)和纯度更低(93.6%),消耗的naoh和中和该naoh所需要的盐酸更多,从而导致产生的废盐nacl也更多。而实施例1的产品收率可达96.5%,纯度高达98.5%,电解过程中的电压低(4.4-5.1v)且稳定(电压变化范围为0.7v)。
[0115]
通过图2可知,电解过程中产生的未知副产物1在水洗过程中能去除,但未知副产物2难以在纯化过程中去除,通过图2的c、d对比可知,实施例1中的未知副产物2更少,因此最终的产品纯度更高。
[0116]
对比例2
[0117]
以碱性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0118]
与实施例1的电解条件相比,区别仅在于采用1.0m naoh的水溶液为阳极液,其余电解条件相同。
[0119]
电解过程中,观察到阴极和阳极之间的电压稳定在7.1-10.4v范围内。
[0120]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.1%,4-氨基-3,6-二氯吡啶甲酸收率为90.2%;以酚酞为指示剂酸碱滴定阳极液中naoh的浓度为0.1m,阴极液中naoh的浓度为1.8m。
[0121]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为92.5%。
[0122]
同等电解条件下,实施例2与对比例2的区别仅在于阳极液不同,对比例2使用碱性水溶液作为阳极液,电解电压更高且不稳定,产品收率和纯度更低,消耗的naoh更多,中和
需要的盐酸也更多,从而导致产生的废盐nacl也更多。
[0123]
对比例3
[0124]
以非活化银碱性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸的方法:
[0125]
与实施例1的电解条件相比,区别仅在于采用未活化银网作为阴极。
[0126]
电解过程中,观察到阴极和阳极之间的电压稳定在4.6-5.4v范围内。电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为53.1%,4-氨基-3,6-二氯吡啶甲酸收率为36.6%。
[0127]
对比例4
[0128]
以中性阳极液电解脱氯合成4-氨基-3,6-二氯吡啶甲酸
[0129]
电解条件为:以nafion 324阳离子膜为隔膜的h型电解槽中(如图1);以实施例1方法制备的活化银网电极为阴极,相同面积的钛基氧化铱涂层片(几何尺寸为0.5cm
×
4.0cm
×
6.0cm)为阳极,阴阳极之间的距离为6cm;200ml含2.0m na2so4水溶液为阳极液,100ml含0.3m 4-氨基-3,5,6-三氯吡啶甲酸 0.6m naoh的水溶液为阴极液。在30-35℃下,搅拌阴极液,通入2a/dm2的电流,反应6小时后停止电解。
[0130]
电解过程中,观察到阴极和阳极之间的电压稳定在4.2-4.8v范围内。
[0131]
电解结束后,用高效液相色谱测定阴极液中4-氨基-3,5,6-三氯吡啶甲酸的转化率为99.2%,4-氨基-3,6-二氯吡啶甲酸收率为92.5%;以酚酞为指示剂酸碱滴定阳极液中h2so4的浓度为1.0m,阴极液中naoh的浓度为1.1m。
[0132]
然后对阴极液进行如下处理:首先,在85~90℃下用浓盐酸将阴极液ph调到~1,过夜自然冷却析出白色固体;然后,对白色固体进行过滤、水洗、烘干处理,得到白色固体粉末;最后,用高效液相色谱测定白色固体粉末中4-氨基-3,6-二氯吡啶甲酸的纯度为96.5%。
[0133]
与现有技术相比,本发明的有益效果主要体现在:(1)4-氨基-3,6-二氯吡啶甲酸产品收率提高了4.7-7.0%,收率可达到96.5%以上,不仅能大幅度提高产率,带来更大的商业利润;(2)本发明能大大减少未知副产物2的产生,提高产品纯度,能提高4.9-6.0%,纯度可达98.5%甚至更高,而且能大大提高产品的应用潜力和应用范围,商业利润能提高到产品价格10万元/吨产品左右;(2)每生产一吨4-氨基-3,6-二氯吡啶甲酸产品可减少碱金属氢氧化物(naoh为例)用量500kg以上、减小碱金属氯化物废盐(nacl废盐为例)排放800kg以上;(3)电解电压更低(平均电压从7.5v下降到了4.75v)且更稳定(电压变化范围从~2.7v下降到~0.7v),能耗大大降低。
[0134]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。