1.本发明涉及医药技术领域,特别是涉及一种复合树脂及其制备方法和用途。
背景技术:
2.我国成人患龋率高达90%,龋病有效治疗为是口腔医学领域的重点和难点。其中,对于中度或深度龋损的患牙通常采取在去除龋坏组织后,利用充填材料进行窝洞充填修复。而复合树脂由于具有操作简便、色泽美观、可操作性强及粘接固位效果好等优势,成为目前临床上最常用的牙体充填材料。然而,研究表明复合树脂充填物平均寿命只有6年且年失败率到达15%,其中复合树脂的继发龋是造成复合树脂充填修复失败的最主要原因,这主要是由于复合树脂修复体表面易积聚菌斑,菌斑分泌物造成牙齿表面发生脱矿,继而破坏牙齿结构并为细菌的侵入提供通道。因此,如何赋予复合树脂兼具抗菌性能和再矿化性能,从而有效降低复合树脂因继发龋导致的修复失败已经成为复合树脂研究领域对重点和难点问题。
3.如何降低继发龋、并能主动诱导脱矿的牙体组织再矿化是当前牙体充填材料领域的研究热点。目前,较多研究集中在对复合树脂的无机填料、树脂基质等方面进行改进:例如通过添加磷酸氢钙纳米颗粒、磷酸四钙纳米颗粒、或氟化钙纳米颗粒作为纳米填料,促进牙体缺损部分的再矿化;通过引入抗菌纳米颗粒填料、抗菌剂或抗菌单体来赋予复合树脂抗菌性能。然而,目前研究引入抗菌性能的手段大多数是通过掺杂抗菌剂或银离子,随着时间延长抗菌性能会迅速下降,还会降低复合树脂机械性能甚至增加细胞毒性风险。此外,目前的研究大多针对复合树脂抗菌性或再矿化性的单独研究,如何获得兼具抗菌性和再矿化性能对复合树脂却鲜有报道。
技术实现要素:
4.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种复合树脂及其制备方法和用途,其为兼具抗菌和再矿化性能的光固化复合树脂,为降低继发龋和牙体组织脱矿导致的复合树脂修复失败提供技术保障,用于解决现有技术中的问题。
5.为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
6.本发明提供一种复合树脂,所述复合树脂的原料组分包括生物活性玻璃微球、无机填料和树脂基质。
7.优选地,所述无机填料包括玻璃粉和气相二氧化硅纳米颗粒。
8.优选地,所述树脂基质包括双酚a
‑
甲基丙烯酸缩水甘油酯(bis
‑
gma)、二甲基丙烯酸二缩三乙二醇酯(tegdma)、光引发剂和二甲基氨基苯甲酸乙酯。
9.优选地,所述光引发剂为樟脑醌(cq)。
10.二甲基丙烯酸二缩三乙二醇酯(tegdma)为稀释剂。
11.优选地,所述生物活性玻璃微球的制备方法,包括:
12.将二氧化硅、碳酸钙、碳酸钠和五氧化二磷混合后烧结获得生物活性玻璃;
13.然后研磨并过滤得到所述生物活性玻璃微球。
14.优选地,研磨时采用球磨仪器。
15.优选地,生物活性玻璃微球的制备方法还包括:将研磨后的产物加入碱的水溶液中加热形成凝胶,然后将硬化的凝胶粉碎过滤。
16.更优选地,所述碱为氢氧化钠。更优选地,所述碱的水溶液的ph为10~14。更优选地,加热形成凝胶的加热温度为80~90℃。
17.更优选地,所述二氧化硅、碳酸钙、碳酸钠和五氧化二磷的质量比为(10~15):(5~7):(5~10):1。
18.更优选地,所述烧结的温度为900~1300℃。更优选地,900~1300℃下烧结1~4h。
19.更优选地,所述烧结工艺中先升温至900~1300℃,然后烧结。升温速率为400℃~500℃/h。
20.优选地,以所述复合树脂的原料组分的总质量为基准计,所述生物活性玻璃微球为2wt%~24wt%,所述树脂基质的含量为20~30wt%,所述无机填料的含量为46~78wt%。
21.优选地,所述生物活性玻璃微球的质量为8wt%~24wt%。
22.优选地,以所述树脂基质的总质量为基准计,所述双酚a
‑
甲基丙烯酸缩水甘油酯为58wt%~65wt%,二甲基丙烯酸二缩三乙二醇酯为28wt%~35wt%,光引发剂为1.5~2.5wt%,二甲基氨基苯甲酸乙酯为3.5~4.5wt%。
23.优选地,所述生物活性玻璃微球和所述玻璃粉表面均经过硅烷偶联剂进行偶联化处理。硅烷偶联化处理为参照现有技术,如分别将所述生物活性玻璃微球和/或所述无机填料与硅烷偶联剂在有机溶剂中接触并反应。更优选地,反应温度为50~70℃。
24.更优选地,所述硅烷偶联剂为γ
‑
mps。
25.优选地,所述无机填料包括玻璃粉和气相二氧化硅纳米颗粒时,所述玻璃粉与所述气相二氧化硅纳米颗粒的质量比为(15~24):1。
26.更优选地,以复合树脂的原料组分的总质量为基准计,所述复合树脂的原料组分包括2~24wt%生物活性玻璃微球,49~71wt%玻璃粉,1~3wt%气相二氧化硅纳米颗粒,树脂基质24wt%。
27.本发明还公开了如上述所述的复合树脂的制备方法,具体步骤如下:
28.在避光条件下,混合树脂基质的各组分形成树脂膏,然后加入生物活性玻璃微球和无机填料混合均匀。
29.优选地,采用离心搅拌实现混合均匀。
30.优选地,先将生物活性玻璃微球与所述无机填料混合均匀,然后加入树脂膏中。
31.本发明还公开了如上述所述的复合树脂用于作为牙体充填材料的用途。
32.优选地,在用于牙体充填材料时,先将所述复合树脂填充至牙体缺损部分,然后进行光固化。
33.优选地,所述光固化是指采用可见光进行照射,以引发所述复合树脂进行固化交联。
34.更优选地,可见光的波长范围为360~520nm。更优选地,可见光的波长范围为465~470nm。
35.本发明还公开了一种牙体充填材料,其采用如上述所述的复合树脂经光固化后获得。
36.本发明上述技术方案的有益效果为:
37.(1)本发明提供的含有生物活性玻璃微球的复合树脂能够抑制变形链球菌,降低菌落数量比例为32.54%~89.96%,表明该复合树脂具有良好的抗菌性能;
38.(2)本发明提供的含有生物活性玻璃微球的复合树脂可诱导脱矿牙本质表面形成片层状或较厚的再矿化层,几乎完全封闭牙本质小管,提示该复合树脂具有良好的诱导牙体组织再矿化性能;
39.(3)本发明提供的含有生物活性玻璃微球的复合树脂的弯曲强度可达103.33mpa~105.41mpa,高于iso4049:2019《牙科聚合物基修复材料》和yy 1042
‑
2011《牙科学聚合物基修复材料》标注规定,且压缩强度209.54mpa~232.16mpa,有效满足临床应用需求。
附图说明
40.图1为对比例1的直接抗菌效应结果图。
41.图2为实施例1直接抗菌效应结果图。
42.图3为实施例2直接抗菌效应结果图。
43.图4为实施例3直接抗菌效应结果图。
44.图5为实施例4直接抗菌效应结果图。
45.图6为实施例5直接抗菌效应结果图。
46.图7为实施例5直接抗菌效应结果图。
47.图8为对比例1诱导脱矿牙本质再矿化sem图。
48.图9为实施例1诱导脱矿牙本质再矿化sem图。
49.图10为实施例2诱导脱矿牙本质再矿化sem图。
50.图11为实施例3诱导脱矿牙本质再矿化sem图。
51.图12为实施例4诱导脱矿牙本质再矿化sem图。
52.图13为实施例5诱导脱矿牙本质再矿化sem图。
具体实施方式
53.以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
54.在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
55.当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实
现本发明。
56.生物活性玻璃为一种硅酸盐基生物材料,由二氧化硅、氧化钠、氧化钙、五氧化二磷等氧化物组成,其生物活性较高,在体液环境下能释放离子,一方面促进羟基磷灰石层的形成,另一方面可使局部环境的ph值升高,二者为杀灭细菌和诱导局部矿化提供了基础。
57.本技术中申请人提供一种复合树脂,所述复合树脂的原料组分包括生物活性玻璃微球、无机填料和树脂基质。
58.在一个优选的实施方式中,所述无机填料包括玻璃粉和气相二氧化硅纳米颗粒
59.在一个优选的实施方式中,所述树脂基质包括双酚a
‑
甲基丙烯酸缩水甘油酯(bis
‑
gma)、二甲基丙烯酸二缩三乙二醇酯(tegdma)、光引发剂和二甲基氨基苯甲酸乙酯。
60.在一个优选的实施方式中,所述光引发剂为樟脑醌(cq)。
61.二甲基丙烯酸二缩三乙二醇酯(tegdma)为稀释剂。
62.在一个优选的实施方式中,所述生物活性玻璃微球的制备方法,包括:
63.将二氧化硅、碳酸钙、碳酸钠和五氧化二磷混合后烧结获得生物活性玻璃;
64.然后研磨。在一个优选的实施方式中,研磨时采用球磨仪器。
65.在一个优选的实施方式中,生物活性玻璃微球的制备方法还包括:将研磨后的产物加入碱的水溶液中加热形成凝胶,然后将硬化的凝胶粉碎过滤。在一个更优选的实施方式中,所述碱为氢氧化钠。在一个更优选的实施方式中,所述碱的水溶液的ph为10~14。在一个更优选的实施方式中,加热形成凝胶的加热温度为80~90℃。
66.在一个优选的实施方式中,所述生物活性玻璃微球的粒径为45~75μm。
67.在一个优选的实施方式中,所述玻璃粉的粒径为0.7~~1μm。
68.在一个优选的实施方式中,所述二氧化硅、碳酸钙、碳酸钠和五氧化二磷的质量比为(10~15):(5~7):(5~10):1。
69.在一个优选的实施方式中,所述烧结的温度为900~1300℃。更优选地,900~1300℃下烧结1~4h。
70.在一个优选的实施方式中,所述烧结工艺中先升温至900~1300℃,然后烧结。升温速率为400℃~500℃/h。
71.在一个具体的实施方式中,0~400℃升温烧结1小时(升温速率为400℃/h),400~900℃升温烧结1小时(升温速率为500℃/h),900~1300℃升温烧结1小时(升温速率为500℃/h),1300℃恒温2小时,获得生物活性玻璃。
72.在一个优选的实施方式中,以所述复合树脂的原料组分的总质量为基准计,所述生物活性玻璃微球为2wt%~24wt%,所述树脂基质的含量为20~30wt%,所述无机填料的含量为46~78wt%。
73.在一个优选的实施方式中,所述生物活性玻璃微球的质量为8wt%~24wt%。
74.在一个优选的实施方式中,以所述树脂基质的总质量为基准计,所述双酚a
‑
甲基丙烯酸缩水甘油酯为58wt%~65wt%,二甲基丙烯酸二缩三乙二醇酯为28wt%~35wt%,光引发剂为1.5~2.5wt%,二甲基氨基苯甲酸乙酯为3.5~4.5wt%。
75.在一个优选的实施方式中,所述生物活性玻璃微球和所述无机填料表面均经过硅烷偶联剂进行偶联化处理。硅烷偶联化处理为参照现有技术,如分别将所述生物活性玻璃微球和/或所述无机填料与硅烷偶联剂在有机溶剂中接触并反应。更优选地,反应温度为50
~70℃。
76.在一个优选的实施方式中,所述硅烷偶联剂为γ
‑
mps。
77.在一个优选的实施方式中,所述无机填料包括玻璃粉和气相二氧化硅纳米颗粒时,所述玻璃粉与所述气相二氧化硅纳米颗粒的质量比为(15~24):1。
78.在一个优选的实施方式中,以复合树脂的原料组分的总质量为基准计,所述复合树脂的原料组分包括2~24wt%生物活性玻璃微球,49~71wt%玻璃粉,1~3wt%气相二氧化硅纳米颗粒,树脂基质24wt%。
79.在一个更优选的实施方式中,所述生物活性玻璃微球的含量为8~24wt%。在一个最优选的实施方式中,所述生物活性玻璃微球的含量为20~24wt%。
80.在一个更优选的实施方式中,所述玻璃粉的含量为49~65wt%。
81.在一个更优选的实施方式中,所述气相二氧化硅纳米颗粒的含量还可以为1wt%、2wt%或3wt%。
82.本发明还公开了如上述所述的复合树脂的制备方法,具体步骤如下:
83.在避光条件下,混合树脂基质的各组分形成树脂膏,然后加入生物活性玻璃微球和无机填料混合均匀。
84.在一个优选的实施方式中,先将生物活性玻璃微球与所述无机填料混合均匀,然后加入树脂膏中。
85.本发明还公开了如上述所述的复合树脂用于作为牙体充填材料的用途。
86.在一个优选的实施方式中,在用于牙体充填材料时,先将所述复合树脂填充至牙体缺损部分,然后进行光固化。
87.在一个优选的实施方式中,所述光固化是指采用可见光进行照射,以引发所述复合树脂进行固化交联。在一个更优选的实施方式中,可见光的波长范围为360~520nm。在一个更优选的实施方式中,可见光的波长范围为465~470nm。
88.本技术中的复合树脂具有良好的相容性和反应性。用于作为牙体充填材料能够有效的抑制变形链球菌,诱导牙体组织再矿化性能,而且其强度也不符合临床应用需求。
89.以下通过具体的试验及试验效果对本技术上述技术方案及其达到的技术效果进行进一步解释和说明。
90.在下述具体的试验中采用的生物活性玻璃微球的制备方法为:
91.称取9g二氧化硅、4.8g碳酸钙、4.9g碳酸钠、0.8g五氧化二磷,震荡混匀后转入坩埚备用;将上述坩埚移入马弗炉中,并按照下列程序进行烧结:0~400℃升温烧结1小时(400/℃
·
h
‑1),400~900℃升温烧结1小时(500/℃
·
h
‑1),900~1300℃升温烧结1小时(500/℃
·
h
‑1),1300℃恒温2小时,获得生物活性玻璃;采用研磨仪研磨;在1m naoh水溶液中,伴随在800rpm条件下机械搅拌在85℃下成胶24小时,并过滤制得生物活性玻璃微球。生物活性玻璃微球的粒径为45~75μm。
92.在下述具体的试验中对生物活性玻璃微球和对玻璃粉硅烷化处理的过程为:
93.将所述生物活性玻璃微球和所述无机填料加入到含有甲基丙烯酰氧基丙基三甲氧基硅烷(γ
‑
mps)的环已烷溶液中,并在室温下搅拌30分钟。然后在65℃条件下搅拌30分钟。反应完成后,利用旋转蒸发器除去溶剂,无水乙醇洗涤3次后在真空干燥条件制得硅烷化的生物活性玻璃微球/无机填料。
94.在下述具体的试验中对比例1和实施例1~6中采用的树脂胶液的各原料组分的含量百分含量为:
95.以所述树脂基质的总质量为基准计,所述双酚a
‑
甲基丙烯酸缩水甘油酯为62.5wt%,二甲基丙烯酸二缩三乙二醇酯为31.2wt%,光引发剂为2.1wt%,二甲基氨基苯甲酸乙酯为4.2wt%。
96.对比例1
97.本对比例中复合树脂没有采用生物活性玻璃微球。
98.包括如下制备方法:
99.将树脂单体双酚a
‑
甲基丙烯酸缩水甘油酯和稀释剂二甲基丙烯酸二缩三乙二醇酯按照所述比例混合均匀,并在严格避光条件下加入樟脑醌和二甲基氨基苯甲酸乙酯,充分搅拌均匀后,得到粘稠的树脂胶液;
100.将73wt%表面硅烷偶联化处理的玻璃粉、3wt%气相二氧化硅混合均匀后,逐次添加到24wt%粘稠树脂胶液中,在真空搅拌机中充分搅拌均匀,消除气泡,完成对比例复合树脂制备。
101.实施例1
102.将树脂单体双酚a
‑
甲基丙烯酸缩水甘油酯和稀释剂二甲基丙烯酸二缩三乙二醇酯按照所述比例混合均匀,并在严格避光条件下加入樟脑醌和二甲基氨基苯甲酸乙酯,充分搅拌均匀后,得到粘稠的树脂胶液;
103.将8wt%硅烷偶联化生物活性玻璃微球、65wt%硅烷偶联化玻璃粉、3wt%气相二氧化硅混合均匀后,逐次添加到24wt%粘稠树脂胶液中,在真空搅拌机中充分搅拌均匀,消除气泡,完得到具备抗菌和再矿化性能的复合树脂。
104.实施例2
105.如实施例1所述,不同的是:
106.硅烷偶联化生物活性玻璃微球添加量为12wt%,硅烷偶联化玻璃粉添加量为61wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
107.实施例3
108.如实施例1所述,不同的是:
109.硅烷偶联化生物活性玻璃微球添加量为16wt%,硅烷偶联化玻璃粉添加量为57wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
110.实施例4
111.如实施例1所述,不同的是:
112.硅烷偶联化生物活性玻璃微球添加量为20wt%,硅烷偶联化玻璃粉添加量为53wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
113.实施例5
114.如实施例1所述,不同的是:
115.硅烷偶联化生物活性玻璃微球添加量为24wt%,硅烷偶联化玻璃粉添加量为49wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
116.实施例6
117.如实施例1所述,不同的是:
118.硅烷偶联化生物活性玻璃微球添加量为2wt%,硅烷偶联化玻璃粉添加量为71wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
119.对比例2
120.如实施例1所述,不同的是:
121.硅烷偶联化生物活性玻璃微球添加量为28wt%,硅烷偶联化玻璃粉添加量为45wt%,气相二氧化硅添加量为3wt%,即上述无机填料占复合树脂质量比保持76wt%。
122.对比例3
123.如实施例1所示,不同的是:采用未经硅烷化处理的生物活性玻璃微球。
124.对比例4
125.如实施例1所示,不同的是:
126.采用的树脂胶液的各原料组分的含量百分含量为:以所述树脂基质的总质量为基准计,所述双酚a
‑
甲基丙烯酸缩水甘油酯为55wt%,二甲基丙烯酸二缩三乙二醇酯为38.7wt%,光引发剂为2.1wt%,二甲基氨基苯甲酸乙酯为4.2wt%。
127.对比例5
128.如实施例1所示,不同的是:
129.采用的树脂胶液的各原料组分的含量百分含量为:以所述树脂基质的总质量为基准计,所述双酚a
‑
甲基丙烯酸缩水甘油酯为68wt%,二甲基丙烯酸二缩三乙二醇酯为25.7wt%,光引发剂为2.1wt%,二甲基氨基苯甲酸乙酯为4.2wt%。
130.试验例1
131.利用直接接触法检测对比例1和实施例1~5的抑菌效应。
132.将对比例1~2和实施例1~5的复合树脂制作直径6mm、高度4mm的圆柱形试样(n=3),消毒备用。细胞培养板每孔中加入含0.2ml变形链球菌(1
×
105cfu/ml)2ml的bhi培养基,并分别于对比例和实施例1~5的复合树脂试样接触,37℃厌氧孵育24h后将试件取出,将粘附于试件表面的细菌完全洗脱,将洗脱液梯度稀释后,取100μl稀释液接种于bhi琼脂平板,拍照后利用image
‑
j软件进行菌落计数并分析。
133.图1~图7依次分别为对比例1和实施例1~6的直接抗菌效应图。
134.对比例1和实施例1~6菌落抑制数量和抑制比例如表1所示。
135.表1
[0136] 菌落数(个)抑制比例(%)对比例11178.67
±
7.50/实施例1795.15
±
6.2832.54
±
2.15实施例2748.33
±
7.3736.51
±
2.71实施例3416.33
±
9.2464.68
±
4.87实施例4266.16
±
8.2177.42
±
4.26实施例5181.33
±
1.5889.96
±
0.37实施例61166.78
±
9.241.06
±
0.59
[0137]
*
p<0.05vs.对比例1
[0138]
结合对比例1和实施例1~6抗菌效应的结果证明(图1~7,表1),引入较低含量的生物活性玻璃微球(2wt%)并不能显著抑制菌落数量(p>0.05);当生物活性玻璃微球含量
在8wt~24wt%时,,复合树脂可显著降低变形链球菌的菌落数量(p<0.05),提升复合树脂抑菌比例,且随着生物玻璃微球含量的增加,复合树脂的抑菌效果逐渐升高,具有浓度依赖性关系,提示引入适宜质量分数的生物活性玻璃微球可有效提高复合树脂的抗菌性能。
[0139]
试验例2
[0140]
对比例1和实施例1~5的脱矿牙本质再矿化试验。
[0141]
分别将对比例和实施例1~5制成直径6mm、厚度4mm的试样圆片备用。
[0142]
选择因正畸需要拔除的第一前磨牙,利用环氧树脂包埋固定,用硬组织切片机在每颗牙齿的釉牙骨质界下方沿水平方向切成厚度为1mm牙齿切片。将上述牙齿切片用0.5%次氯酸钠清洗5分钟,然后用蒸馏水和70%酒精交替冲洗后,浸入10%磷酸溶液12h进行脱矿。将上述脱矿后的牙齿切片分别与对比例和实施例1~5试样圆片浸入到模拟体液(sbf)中【各组sbf加入量根据公式vs=sa/10计算,其中vs=spf加入体积(ml),sa=浸泡样品表面积(mm2)】,上述各实验组在37℃、sbf中连续浸泡21天后,取出样品常温干燥,利用扫描电镜观察牙体切片的再矿化情况。
[0143]
图8~图13依次分别为对比例1和实施例1~5的诱导脱矿牙本质再矿化sem图。
[0144]
实施例1~5诱导脱矿牙本质再矿化的结果(图9~13)显示:脱矿牙本质片在与对比例接触后,其牙本质小管清晰可见,且未见磷灰石晶体沉积;脱矿牙本质片在分别与实施例1~5接触后,各组牙本质表面均可见部分矿化沉积,其中脱矿牙本质片分别在与实施例4、实施例5组接触后,可见牙本质表面出现大量矿化沉浸颗粒融合成片、矿化层逐渐便后、甚至是牙本质小管被完全封闭,表明生物活性玻璃微球可有效提升复合树脂的再矿化性能。
[0145]
试验例3
[0146]
对比例1~4和实施例1~5的力学性能测定
[0147]
根据iso 4049:2020要求,将对比例和实施例1~5按照:长(l)
×
高(h)
×
宽(b)=25mm
×
2mm
×
2mm的尺寸制成标准测试试件(n=5)。待试件完全固化后,利用万能力学试验机检测试样的弯曲强度,实验参数设定:跨距(l)=20mm,加载速度为0.5mm/min,直至试件断裂,并记录断裂时的最大载荷pmax(n)。弯曲强度(s)根据公式:s(mpa)=3pmaxl/(2bh2),计算对比例和实施例1~5的弯曲强度。
[0148]
根据iso 4049:2020要求,将对比例和实施例1~5制成直径4mm、高6mm圆柱形试件(n=5)。待试件完全固化后,在万能材料试验机上装夹试件,进行压缩强度测试,其中加载头加载速度为1mm/min,记录试件破裂时的破坏载荷f,其中压缩强度(cs)根据公式:cs(mpa)=f/πr2,计算对比例和实施例1~5的压缩强度。
[0149]
对比例1~4和实施例1~5的弯曲强度和压缩强度结果如表2所示。
[0150]
表2
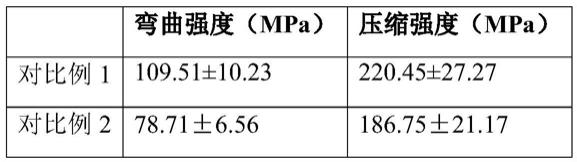
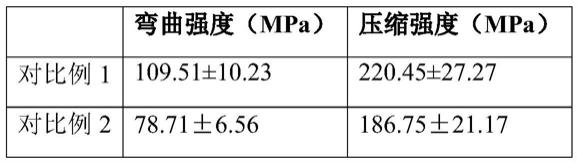
[0151]
[0152][0153]
结果显示含适宜比例(8~24wt%)的硅烷化处理生物活性玻璃微球,可有效均匀分散到复合树脂中,并通过自身硅烷化保证了复合树脂力性能的临床要求。而当生物活性玻璃微球未经硅烷化处理或硅烷化处理的生物活性玻璃微球比例过高时,导致其不能有效分散复合树脂的有机基质中,造成复合树脂的弯曲强度和压缩强度发生显著下降,且低于标准要求的80mpa。此外,对比例4~5也显示,当调整树脂胶液的比例时,也会造成复合树脂的力学性能的下降,不符合临床应用的要求。
[0154]
注:iso4049:2019《牙科聚合物基修复材料》和yy 1042
‑
2011《牙科学聚合物基修复材料》标注规定,对于适用于牙合面修复的聚合物基充填和修复材料,其挠曲强度不应低于80mpa。
[0155]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。