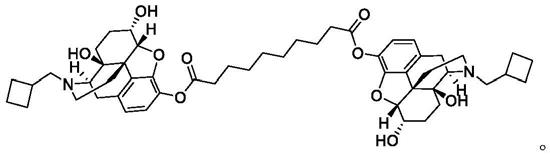
1.本发明属于医药领域,具体涉及一种塞纳布啡的储库制剂,其制备方法以及在制药工业中的应用。
技术背景
2.纳布啡癸二酸酯(dinalbuphine sebacate,dns)又叫塞纳布啡,是纳布啡的二聚体,其化学结构如下所示,该化合物是通过癸二酸连接基连接的纳布啡 (nalbuphine)的二酯,为纳布啡的前药,主要用于治疗治疗中度至重度术后疼痛。因纳布啡必须每4至6小时注射一次,而dns作为纳布啡前药能够缓慢降解,具备制备成缓释制剂的潜力,
[0003][0004]
阿片药物因存在依赖和成瘾风险,因此施用的阿片药物的药物浓度需要恰到好处的达到镇痛效果但尽量避免带来药物依赖风险。此外,因术后痛、癌痛或其他较为剧烈的慢性或急性疼痛往往需要在较长的时间应用,为避免反复注射,减少疼痛管理负担,具有平稳血药浓度的长效缓释阿片药物注射制剂一直是开发的热点。目前纳布啡癸二酸酯的长效缓释制剂已上市,使剧烈或长期疼痛的患者无需频繁施用纳布啡制剂,其处方工艺在wo2016189393a中公开,为一种注射用油混溶性制剂,该制剂通过将dns溶于比例为0.8~1.2:1的苯甲酸苄酯与芝麻油、蓖麻油等植物溶剂的混合溶剂中制备得到,其载药量较小,dns约为70~100mg/ml,载药量约为7~10重量%,因此注射体积较大。此外,患者在手术后仍然需要口服酮咯酸来缓解疼痛,且注射用油溶液存在容易氧化,易污染,不易保存等缺点,芝麻油、蓖麻油等植物性溶剂易导致过敏反应,在临床肌肉注射时病人痛苦感比较强、易导致肌肉结块;另外苯甲酸苄酯还具有包括皮肤刺激和过敏反应等不良反应。
[0005]
专利cn104968338a公开了一种长效止痛药癸二酰二纳布啡酯
‑
plga控释剂型,采用聚乳酸、聚乳酸
‑
聚乙二醇共聚物等作为载体,制备成微球。但微球过程制备复杂,有机溶剂残留及无菌保障等均是一大挑战。
[0006]
溶致液晶(lyotropic liquid crystal,llc)是两亲性分子在溶剂中,由胶束进一步缔合而形成,其相变可由水分的增加引起,常见的3种晶型为层状、六角相和立方相,后2种又分为正向和反向结构。作为药物载体时,溶致液晶可以为亲水性、亲脂性和两亲性药物提供一个多功能的传递平台,同时能够提高药物的稳定性,并实现药物的持续释放。
[0007]
专利cn101014319a公开了一种包含一种二酰基甘油和/或至少一种生育酚,至少一种磷脂酰甘油,至少一种生物相容溶剂的通用低粘度非液晶混合物的预制剂,适用于多种给药途径,但其载药量不高,说明书中记载奥曲肽药物负载量约为 0.5~3%(w/w),氯己定的药物负载量约为5%(w/w),抗炎/镇痛药物苄达明的药物负载量约为3.2~4.5%(w/w),维思通的药物负载量约为5%(w/w),因此对于缓释长期给药和/或需要药物负载量较大
的制剂并不适用。
[0008]
专利cn101123949a公开了一种包含10~40%的至少一种磷脂酰胆碱,30~75%的至少二酰基甘油、至少一种生育酚或其混合物,5~30%的至少一种分子量低于 8000道尔顿的表面活性剂的离子组合物。但是该组合物最多只能引入20wt%的活性剂。此外为了增加载药量和降低注射组合物的粘度,其所表面活性剂含量较高,对于长效缓释的注射制剂而言存在着一定的溶血风险。
[0009]
综上所述,目前纳布啡癸二酸酯长效注射剂尚不能满足临床需求,对于载药量的提高,减少患者注射痛,避免产生溶血及过敏反应方面仍亟需改善。
技术实现要素:
[0010]
本发明提供一种纳布啡癸二酸酯的储库前体组合物,与目前现有技术中的纳布啡癸二酸酯的注射组合物相比,采用非油性制剂配方,具有更高的载药量和更少的注射体积,减少注射部位疼痛,减少过敏反应的发生,提高疼痛管理中患者用药顺应性;此外具有更低的表面活性剂成分,减少血管刺激性和溶血风险。
[0011]
本发明提供一种低粘度储库前体组合物,其包含下列组分:
[0012]
(a)、酰基甘油,所述酰基甘油为25~50重量%;
[0013]
(b)、磷脂酰胆碱(pc),所述磷脂酰胆碱为15~40重量%;
[0014]
(c)、至少一种含氧有机溶剂,所述含氧有机溶剂为15~40重量%;
[0015]
(d)、至少一种非离子型稳定性两亲物质,所述非离子型稳定性两亲物质为 0.1~1.5重量%;
[0016]
(e)、纳布啡癸二酸酯,所述纳布啡癸二酸酯至少20重量%;
[0017]
所述低粘度储库前体组合物的粘度为0.01~1pa
·
s(20℃);所述储库前体组合物的ph值范围为9~12;各组分(a)、(b)、(c)、(d)和(e)的重量百分比为相对于(a)、(b)、 (c)、(d)和(e)的总重量而言。
[0018]
本发明的一个实施例方案中,所述储库前体组合物中,包含下列组分:酰基甘油,磷脂酰胆碱(pc),至少一种含氧有机溶剂,至少一种非离子型稳定性两亲物质,纳布啡癸二酸酯;其中,所述酰基甘油为25~35重量%,所述pc为15~25重量%,所述含氧有机溶剂为20~40重量%;所述非离子型稳定性两亲物质为0.5~1重量%;所述纳布啡癸二酸酯为20~30重量%。
[0019]
在本发明的一个实施例方案中,所述(a)酰基甘油与(b)磷脂酰胆的总重占(a)、 (b)、(c)、(d)和(e)的总重比例为25~70重量%,优选30~60重量%,更优选35~55重量%。
[0020]
本发明的一个实施例方案中,所述酰基甘油包括至少50重量%的具有c12
‑
c18 酰基基团部分,所述酰基基团具有0~2个不饱和度。
[0021]
本发明的一个实施例方案中,所述低粘度储库前体组合物在20℃具有 0.01~1pa
·
s的粘度,优选具有0.01~0.5pa
·
s的粘度,更优选具有0.01~0.3mpa
·
s的粘度。
[0022]
本发明的一个实施例方案中,所述的储库前体组合物中,组分(b)磷脂酰胆碱与组分(a)酰基甘油的质量比例可在较宽的范围内配制前体组合物,组分(b)磷脂酰胆碱与组分(a)酰基甘油的质量比例范围可在0.3:1~1:1的范围内,优选0.4:1~0.8:1的范围内。更优选0.5:1~0.7:1的范围内。
[0023]
本发明的一个实施例方案中,所述的储库前体组合物中,其在适宜的ph范围内具有良好的稳定性,在ph值范围为9~12是适宜的,优选10~12,更优选10
‑
11。
[0024]
本发明的一个实施例方案中,含氧有机溶剂可显著降低储库前体组合物的粘度,所述含氧有机溶剂选自酰胺类,c1
‑
c3的醇类中的至少一种,所述酰胺类选自 n
‑
甲基吡咯烷酮(nmp)、二甲基甲酰胺、2
‑
吡咯烷酮和二甲基乙酰胺中的至少一种,优选n
‑
甲基吡咯烷酮;所述c1
‑
c3的醇类选自乙醇或异丙醇中的至少一种,优选乙醇。
[0025]
本发明的一个实施例方案中,所述含氧有机溶剂选自nmp或乙醇中的任意一种,优选nmp。
[0026]
本发明的一个实施例方案中,通过研究发现,纳布啡癸二酸酯在nmp中稳定性优于其他含氧有机溶剂,其中nmp的优选为25~40重量%,更优选30~40重量%。
[0027]
本发明的一个实施例方案中,组分(a)酰基甘油包括“头部基团”甘油部分和独立地2~3个“尾部基团”酰基基团部分,在本发明中,所述酰基甘油为二酰基甘油、三酰基甘油或二者的混合物;特别地,优选二酰基甘油。
[0028]
本发明的一个实施例方案中,组分(a)酰基甘油可以包括至少50重量%的具有这样的酰基基团的部分。
[0029]
本发明的一个实施例方案中,所述酰基甘油包括至少50重量%的具有c12
‑
c24 酰基基团部分,所述酰基基团具有0~9个不饱和度。
[0030]
本发明的一个实施例方案中,所述酰基甘油包括至少50重量%的具有c12
‑
c18 酰基基团部分,所述酰基基团具有0~2个不饱和度。
[0031]
本发明的一个实施例方案中,所述酰基甘油包括至少50重量%的具有c16
‑
c18 酰基基团部分,所述酰基基团具有0~2个不饱和度。
[0032]
本发明的一个实施例方案中,所述“尾部基团”的选择是独立的,即酰基基团可具有相同或不同的碳原子,且各自独立地是饱和或不饱和的,在本发明中,所述酰基甘油的酰基基团选自月桂酰基(c12:0)、肉豆蔻酰基(c14:0)、棕榈酰基(c16:0)、植烷酰基(phytanoly)(c16:0)、棕榈油酰基(c16:1)、硬脂酰基(c18:0)、油酰基(c18:1)、反油酸酰基(elaidoyl)(c18:1)、亚油酰基(linoleoyl)(c18:2)、亚麻酰基 (linolenoyl)(c18:3)、花生四烯酰基(c20:4)、山嵛酰基(c22:0)和二十四碳酰基(c24:9),优选油酰基。
[0033]
本发明的一个实施例方案中,所述酰基基团基于天然脂肪酸,包括但不限于月桂酸、肉豆蔻酸、棕榈酸、植烷酸、棕榈炔酸、硬脂酸、油酸、反油酸、亚油酸、亚麻酸、花生四烯酸、山嵛酸或二十四碳酸。优选的酰基基团的来源是棕榈酸、硬脂酸、油酸和亚麻酸,特别地,优选油酸。
[0034]
本发明的一个实施例方案中,所述酰基甘油至少包括一部分二酰基甘油,也可以是单一的二酰基甘油;其中二酰基甘油包含至少50%的二油酸甘油酯(gdo),优选包含至少80%的gdo,更优选基本上全部都为gdo的二酰基甘油。
[0035]
本发明的一个实施例方案中,所述非离子型两亲物质包括但不限于脂肪酸甘油酯类(例如脂肪酸单甘油酯、脂肪酸二甘油脂),多元醇类(例如蔗糖脂肪酸酯类、脂肪酸山梨坦类、聚山梨酯类、烯基乙二醇酯类),聚氧乙烯类(例如聚氧乙烯脂肪酸酯类、聚氧乙烯脂肪醇醚类、聚氧乙烯脂肪胺类),聚氧乙烯
‑
聚氧丙烯共聚物类中的至少一种。
[0036]
本发明的一个实施例方案中,所述非离子型两亲物质包括但不限于司盘类(山梨
醇酐脂肪酸酯类,span),吐温类(聚山梨酯类,tween),卖泽类(聚氧乙烯脂肪酸酯类,myrij),苄泽类(聚氧乙烯与月桂醇缩合物,brij),西土马哥类(聚乙二醇与十六醇缩合物,cetomacrogol),平平加o类(氧乙烯与油醇缩合物,preogolo),埃莫尔弗类(聚氧乙烯蓖麻油类,emlphor),泊洛沙姆类(聚氧乙烯与聚氧丙烯嵌段共聚物类,poloxamer),聚乙二醇类(peg)中的至少一种。
[0037]
本发明的其他实施例方案中,所述非离子型两亲物质选自司盘类、吐温类、聚乙二醇类、泊洛沙姆类、苄泽类、卖泽类或聚氧乙烯蓖麻油类中的至少一种。
[0038]
本发明的一个实施例方案中,所述非离子型两亲物质选自司盘类、吐温类、泊洛沙姆类或聚氧乙烯蓖麻油类中的至少一种,优选司盘类或吐温类中的至少一种。
[0039]
本发明的一个实施例方案中,所述司盘类包括但不限于司盘20、司盘40、司盘 60、司盘65、司盘80或司盘85;所述吐温类包括但不限于吐温20、吐温40、吐温60、吐温80或吐温85;泊洛沙姆类包括但限于泊洛沙姆101、泊洛沙姆105、泊洛沙姆108、泊洛沙姆122、泊洛沙姆123、泊洛沙姆124、泊洛沙姆181、泊洛沙姆182、泊洛沙姆183、泊洛沙姆184、泊洛沙姆185、泊洛沙姆188、泊洛沙姆212、泊洛沙姆215、泊洛沙姆217、泊洛沙姆231、泊洛沙姆234、泊洛沙姆235、泊洛沙姆237、泊洛沙姆238、泊洛沙姆282、泊洛沙姆284、泊洛沙姆288、泊洛沙姆331、泊洛沙姆333、泊洛沙姆334、泊洛沙姆335、泊洛沙姆338、泊洛沙姆401、泊洛沙姆402、泊洛沙姆403或泊洛沙姆407;聚氧乙烯蓖麻油类选自聚氧乙烯蓖麻油el
‑
10、聚氧乙烯蓖麻油el
‑
20、聚氧乙烯蓖麻油el
‑
40、聚氧乙烯蓖麻油el
‑
60或聚氧乙烯蓖麻油c
‑
125。
[0040]
本发明的一个实施例方案中,所述所述司盘类选自司盘20、司盘40、司盘60、司盘65、司盘80或司盘85中的至少一种,优选司盘20或司盘80中的至少一种;所述吐温类选自吐温20、吐温40、吐温60、吐温80或吐温85中的至少一种,优选吐温20 或吐温80中的至少一种。
[0041]
本发明的一个实施例方案中,可以来自天然来源,也可以是人工合成,但优选天然来源。适当的磷脂来源包括蛋、心脏(例如牛心等)、脑、肝(例如牛肝)和植物来源,包括大豆。这些来源可以提供一种或多种磷脂酰胆碱,可以是单一使用的也可以混合使用。
[0042]
本发明的一个实施例方案中,所述磷脂酰胆碱(磷脂,pc)选自卵pc、心pc、脑pc、肝pc、蛋黄pc或大豆pc中的至少一种,优选卵pc、蛋黄pc或大豆pc中的至少一种,更优选大豆pc。
[0043]
本发明提供一种低粘度储库前体组合物,其包含下列组分:
[0044]
(a)、二油酸甘油酯,所述二油酸甘油酯为25~35重量%;
[0045]
(b)、大豆pc,所述大豆pc为15~25重量%;
[0046]
(c)、n
‑
甲基吡咯烷酮,所述n
‑
甲基吡咯烷酮为25~40重量%;
[0047]
(d)、吐温,所述吐温为0.1~1重量%;
[0048]
(e)、纳布啡癸二酸酯,所述纳布啡癸二酸酯20~30重量%;
[0049]
其中,大豆pc与二油酸甘油酯的质量比选自0.5:1~0.7:1,所述吐温选自吐温20 或吐温80中的至少一种;所述低粘度储库前体组合物的粘度为0.01~0.3pa
·
s(20℃);各组分(a)、(b)、(c)、(d)和(e)的重量百分比为相对于(a)、(b)、(c)、(d)和(e)的总重量而言。
[0050]
本发明提供一种低粘度储库前体组合物,其包含下列组分:
[0051]
(a)、二油酸甘油酯,所述二油酸甘油酯为25~35重量%;
[0052]
(b)、大豆pc,所述大豆pc为15~25重量%;
[0053]
(c)、n
‑
甲基吡咯烷酮,所述n
‑
甲基吡咯烷酮为30~40重量%;
[0054]
(d)、吐温,所述吐温为0.5~1重量%;
[0055]
(e)、纳布啡癸二酸酯,所述纳布啡癸二酸酯20~30重量%;
[0056]
其中,大豆pc与二油酸甘油酯的质量比选自0.5:1~0.7:1,所述吐温选自吐温20 或吐温80中的至少一种;所述低粘度储库前体组合物的粘度为0.01~0.3pa
·
s(20℃);各组分(a)、(b)、(c)、(d)和(e)的重量百分比为相对于(a)、(b)、(c)、(d)和(e)的总重量而言。
[0057]
本发明的实施例方案中,参数范围选择包括端点值,例如25~50重量%包括端点值“20%”和“50%”;比值范围0.3:1~1:1包括“0.3:1”和“1:1”。
[0058]
本发明所提供的低粘度储库前体组合物,关键在于可减少特定剂量的纳布啡癸二酸酯的注射体积,在一个实施例方案中,本发明所述的低粘度储库前体组合物至少包括150mg/ml的纳布啡癸二酸酯,优选150~500mg/ml的纳布啡癸二酸酯,更优选200~300mg/ml的纳布啡癸二酸酯,高载药量的储库前体组合物有利于减少注射体积。
[0059]
本发明所提供的储库前体组合物,优选作为一种预制剂进行使用,本发明的储库前体组合物优选通过胃肠外施用。该施用通常通过皮下注射或肌肉注射进行施用。
[0060]
本发明所提供的储库前体组合物,因为独特的高载药量的特性,注射体积优选不超过3ml/每次施用,更优选不超过2ml/每次施用,最优选不超过1ml/每次施用。
[0061]
本发明所提供的储库前体组合物,一般充填于预填充装置进行临床应用,例如注射器。在一个实施例方案中,因本发明所提供的储库前体组合物具有较低的粘度,因而具有良好的通针性。本发明的储库前体组合物可以顺利通过国际通用标准注射器针头,也可通过各种细针孔,例如18g或更小的针孔,23g或更小的针孔,27g 或更小的针孔。
[0062]
本发明的一个实施例方案中,所述预填充装置包括但不限于具有1个或多个填充腔室的装置,优选使用1个或2个填充腔室的预填充装置,具有2个填充腔室的预填充装置如爱尔特株式会社的注射填充装置可以考虑使用。
[0063]
在本发明中,国际通用标准注射器针头18g、23g、27g尺寸如下所示:
[0064][0065]
本发明进一步提供储库前体组合物的制备方法,包含以下方法中的任意一种:
[0066]
方法一
[0067]
将处方比例的塞纳布啡、磷脂酰胆碱、酰基甘油、非离子型稳定性两亲物质溶解于含氧有机溶剂中,制备得到纳布啡癸二酸酯储库前体组合物。
[0068]
本发明的一个实施例方案中,按照上述制方法一制备得到的纳布啡癸二酸酯储库前体组合物一般呈混悬状。
[0069]
方法二
[0070]
(1)取处方比例的磷脂酰胆碱和酰基甘油混合,取约一半处方量的含氧有机溶剂,将磷脂酰胆碱和酰基甘油混合物溶于含氧有机溶剂;
[0071]
(2)取处方比例的塞纳布啡,非离子型稳定性两亲物质,溶解于剩余处方量的含氧有机溶剂;
[0072]
(3)临用前将(1)和(2)制备所得物混匀得到纳布啡癸二酸酯储库前体组合物。
[0073]
在本发明中,所述方法二中的(1)和(2)的制备不分先后。
[0074]
在本发明的一个实施例方案中,按照方法二中所述方法制备得到的纳布啡癸二酸酯储库前体组合物可在至少2小时内维持澄清透明溶液状态,优选至少5小时内维持澄清透明溶液状态,更优选至少10小时内维持澄清透明溶液状态。
[0075]
在本发明所述的制备方法一和方法二中,所述磷脂酰胆碱选自卵pc、心pc、脑pc、肝pc、蛋黄pc或大豆pc中的至少一种,优选卵pc、蛋黄pc或大豆pc中的至少一种,更优选大豆pc。
[0076]
在本发明所述的制备方法一和方法二中,所述酰基甘油优选二酰基甘油,更优选二油酸酰基甘油。
[0077]
在本发明所述的制备方法一和方法二中,所述含氧有机溶剂选自酰胺类,c1
‑ꢀ
c3醇类溶剂中的至少一种,所述酰胺类选自n
‑
甲基吡咯烷酮(nmp)、二甲基甲酰胺、2
‑
吡咯烷酮和二甲基乙酰胺中的至少一种,优选n
‑
甲基吡咯烷酮;所述c1
‑
c3 醇类选自乙醇或异丙醇中的至少一种,优选乙醇。
[0078]
在本发明所述的制备方法一和方法二中,所述含氧有机溶剂优选nmp。
[0079]
本发明的一些实施例方案中,本发明的制备方法还包含将所述制备得到的组合物以通过0.22μm无菌过滤膜的方式进行过滤除菌。
[0080]
本发明进一步提供一种纳布啡癸二酸酯储库前体组合物用于制备治疗疼痛药物中的用途。
[0081]
本发明的一个实施例方案中,所述疼痛选自外科手术疼痛或长期慢性疼痛。所述外科手术包括但不限于常见类型的普通外科手术,如疝气手术、痔疮手术、腹部手术、整形科手术、骨科手术、耳鼻喉科手术等。
[0082]
本发明的一个实施例方案中,所述长期慢性疼痛包括但不限于癌痛、慢性后背疼痛和慢性关节疼痛等。
[0083]
本发明有益的技术效果
[0084]
本发明提供一种纳布啡癸二酸酯的低粘度储库前体组合物,与目前现有技术中的纳布啡癸二酸酯的注射组合物相比,采用非油性制剂配方,具有更高的载药量和更少的注射体积,减少注射部位疼痛,减少过敏反应的发生,提高疼痛管理中患者用药顺应性;此外具有更低的表面活性剂成分,减少血管刺激性和溶血风险。
附图说明
[0085]
图1为纳解疼、处方f1
‑
f3、v8和v11的溶出曲线对比图。
实施例
[0086]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。除非另外指明,本文所指的比例、百分比等均以重量计。
[0087]
试剂
[0088]
塞纳布啡(来源:自制);纳布啡(来源:赛诺菲);大豆pc(来源:沈阳天峰生物制药有限公司);二油酸甘油酯(来源:英国croda公司);吐温20(来源:英国croda公司);nmp(来源:印度finar公司);纳疼解(来源:顺天医药生技股份有限公司)
[0089]
设备
[0090]
粘度计:snb
‑
1,上海天美;
[0091]
溶出仪:logan 850dl全自动溶出取样系统;
[0092]
高效液相色谱:agilent 1260 infinity液相色谱仪;
[0093]
ph计:mettler toledo fiveeasy plus
[0094]
实施例1溶解度测试
[0095]
在本发明中,本发明经过研究发现(参照中国药典2015版四部<凡例>溶解度测定方法),所述的储库前体组合物中的活性成分纳布啡癸二酸酯与其代谢物纳布啡差异较大,纳布啡癸二酸酯与纳布啡在不同缓冲液中的溶解度不同,具体溶解度差异如下表1所示。
[0096]
表1 塞纳布啡与纳布啡溶解度差异
[0097][0098]
结果
[0099]
从上述表1内容可知,纳布啡和塞纳布啡在模拟人体缓冲液中,溶解度差异较大,而活性成分的溶解度对于制剂处方研究中具有重要意义,因此在研究相关制剂处方时纳布啡和塞纳布啡之间的可参考意义不大。
[0100]
实施例2溶剂对塞纳布啡稳定性的影响
[0101]
称量适量塞纳布啡,用ph3.8的醋酸盐缓冲液配置成约3μg/ml,加入2%(v/v) 乙醇或nmp,分别在0min、10min、30min、60min进行hplc检测,hplc方法同如下所述。
[0102]
液相色谱检测(安捷伦1260),色谱柱zorbax sb
‑
aq,4.6
×
150,5μm。色谱条件:a相(称取3.85g乙酸铵加入到1000ml超纯水中,超声溶解,加入3ml 乙酸,过0.45μm滤膜,超声15min后待用)
‑
b相(乙腈)=40%:60%;柱温箱: 40℃;进样器:25℃;流速:1.5ml/min;波长:278nm。进样量10μl,按外标法,以不同取样时间点的峰面积相对于0min的峰面积百分比,通过峰面积计算塞纳布啡在不同时间点的含量。结果如下表2和表3所示。
[0103]
表2 塞纳布啡在nmp中的含量变化
[0104]
时间塞纳布啡峰面积(nmp)峰面积百分比0min917.312100.0%10min910.23799.2%30min923.139100.6%60min915.09199.8%
[0105]
表3 塞纳布啡在乙醇中的含量变化
[0106]
时间塞纳布啡峰面积(乙醇)峰面积百分比0min919.367100.0%10min910.12399.0%30min891.65997.0%60min844.87091.9%
[0107]
结果
[0108]
通过上述表2和表3的结果可知,塞纳布啡在乙醇中不稳定,第60分钟后含量下降显著,仅为91.9%,提示塞纳布啡在乙醇中容易降解产生杂质;而塞纳布啡在nmp中的含量稳定,第60分钟的测定值几乎与最初测定值(第0分钟)无任何变化,提示塞纳布啡在nmp中稳定,nmp为适宜塞纳布啡的溶剂。
[0109]
实施例3塞纳布啡储库前体组合物的制备方法
[0110]
方法1
[0111]
称量处方比例的塞纳布啡、大豆磷脂、二油酸甘油酯和吐温20溶解在溶剂nmp 中,制备得到塞纳布啡储库前体组合物,此法配置的液体呈混悬状。
[0112]
方法2
[0113]
(1)称量处方比例的大豆磷脂、二油酸甘油酯,并混合,取约一半处方量的 nmp,将大豆磷脂、二油酸甘油酯混合物溶于nmp;
[0114]
(2)称量处方比例的塞纳布啡和吐温20、与剩余处方量的nmp混合,溶解;
[0115]
(3)使用前将(1)和(2)的所得液体混匀得到塞纳布啡储库前体组合物,此液体为澄清透明溶液状态。临用前混匀(1)和(2)进行配制得到塞纳布啡储库前体组合物,澄清透明溶液状态可保持10个小时。
[0116]
处方如下表4和表5所示。
[0117]
表4 系列处方1
[0118][0119]
[0120]
表5 系列处方2
[0121][0122]
结果
[0123]
表4处方用方法2配置成塞纳布啡储库前体组合物,表5的处方分别用方法1 和2配置成塞纳布啡储库前体组合物,结果发现按照方法1配置的全部为混悬液;用方法二配置全部为澄清透明状溶液,且可在10小时内保持澄清透明。
[0124]
实施例4通针性、粘度及载药量测试
[0125]
将按照实施例3制备得到的塞纳布啡储库前体组合物进行通针性测试,具体方法为将注射器放置于注射器支撑装置上,通过与力量感应元连接的柱型探头对注射器活塞施加压力,推动溶液通过注射器和针头,直到将溶液推出针头,计算溶液流出针头的平均力。可顺利通过者标记为“√”,不能通过者标记为
“×”
。系列处方2中f1
‑
f3的测试结果如下表6所示。
[0126]
表6 f1
‑
f3的通针测试结果
[0127][0128]
结果
[0129]
经过测试,在系列处方1中,处方v1
‑
v5过于粘稠,不能通过国际通用标准注射器针头27g;处方v6
‑
v11粘度较低,均可通过国际通用标准注射器针头27g。
[0130]
此外,在系列处方2中,如表6结果所示,无论按照方法1制备还是按照方法 2制备,处方f2均具有良好通针性,能通过27g针头;处方f3在方法1中未能通过27g,但在方法2下均能通过;而处方f1按照方法1制备不能通过27g和23g,在方法2下不能通过27g;可见包含吐温20可明显降低储库前体组合物粘度,降低注射难度,提高临床使用的顺应性。
[0131]
通过上述表4的结果也可知,虽然较高含量的nmp能够降低组合物的粘度,但如果处方中无非离子型两亲物质,则无法减低组合物的粘度到令人满意的程度 (v4 vs v5)。通过表4和表5的内容还可发现,为了提高载药量,大豆pc和 gdo的总重占组合物总重需要达到适宜的比例(v3
‑
a vs v5),一般而言为了使载药量不小于20%,大豆pc和gdo的总重占组合物总重比例不大于60%,nmp 的含量一般不低于20%。
[0132]
实施例5不同处方对溶出度影响的测试
[0133]
对按照处方f1、f2、f3、v8和v11制备所得产品(均按照实施例3中的方法2进行制备)进行溶出实验测试,具体方法为按照中国药典2015版四部《0931 溶出度与释放度测定
法》第一法(篮法)开展体外溶出实验。ph3.8醋酸盐缓冲液,溶出介质体积900ml,介质温度37
±
0.5℃,将相当于7.5mg塞纳布啡的制剂,取样量约50
‑
150μl,加入到溶出介质中,转速250rpm,取样时间点5min,30min, 60min,120min,180min,240min、360min,分别取释放介质10ml,用0.45μm孔径的滤膜滤过,取续滤液1ml作为供试品溶液。
[0134]
液相色谱检测(安捷伦1260),色谱柱zorbax sb
‑
aq,4.6
×
150,5μm。
[0135]
色谱条件:a相(称取3.85g乙酸铵加入到1000ml超纯水中,超声溶解,加入3ml乙酸,过0.45μm滤膜,超声15min后待用)
‑
b相(乙腈)=40%:60%;柱温箱:40℃;进样器:25℃;流速:1.5ml/min;波长:278nm。进样量10μl,按外标法,以峰面积计算塞纳布啡在不同时间点的溶出量。各时间点取样的供试品溶液的溶出量(wt%)具体结果见下表7所示。
[0136]
表7 f1
‑
f3、v8和v11处方的塞纳布啡溶出度
[0137]
编号5min30min60min120min240min360min纳疼解12.2%48.7%67.3%81.7%90.0%90.7%f17.2%23.7%45.6%60.1%70.9%74.1%f216.3%53.6%71.1%86.4%92.9%95.9%f35.2%33.7%55.6%70.1%80.9%84.1%v818.0%55.5%73.9%87.5%92.4%93.4%v119.0%45.5%61.9%76.5%84.4%90.9%
[0138]
结果
[0139]
通过上述表7和图1结果可知,处方f2、f3、v8和v11溶出度较好,在 360分钟时溶出度均可达到80%以上,和市售制剂纳疼解相当;尤其是处方f2、 v8和v11能够达到90%以上,甚至优于纳疼解,溶出曲线均匀平缓,无明显突释现象发生,有利于维持平稳的血药浓度。
[0140]
实施例6纳布啡在处方中的溶出度测试
[0141]
本发明为了进一步测试活性成分塞纳布啡和纳布啡区别、以及溶剂对溶出度的影响,对以下处方进行制备并测试纳布啡的溶出度,具体处方见下表8。
[0142]
表8 测试处方n1
‑
n2
[0143][0144]
上述处方n1
‑
n2按照实施例3中方法1进行制备。
[0145]
对按照处方n1和n2制备所得产品进行溶出实验测试,具体方法为按照中国药典2015版四部《0931溶出度与释放度测定法》第一法(篮法)开展体外溶出实验。ph3.8醋酸盐缓冲液,溶出介质体积900ml,介质温度37
±
0.5℃,将相当于 7.5mg纳布啡的制剂,取样量
约50
‑
150μl,加入到溶出介质中,转速250rpm,取样时间点5min,30min,60min,120min,180min,240min、360min,分别取释放介质10ml,用0.45μm孔径的滤膜滤过,取续滤液1ml作为供试品溶液。
[0146]
液相色谱检测(安捷伦1260),色谱柱zorbax sb
‑
aq,4.6
×
150,5μm。
[0147]
色谱条件:a相(称取3.85g乙酸铵加入到1000ml超纯水中,超声溶解,加入3ml乙酸,过0.45μm滤膜,超声15min后待用)
‑
b相(乙腈)=40%:60%;柱温箱:40℃;进样器:25℃;流速:1.5ml/min;波长:278nm。进样量10μl,按外标法,以峰面积计算纳布啡在不同时间的溶出量。各时间点取样的供试品溶液的溶出量(wt%)具体结果见下表9所示。
[0148]
表9 n1和n2处方的纳布啡溶出度
[0149]
编号5min30min60min120min240min360minn13.2%15.7%45.6%60.1%62.3%62.0%n24.0%23.7%52.1%77.3%76.2%76.0%
[0150]
结果
[0151]
通过上述表9结果所示,处方n1和n2中的纳布啡在360min内无法完全溶出,可能原因是处方对纳布啡具有包裹作用,导致其无法彻底释放,因此纳布啡与塞纳布啡相比,纳布啡并不合适于本发明的储库前体组合物的配制;另外,处方n 2的纳布啡在各个采样时间点的溶出度要明显好于处方n1,可见nmp对于组合物中纳布啡的溶出度的提高明显优于乙醇。
[0152]
本领域技术人员会清楚,可以进行本发明的许多修改和变化而不背离其精神和范围。本文所述的具体实施方案仅通过实例的方式提供,并不意味着以任何方式限制。本发明的真正范围和精神通过所附权利要求书示出,说明书和实施例仅是示例性的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。