利用ii类mhc模型鉴别新抗原
背景技术:
1.基于肿瘤特异性新抗原的治疗性疫苗和t细胞疗法作为下一代个性化癌症免疫疗法具有广阔的前景。1–3鉴于产生新抗原的可能性相对较高,具有高突变负荷的癌症,如非小细胞肺癌(nsclc)和黑素瘤成为此类疗法的特别值得关注的靶标。
4,5
早期有证据显示,基于新抗原的疫苗接种能够引起t细胞应答6并且靶向新抗原的t细胞疗法在某些情况下能够在选择的患者中引起肿瘤消退。72.特别地,用于基于新抗原的疫苗接种和新抗原靶向性t细胞疗法的ii类mhc呈递的新抗原的鉴别是一种有希望的治疗,因为高达50%的新抗原反应性til包含cd4细胞,所述细胞对由ii类mhc等位基因呈递的新抗原具有响应。这些cd4细胞已被证明有助于cd8细胞的抗肿瘤反应,并在一些情况下直接攻击肿瘤细胞。尽管存在ii类mhc呈递的新抗原用于癌症治疗的该很有前景的潜力,但ii类mhc呈递的新抗原的阳性预测值(ppv)低于cd8细胞识别的mhc i类呈递的新抗原的ppv。
3.ii类mhc呈递的新抗原的这些相对较差的呈递预测结果可能部分归因于ii类mhc分子相对于i类mhc分子的结构。具体地,相对于i类mhc分子,ii类mhc分子往往具有更开放的肽结合沟。由于这种结构上的差异,i类mhc分子倾向于结合长度为8
‑
11个氨基酸的肽,而ii类mhc分子结合长度更加可变的肽(图14f)。由于ii类mhc分子呈递的肽长度的可变性,相对于i类mhc分子呈递的肽,ii类mhc分子呈递的肽可能更难预测。
4.因此,ii类mhc呈递的新抗原和识别新抗原的t细胞已成为评估肿瘤响应
77,110
、检查肿瘤进展
111
和设计下一代个性化疗法
112
的主要挑战。目前的新抗原鉴别技术是费时和费力的
84,96
,或者不够精确87,
91
–
93
。此外,尽管最近已证明识别新抗原的t细胞是til的主要成分
84,96,113,114
并且在癌症患者的外周血中循环
107
,但目前用于鉴别新抗原反应性t细胞的方法具有以下三个局限性的组合:(1)其依赖于难以获得的临床试样,例如til
97,98
或白细胞分离术(leukaphereses)
107
(2)其需要筛选不切实际的大肽库
95
或(3)其依赖于mhc多聚体,这实际上只对很小数量mhc等位基因数可用。
5.另外,提出的初步方法并入了使用下一代测序的基于突变的分析、rna基因表达及候选新抗原肽的mhc结合亲和力预测8。然而,提出的这些方法都无法模拟整个表位产生过程,该过程除含有基因表达和mhc结合外,还含有许多步骤(例如tap转运、蛋白酶体裂解、mhc结合、将肽
‑
mhc复合物转运至细胞表面和/或tcr对mhc的识别;内吞或自噬、通过细胞外或溶酶体蛋白酶(例如组织蛋白酶)裂解、并且/或者与clip肽竞争hla
‑
dm催化的hla结合)9。因此,现有的方法可能会有低阳性预测值(ppv)降低的问题。(图1a)
6.事实上,多个研究团队所进行的关于由肿瘤细胞呈递的肽的分析显示,预计使用基因表达和mhc结合亲和力呈递的肽中不到5%可以在肿瘤表面mhc上发现
10,11
(图1b)。近期观察到的仅针对突变数量的检查点抑制剂反应无法提高对结合受限的新抗原的预测准确性进一步支持了结合预测与mhc呈递之间的这一低相关性。
12
这些呈递预测的失败在ii类mhc等位基因呈递的新抗原的情况下尤其如此。
7.现有的呈递预测方法的这一低阳性预测值(ppv)提出了有关基于新抗原的疫苗设
计和基于新抗原的t细胞疗法的问题。如果使用ppv低的预测方法来设计疫苗,则大多数患者不太可能接受治疗性新抗原,且少数患者可能要接受一种以上新抗原(即使假设所有呈递的肽都具有免疫原性)。同样,如果治疗性t细胞是基于低ppv的预测设计的,则大多数患者不太可能接受对肿瘤新抗原具有反应性的t细胞,以及使用下游实验室技术在预测后鉴别预测性新抗原的时间和物理资源成本可能过高。因此,用当前方法进行新抗原疫苗接种和t细胞疗法不太可能在众多具有肿瘤的受试者中取得成功。(图1c)
8.此外,先前的方法仅使用顺式作用突变来产生候选新抗原,而在很大程度上忽视了考虑neo
‑
orf的其它来源,包括在多种肿瘤类型中出现且导致许多基因异常剪接的剪接因子突变
13
,及产生或移除蛋白酶裂解位点的突变。
9.最后,由于文库构建、外显子组和转录组捕捉、测序或数据分析的条件并非最佳条件,故肿瘤基因组和转录组分析的标准方法可能会遗漏产生候选新抗原的体细胞突变。同样,标准肿瘤分析方法可能会无意中促成序列伪影或生殖系多态现象作为新抗原,而分别导致疫苗能力的低效使用或自身免疫的风险。
技术实现要素:
10.本文公开了一种鉴别和选择用于个性化癌症疫苗、用于t细胞疗法或其二者的由ii类mhc等位基因呈递的新抗原的优化方法。首先,提出了使用下一代测序(ngs)鉴别新抗原候选物的优化的肿瘤外显子组和转录组分析方法。这些方法建立在标准ngs肿瘤分析方法的基础之上,以确保在所有类别的基因组变化内推进最高敏感性和特异性的新抗原候选物。其次,提出了选择高ppv ii类mhc等位基因呈递的新抗原的新颖方法来克服特异性问题并确保打算包括在疫苗中和/或作为t细胞疗法的靶标的ii类mhc等位基因呈递的新抗原较大可能地引发抗肿瘤免疫。取决于实施方案,这些方法包括训练的统计回归或非线性深度学习ii类mhc模型,这些模型共同地模拟肽
‑
ii类mhc等位基因定位以及多种长度的肽的独立ii类mhc等位基因基元(per
‑
mhc class ii allele motif),在不同长度的肽中共有统计强度。非线性ii类mhc深度学习模型可以专门设计和训练用于将同一细胞中的不同mhc等位基因处理为独立的,由此解决了线性模型所具有的不同mhc等位基因会相互干扰的问题。最后,解决了基于ii类mhc等位基因呈递的新抗原的个性化疫苗设计和制造和用于t细胞疗法的个性化ii类mhc等位基因呈递的新抗原特异性t细胞产生的其它需要考虑的问题。
11.本文公开的模型优于在结合亲和力上训练的最新预测器和基于ms肽数据的早期预测器多达一个数量级。通过更可靠地预测ii类mhc等位基因的肽呈递,该模型可以使用临床实践方法以更具时间和成本效益的方式鉴别用于个性化疗法的ii类mhc等位基因呈递的新抗原特异性或肿瘤抗原特异性t细胞,其使用有限量的患者外周血,每位患者筛查少量的肽,并且不一定依赖于mhc多聚体。然而,在另一个实施方案中,本文公开的模型可通过减少为了鉴别ii类mhc等位基因呈递的新抗原或肿瘤抗原特异性t细胞而需要筛选的与mhc多聚体结合的肽的数目,来使用mhc多聚体以更具时间和成本效益的方式鉴别ii类mhc等位基因呈递的肿瘤抗原特异性t细胞。
12.本文公开的ii类mhc模型在til新表位数据集上的预测性能和前瞻性新抗原反应性t细胞鉴别任务证明,现在有可能通过对ii类mhc等位基因处理和呈递进行建模来获得治疗上有用的ii类mhc等位基因呈递的新表位预测。总而言之,这项工作为ii类mhc等位基因
呈递的抗原靶向免疫治疗提供了实用的计算机模拟ii类mhc等位基因呈递的抗原鉴别,从而加快了治愈患者的进程。
附图说明
13.参照以下描述和附图将更好地理解本发明的这些和其它特征、方面及优势,在附图中:
14.图1a显示当前用于鉴别新抗原的临床方法。
15.图1b显示<5%的预测结合肽被呈递在肿瘤细胞上。
16.图1c显示新抗原预测特异性问题的影响。
17.图1d显示结合预测不足以进行新抗原鉴别。
18.图1e显示mhc
‑
i呈递的机率随肽长度的变化。
19.图1f显示由promega动态范围标准(dynamic range standard)生成的示例性肽谱。
20.图1g显示添加特征如何增加模型阳性预测值。
21.图2a是根据一个实施方案,用于鉴别患者体内肽呈递的可能性的环境的概述。
22.图2b和2c示出了根据一个实施方案的获得呈递信息的方法。
23.图3是一个高级框图,示出了根据一个实施方案的呈递鉴别系统的计算机逻辑组件。
24.图4示出了根据一个实施方案的一组示例训练数据。
25.图5示出了与mhc等位基因相关联的示例网络模型。
26.图6a示出了根据一个实施方案的mhc等位基因共享的示例网络模型nn
h
(
·
)。
27.图6b示出了根据另一个实施方案的mhc等位基因共享的示例网络模型nn
h
(
·
)。
28.图7示出了使用示例网络模型生成与一个mhc等位基因相关联的肽的呈递可能性。
29.图8示出了使用示例网络模型生成与一个mhc等位基因相关联的肽的呈递可能性。
30.图9示出了使用示例网络模型生成与多个mhc等位基因相关联的肽的呈递可能性。
31.图10示出了使用示例网络模型生成与多个mhc等位基因相关联的肽的呈递可能性。
32.图11示出了使用示例网络模型生成与多个mhc等位基因相关联的肽的呈递可能性。
33.图12示出了使用示例网络模型生成与多个mhc等位基因相关联的肽的呈递可能性。
34.图13a示出了nsclc患者中突变负荷的样本频率分布。
35.图13b示出了根据一个实施方案的基于患者是否满足最小突变负荷的纳入标准选择的患者的模拟疫苗中呈递的新抗原的数量。
36.图13c根据一个实施方案比较了与包含基于呈递模型鉴别的治疗子集的疫苗相关的所选患者和与包含通过现有技术水平模型鉴别的治疗子集的疫苗相关的所选患者之间的模拟疫苗中呈递的新抗原的数量。
37.图13d比较了与包含基于hla
‑
a*02:01的单独立等位基因呈递模型鉴别的治疗子集的疫苗相关的所选患者和与包含基于hla
‑
a*02:01和hla
‑
b*07:02的双独立等位基因呈
递模型鉴别的治疗子集的疫苗相关的所选患者之间的模拟疫苗中呈递的新抗原的数量。根据一个实施方案,疫苗容量设置为v=20个表位。
38.图13e根据一个实施方案比较了基于突变负荷选择的患者和通过期望效用得分选择的患者之间的模拟疫苗中呈递的新抗原的数量。
39.图14a是使用质谱法从人肿瘤细胞和肿瘤浸润淋巴细胞(til)上的ii类mhc等位基因洗脱的肽的长度的直方图。
40.图14b示出了两个示例性数据集的mrna定量与每残基呈递的肽之间的依赖性。
41.图14c比较使用两个示例性数据集训练和测试的示例性呈递模型的性能结果。
42.图14d是描绘了在总共73个包含人ii类hla分子的样品中,每个样品使用质谱法测序的肽的数量的直方图。
43.图14e是描绘了其中特定的ii类mhc分子等位基因被鉴别的样品的数量的直方图。
44.图14f是描绘了在总共73个样品中,对于一系列肽长度中的每个肽长度,由ii类mhc分子呈递的肽的比例的直方图。
45.图14g是描绘73个样品中存在的基因的基因表达与ii类mhc分子对基因表达产物的呈递普遍性之间的关系的线形图。
46.图14h是比较具有不同输入的同一模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能的线形图。
47.图14i是比较三种不同模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能的线形图。
48.图14j描绘了图14i的bi
‑
lstm模型的示例性实施方案,所述bi
‑
lstm模型被配置成预测hla
‑
drb(ii类mhc基因)的肽呈递。
49.图14k是描绘图14i的bi
‑
lstm、mlp、rnn和结合亲和力模型的全精度召回率曲线(full precision
‑
recall curve)的线形图。
50.图14l是线形图,其比较使用两种不同标准的同类最佳现有技术模型与利用两种不同输入的本文公开的呈递模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能。
51.图14m是直方图,其描绘了在q值小于0.1的情况下,对总共230个样品中的每个样品使用质谱法获得的测序肽的数量,所述样品包含含有ii类hla分子的人肿瘤(nsclc、淋巴瘤和卵巢癌)和细胞系(ebv)。
52.图14n是描绘其中特定的ii类mhc分子等位基因被鉴别的样品的数量的直方图。
53.图14o描绘了结合到i类mhc分子的肽和结合到ii类mhc分子的肽。
54.图14p描绘了图14q的初始模型的初始神经网络的示例性实施方案,所述初始模型被配置成预测ii类mhc分子的肽呈递。
55.图14q是比较“bi
‑
lstm”呈递模型与“初始”呈递模型在预测肽测试数据集中的肽将由测试数据集中存在的ii类mhc分子中的至少一种呈递的可能性方面的性能的线形图。
56.图15比较了“ms模型”、“netmhciipan排名”:netmhciipan3.1
77
(取hla
‑
drb1*15:01和hla
‑
drb5*01:01中的最低netmhciipan百分比排名)和“netmhciipan nm”:netmhciipan3.1(取hla
‑
drb1*15:01和hla
‑
drb5*01:01中的最强亲和力(单位为nm))在对hla
‑
drb1*15:01/hla
‑
drb5*01:01测试数据集中肽的排名中的预测性能。
57.图16描绘了用于将tcr引入受体细胞的tcr构建体的示例性实施方案。
58.图17描绘了用于将tcr克隆到表达系统中以进行治疗开发的示例性p526构建体骨架核苷酸序列。
59.图18描绘了用于将患者新抗原特异性tcr克隆型1tcr克隆到表达系统中用于疗法开发的示例性构建体序列。
60.图19描绘了用于将患者新抗原特异性tcr克隆型3克隆到表达系统中用于疗法开发的示例性构建体序列。
61.图20是根据一个实施方案的用于向患者提供定制的新抗原特异性治疗的方法的流程图。
62.图21示出了用于实施图1和3中所示实体的示例计算机。
具体实施方式
63.i.定义
64.一般说来,权利要求书和说明书中使用的术语意图解释为具有与本领域普通技术人员所理解的普通含义。为清楚起见,以下定义某些术语。如果普通含义与所提供的定义之间存在矛盾,应使用所提供的定义。
65.如本文所使用,术语“抗原”是诱导免疫应答的物质。
66.如本文所使用,术语“新抗原”是具有至少一个使其不同于相应野生型亲本抗原的变化的抗原,例如,该变化是肿瘤细胞突变或肿瘤细胞特异性翻译后修饰。新抗原可以包括多肽序列或核苷酸序列。突变可以包括移码或非移码插入缺失、错义或无义取代、剪接位点变化、基因组重排或基因融合,或产生neoorf的任何基因组或表达变化。突变还可以包括剪接变体。肿瘤细胞特异性翻译后修饰可以包括异常磷酸化。肿瘤细胞特异性翻译后修饰还可以包括蛋白酶体产生的剪接抗原。参见liepe等人,a large fraction of hla class i ligands are proteasome
‑
generated spliced peptides;science.2016oct21;354(6310):354
‑
358。
67.如本文所使用,术语“肿瘤新抗原”是存在于受试者的肿瘤细胞或组织中但不存在于受试者的相应正常细胞或组织中的新抗原。
68.如本文所使用,术语“基于新抗原的疫苗”是基于一个或多个新抗原,例如多个新抗原的疫苗构建体。
69.如本文所使用,术语“候选新抗原”是产生可以代表新抗原的新序列的突变或其它异常。
70.如本文所使用,术语“编码区”是基因中编码蛋白质的部分。
71.如本文所使用,术语“编码突变”是在编码区中存在的突变。
72.如本文所使用,术语“orf”是指开放阅读框。
73.如本文所使用,术语“neo
‑
orf”是由突变或其它异常如剪接而产生的肿瘤特异性orf。
74.如本文所使用,术语“错义突变”是导致一个氨基酸被另一个氨基酸取代的突变。
75.如本文所使用,术语“无义突变”是导致一个氨基酸被终止密码子取代的突变。
76.如本文所使用,术语“移码突变”是导致蛋白质框架改变的突变。
77.如本文所使用,术语“插入缺失”是一个或多个核酸的插入或缺失。
78.如本文在两个或更多个核酸或多肽序列的情况下使用的术语“同一性”百分比是指当比较并对准达到最大对应性时,如使用以下描述的序列比较算法(例如blastp和blastn,或技术人员可用的其它算法)之一测量或通过目测检查得到的两个或更多个序列或子序列具有指定百分比的核苷酸或氨基酸残基是相同的。取决于应用,“同一性”百分比可以存在于所比较的序列的某一区域内,例如在功能结构域内,或者存在于待比较的两个序列的全长内。
79.为进行序列比较,通常,一个序列充当参考序列,以与测试序列相比较。当使用序列比较算法时,将测试序列和参考序列输入计算机,必要时指定子序列座标,并且指定序列算法程序参数。然后,序列比较算法基于指定的程序参数计算测试序列相对于参考序列的序列同一性百分比。或者,可以通过组合在所选序列位置(例如序列基元)处特定核苷酸,或对于翻译的序列来说特定氨基酸的存在或不存在来确定序列相似性或不相似性。
80.用于比较的最佳序列比对可以例如通过smith和waterman,adv.appl.math.2:482(1981)的局部同源性算法;needleman和wunsch,j.mol.biol.48:443(1970)的同源性比对算法;pearson和lipman,proc.nat'l.acad.sci.usa 85:2444(1988)的相似性搜索方法;这些算法的计算机化实施(wisconsin genetics软件包中的gap、bestfit、fasta和tfasta;genetics computer group,575science dr.,madison,wis.);或通过目测检查(一般参见ausubel等人,见下文)来进行。
81.适于测定序列同一性和序列相似性百分比的算法的一个实例是altschul等人,j.mol.biol.215:403
‑
410(1990)中描述的blast算法。执行blast分析的软件通过national center for biotechnology information公开可用。
82.如本文所使用,术语“无终止或通读”是导致天然终止密码子移除的突变。
83.如本文所使用,术语“表位”是抗原中通常由抗体或t细胞受体结合的特定部分。
84.如本文所使用,术语“免疫原性”是例如通过t细胞、b细胞或两者引发免疫应答的能力。
85.如本文所使用,术语“hla结合亲和力”、“mhc结合亲和力”意思指特定抗原与特定mhc等位基因之间的结合亲和力。
86.如本文所使用,术语“诱饵(bait)”是用于自样品富集特定dna或rna序列的核酸探针。
87.如本文所使用,术语“变体”是受试者的核酸与用作对照的参考人基因组之间的差异。
88.如本文所使用,术语“变体识别(variant call)”是对通常由测序确定的变体存在的算法确定。
89.如本文所使用,术语“多态现象”是生殖系变体,即,在个体的所有带有dna的细胞中所发现的变体。
90.如本文所使用,术语“体细胞变体”是在个体的非生殖系细胞中产生的变体。
91.如本文所使用,术语“等位基因”是基因的一种形式,或是基因序列的一种形式,或是蛋白质的一种形式。
92.如本文所使用,术语“hla型”是hla基因等位基因的互补序列。
93.如本文所使用,术语“无义介导的衰变”或“nmd”是由过早终止密码子引起的细胞对mrna的降解。
94.如本文所使用,术语“躯干突变”是起源于肿瘤发展早期且存在于大多数肿瘤细胞中的突变。
95.如本文所使用,术语“亚克隆突变”是起源于肿瘤发展后期且仅存在于一小部分肿瘤细胞中的突变。
96.如本文所使用,术语“外显子组”是编码蛋白质的基因组的子组。外显子组可以是基因组的全体外显子。
97.如本文所使用,术语“逻辑回归”是由统计得到的二进制数据的回归模型,其中因变量等于1的机率的分对数被建模为因变量的线性函数。
98.如本文所使用,术语“神经网络”是用于分类或回归的机器学习模型,由多层线性变换,继之以通常通过随机梯度下降和反向传播训练的逐元素非线性组成。
99.如本文所使用,术语“蛋白质组”是由细胞、细胞群或个体表达和/或翻译的所有蛋白质的集合。
100.如本文所使用,术语“肽组”是由mhc
‑
i或mhc
‑
ii呈递于细胞表面上的所有肽的集合。肽组可以指一个细胞或一组细胞(例如肿瘤肽组,意思指构成肿瘤的所有细胞的肽组的联合)的特性。
101.如本文所使用,术语“elispot”意思指酶联免疫吸附斑点测定,这是一种用于监测人和动物的免疫应答的常用方法。
102.如本文所使用,术语“dextramer”是在流式细胞术中用于抗原特异性t细胞染色的基于葡聚糖的肽
‑
mhc多聚体。
103.如本文所用,术语“mhc多聚体”是包含多个肽
‑
mhc单体单元的肽
‑
mhc复合物。
104.如本文所用,术语“mhc四聚体”是包含四个肽
‑
mhc单体单元的肽
‑
mhc复合物。
105.如本文所使用,术语“耐受性或免疫耐受性”是对一种或多种抗原,例如自身抗原免疫无反应性的状态。
106.如本文所使用,术语“中枢耐受性”是通过缺失自身反应性t细胞克隆或通过促进自身反应性t细胞克隆分化成免疫抑制性调控性t细胞(treg)而在胸腺中经历的耐受性。
107.如本文所使用,术语“外周耐受性”是通过使经历中枢耐受性而存活的自身反应性t细胞下调或无反应性(anergizing),或通过促进这些t细胞分化成treg而在外周经历的耐受性。
108.术语“样品”可以包括借助于包括静脉穿刺、排泄、射精、按摩、活组织检查、针抽吸、灌洗样品、刮取、手术切口或干预在内的手段,或本领域中已知的其它手段从受试者获取单个细胞或多个细胞,或细胞碎片,或体液等分试样。
109.术语“受试者”涵盖细胞、组织或生物体、人或非人,无论是体内、离体还是体外,雄性还是雌性的。术语受试者包括含人在内的哺乳动物。
110.术语“哺乳动物”涵盖人和非人两种,并且包括但不限于人、非人灵长类动物、犬科动物、猫科动物、鼠科动物、牛科动物、马科动物及猪科动物。
111.术语“临床因素”是指受试者状况,例如疾病活动性或严重程度的量度。“临床因素”涵盖受试者健康状况的所有标志物,包括非样品标志物,和/或受试者的其它特征,如但
不限于年龄和性别。临床因素可以是能通过在确定条件下评价来自受试者的一个样品(或样品群)或受试者而获得的分数、一个值或一组值。临床因素也可以由标志物和/或如基因表达替代物之类其它参数进行预测。临床因素可以包括肿瘤类型、肿瘤亚型和吸烟史。
112.缩写:mhc:主要组织相容性复合物;hla:人白细胞抗原或人mhc基因座;ngs:下一代测序;ppv:阳性预测值;tsna:肿瘤特异性新抗原;ffpe:福尔马林固定、石蜡包埋;nmd:无义介导的衰变;nsclc:非小细胞肺癌;dc:树突状细胞。
113.除非上下文另外清楚地规定,否则如本说明书和所附权利要求中所使用,单数形式“一个(种)(a/an)”和“所述”包括多个参照物。
114.本文中未直接定义的任何术语应理解为具有与本发明领域内所理解的通常与之相关的含义。本文论述的某些术语是为了向从业人员描述本发明各方面的组合物、装置、方法等以及其制备或使用提供额外的指导。应了解,相同的事物可以按超过一种方式表示。因此,替代性措辞和同义词可以用于本文所论述的任一个或多个术语。无论本文中是否阐述或论述术语都无关紧要。提供了一些同义词或可取代的方法、材料等。除非明确陈述,否则对一个或数个同义词或等效物的叙述不排除其它同义词或等效物的使用。实例,包括术语实例的使用只是出于说明的目的,且并非在本文中限制本发明各方面的范围和含义。
115.说明书正文内引用的所有参考文献、颁布的专利和专利申请都是以引用的方式整体并入本文中用于所有目的。
116.ii.鉴别新抗原的方法
117.本文公开了用于鉴别对来自受试者的肿瘤细胞的可能由ii类mhc等位基因呈递于所述肿瘤细胞表面上的新抗原具有抗原特异性的t细胞的方法。所述方法包括从受试者的肿瘤细胞以及正常细胞获得外显子组、转录组和/或全基因组核苷酸测序数据。该核苷酸测序数据被用于获得新抗原集合中的每种新抗原的肽序列。通过比较来自肿瘤细胞的核苷酸测序数据和来自正常细胞的核苷酸测序数据来鉴别新抗原集合。具体地,新抗原集合中的每种新抗原的肽序列包含至少一个使其不同于从受试者的正常细胞鉴别的相应野生型肽序列的变化。所述方法还包括将新抗原集合中每种新抗原的肽序列编码成相应的数字矢量。每个数字矢量包含描述构成肽序列的氨基酸和肽序列中氨基酸的位置的信息。所述方法还包括将数字矢量输入机器学习呈递模型,以产生对于新抗原集合中每种新抗原的呈递可能性。每个呈递可能性代表了相应新抗原由受试者的肿瘤细胞表面上的由ii类mhc等位基因呈递的可能性。机器学习呈递模型包含多个参数和函数。所述多个参数基于训练数据集鉴别。所述训练数据集包含:对于多个样品中的每个样品,通过质谱测量与被鉴别为存在于所述样品中的ii类mhc等位基因集合中的至少一种ii类mhc等位基因结合的肽的存在获得的标记,及编码为包含描述构成肽的多个氨基酸和肽中氨基酸的位置的信息的数字矢量的训练肽序列。所述函数代表由机器学习呈递模型作为输入接收的所述数字矢量和由机器学习呈递模型根据所述数字矢量和所述参数作为输出生成的所述呈递可能性之间的关系。所述方法还包括基于所述呈递可能性选择所述新抗原集合的子集,以产生选定的新抗原的集合。所述方法还包括鉴别对所述子集中的至少一种新抗原具有抗原特异性的t细胞,以及回收这些鉴别的t细胞。
118.在一些实施方案中,将数字矢量输入机器学习呈递模型包括:将机器学习呈递模型应用于新抗原的肽序列以生成每种ii类mhc等位基因的依赖性分数。ii类mhc等位基因的
依赖性分数基于肽序列的特定位置处的特定氨基酸指示ii类mhc等位基因是否会呈递新抗原。在另一些实施方案中,将数字矢量输入机器学习呈递模型另外包括:变换依赖性分数以得到每一ii类mhc等位基因的相应独立等位基因可能性,由此指示相应ii类mhc等位基因会呈递相应新抗原的可能性;及将独立等位基因可能性组合以产生新抗原的呈递可能性。在一些实施方案中,变换依赖性分数将新抗原的呈递建模为在ii类mhc等位基因之间相互排斥。在替代实施方案中,将数字矢量输入机器学习呈递模型另外包括:变换依赖性分数的组合以产生呈递可能性。在这样的实施方案中,变换依赖性分数的组合将新抗原的呈递建模为在ii类mhc等位基因之间存在干扰。
119.在一些实施方案中,呈递可能性的集合通过至少一个或多个等位基因非相互作用特征进一步鉴别。在这样的实施方案中,所述方法进一步包括将机器学习呈递模型应用于等位基因非相互作用特征,以产生所述等位基因非相互作用特征的依赖性分数。依赖性分数指示相应新抗原的肽序列是否将基于所述等位基因非相互作用特征而被呈递。在一些实施方案中,所述方法进一步包括将每个ii类mhc等位基因的依赖性分数与等位基因非相互作用特征的依赖性分数组合,变换每个ii类mhc等位基因的组合的依赖性分数以产生每个ii类mhc等位基因的独立等位基因可能性,及组合独立等位基因可能性以产生呈递可能性。ii类mhc等位基因的独立等位基因可能性指示ii类mhc等位基因将呈递相应新抗原的可能性。在替代实施方案中,所述方法进一步包括组合ii类mhc等位基因的依赖性分数与所述等位基因非相互作用特征的依赖性分数;及变换组合的依赖性分数以产生呈递可能性。
120.在一些实施方案中,ii类mhc等位基因包括两个或更多个不同的ii类mhc等位基因。
121.在一些实施方案中,被鉴别为存在于训练数据集的样品中的ii类mhc等位基因集中的至少一个ii类mhc等位基因包括两种或更多种不同类型的ii类mhc等位基因。
122.在一些实施方案中,肽序列包含具有9个氨基酸以外的长度的肽序列。
123.在一些实施方案中,编码肽序列包括使用独热编码方案编码所述肽序列。
124.在一些实施方案中,所述多个样品包括以下至少一种:被工程改造成表达单个ii类mhc等位基因的细胞系;被工程改造成表达多个ii类mhc等位基因的细胞系;从多个患者获得或得到的人细胞系;从多个患者获得的新鲜或冷冻的肿瘤样品;以及从多个患者获得的新鲜或冷冻的组织样品。
125.在一些实施方案中,所述训练数据集还包含以下至少一种:与所述肽中的至少一个的肽
‑
mhc结合亲和力测量值相关的数据;及与所述肽中的至少一个的肽
‑
mhc结合稳定性测量值相关的数据。
126.在一些实施方案中,呈递可能性集合进一步通过如由rna
‑
seq或质谱法测量的所述受试者中ii类mhc等位基因的表达水平鉴别。
127.在一些实施方案中,呈递可能性集合通过特征进一步鉴别,所述特征包括以下中的至少一种:预测的所述新抗原集合中的新抗原与ii类mhc等位基因之间的亲和力;及预测的新抗原编码的肽
‑
mhc复合物的稳定性。
128.在一些实施方案中,所述数字可能性集合通过特征进一步鉴别,所述特征包括以下中的至少一种:在其源蛋白质序列内侧接所述新抗原编码肽序列的c端序列;及在其源蛋白质序列内侧接所述新抗原编码肽序列的n端序列。
129.在一些实施方案中,选择所述选定的新抗原的集合包括基于机器学习呈递模型,选择在所述肿瘤细胞表面上呈递的可能性相对于未选择的新抗原有所增加的新抗原。
130.在一些实施方案中,选择所述选定的新抗原的集合包括基于机器学习呈递模型,选择能够在受试者体内诱导肿瘤特异性免疫应答的可能性相对于未选择的新抗原有所增加的新抗原。
131.在一些实施方案中,选择所述选定的新抗原的集合包括基于呈递模型,选择能够被专职抗原呈递细胞(apc)呈递至天然t细胞的可能性相对于未选择的新抗原有所增加的新抗原。在这样的实施方案中,apc任选地是树突状细胞(dc)。
132.在一些实施方案中,选择所述选定的新抗原的集合包括基于机器学习呈递模型,选择经历中枢或外周耐受性抑制的可能性相对于未选择的新抗原有所降低的新抗原。
133.在一些实施方案中,选择所述选定的新抗原的集合包括基于机器学习呈递模型,选择能够在所述受试者体内诱导针对正常组织的自体免疫应答的可能性相对于未选择的新抗原有所降低的新抗原。
134.在一些实施方案中,所述一种或多种肿瘤细胞选自由以下组成的组:肺癌、黑素瘤、乳癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、b细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病和t细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
135.在一些实施方案中,该方法还包括从所述选定的新抗原的集合产生用于构建个性化癌症疫苗的输出。在这样的实施方案中,个性化癌症疫苗的输出可包括编码所述选定的新抗原的集合的至少一个肽序列或至少一个核苷酸序列。
136.在一些实施方案中,机器学习呈递模型是神经网络模型。在这样的实施方案中,神经网络模型可包括用于所述ii类mhc等位基因的多个网络模型,每个网络模型被分配给所述ii类mhc等位基因中的相应ii类mhc等位基因,并且包括布置在一个或多个层中的一系列节点。在这样的实施方案中,可通过更新神经网络模型的参数来训练神经网络模型,并且其中针对至少一个训练迭代,共同更新至少两个网络模型的参数。
137.在这样的实施方案中,每个网络模型还可包括一个或多个卷积神经网络,所述一个或多个卷积神经网络中的每一个包括一系列布置在一层或多层中的节点,并且具有不同大小的过滤器。一个或多个卷积神经网络中的每一个的过滤器的大小可被设定来鉴别每个新抗原的肽序列中氨基酸的位置,所述新抗原包含肽序列的结合核心或结合锚。
138.在一些实施方案中,机器学习呈递示模型可以是包括一个或多个节点层的深度学习模型。
139.在一些实施方案中,鉴别t细胞包括在扩增t细胞的条件下,将t细胞与所述子集中的一种或多种新抗原共同培养。
140.在一些实施方案中,鉴别t细胞包括在允许t细胞和mhc多聚体结合的条件下,使t细胞与包含所述子集中的一种或多种新抗原的mhc多聚体接触。
141.在一些实施方案中,所述方法进一步包括鉴别所鉴别的t细胞的t细胞受体(tcr)。在这样的实施方案中,鉴别t细胞受体可包括对鉴别的t细胞的t细胞受体序列进行测序。在这样的实施方案中,所述方法可以进一步包括对t细胞进行基因工程改造以表达一种或多种鉴别的t细胞受体中的至少一种,在扩增t细胞的条件下培养所述t细胞,及将扩增的t细
胞输注至受试者体内。在这样的实施方案中,对所述t细胞进行基因改造以表达至少一种鉴别的t细胞受体包括:将鉴别的t细胞的t细胞受体序列克隆到表达载体中,及用所述表达载体转染每一个t细胞。
142.在一些实施方案中,该方法进一步包括在扩增鉴别的t细胞的条件下培养鉴别的t细胞,及将扩增的t细胞输注至受试者体内。
143.本文还公开了分离的t细胞,其对上述新抗原子集中的至少一种选定的新抗原具有抗原特异性。
144.国际专利公布第wo 2018/195357号和国际专利公布第wo2019/050994号据此通过引用以其整体并入。国际专利公布第wo2018/195357号描述了通过ii类mhc分子预测抗原呈递的方法。国际专利公布第wo 2019/050994号描述了鉴别对mhc分子呈递的抗原具有抗原特异性的t细胞的方法。虽然在本技术的这一部分中引用了这些公布,但在国际专利公布第wo 2018/195357号和第wo2019/050994号中提供的公开内容在本技术的每一节中据此通过引用以其整体并入。
145.iii.鉴别新抗原中的肿瘤特异性突变
146.本文还公开了用于鉴别某些突变(例如癌细胞中存在的变体或等位基因)的方法。确切地说,这些突变可以存在于患有癌症的受试者的癌细胞的基因组、转录组、蛋白质组或外显子组中,但不存在于受试者的正常组织中。
147.若肿瘤中的基因突变仅导致肿瘤中蛋白质的氨基酸序列改变,则认为这些突变可用于免疫靶向肿瘤。有用的突变包括:(1)导致蛋白质中的氨基酸不同的非同义突变;(2)通读突变,其中终止密码子被修饰或缺失,导致翻译得到在c端具有新肿瘤特异性序列的较长蛋白质;(3)导致在成熟mrna中包括内含子且由此产生独特肿瘤特异性蛋白质序列的剪接位点突变;(4)产生在2种蛋白质的接合处具有肿瘤特异性序列的嵌合蛋白的染色体重排(即,基因融合);(5)产生具有新肿瘤特异性蛋白质序列的新开放阅读框的移码突变或缺失。突变还可以包括非移码插入缺失、错义或无义取代、剪接位点变化、基因组重排或基因融合,或产生neoorf的任何基因组或表达变化中的一种或多种。
148.在肿瘤细胞中具有突变的肽或由例如剪接位点突变、移码突变、通读突变或基因融合突变产生的突变多肽可以通过对肿瘤和正常细胞中的dna、rna或蛋白质进行测序来鉴别。
149.突变还可以包括先前鉴别的肿瘤特异性突变。已知的肿瘤突变可以见于癌症体细胞突变目录(catalogue of somatic mutations in cancer,cosmic)数据库。
150.多种方法可用于检测个体的dna或rna中特定突变或等位基因的存在。本领域中的改进之处在于提供准确、容易且便宜的大规模snp基因分型。举例来说,已描述若干技术,包括动态等位基因特异性杂交(dash)、微板阵列对角线凝胶电泳(microplate array diagonal gel electrophoresis,madge)、焦磷酸测序、寡核苷酸特异性连接、taqman系统以及各种dna“芯片”技术,如affymetrix snp芯片。这些方法通常通过pcr扩增靶基因区。一些其它的方法基于通过侵袭式裂解产生小信号分子,随后进行质谱法或固定化挂锁探针(padlock probe)和滚环扩增。本领域中已知用于检测特定突变的若干方法概述于下。
151.基于pcr的检测手段可以包括同时多重扩增多个标志物。举例来说,本领域中众所周知,选择pcr引物产生尺寸不重叠且可以同时分析的pcr产物。或者,可用以不同方式标记
且由此可以通过不同方式检测的引物扩增不同标志物。当然,基于杂交的检测手段能够以不同方式检测样品中的多个pcr产物。本领域中已知能够多重分析多个标志物的其它技术。
152.已经开发出数种方法来促进基因组dna或细胞rna中单核苷酸多态性的分析。举例来说,可以通过使用专用的核酸外切酶抗性核苷酸检测单碱基多态性,如例如mundy,c.r.(美国专利第4,656,127号)中所公开的。根据该方法,与紧靠多态性位点3'端的等位基因序列互补的引物能够与从特定动物或人获得的靶分子杂交。如果靶分子上的多态性位点含有与存在的特定核酸外切酶抗性核苷酸衍生物互补的核苷酸,则该衍生物将被合并至杂交引物的末端上。此类合并使得引物对核酸外切酶具有抗性,并由此允许其检测。由于样品的核酸外切酶抗性衍生物的身份是已知的,故引物对核酸外切酶产生抗性的发现披露,靶分子多态性位点中存在的核苷酸与反应中使用的核苷酸衍生物互补。该方法的优势在于,它不需要测定大量无关的序列数据。
153.可以使用基于溶液的方法来确定多态性位点的核苷酸的身份。cohen,d.等人(法国专利2,650,840;pct申请第wo91/02087号)。如在美国专利第4,656,127号的mundy方法中所述,采用与紧靠多态性位点3'端的等位基因序列互补的引物。该方法使用标记过的双脱氧核苷酸衍生物来确定该位点的核苷酸的身份,如果与多态性位点的核苷酸互补,则该核苷酸将被合并至引物末端上。goelet,p.等人(pct申请第92/15712号)描述了一种替代性方法,称为遗传位点分析(genetic bit analysis)或gba。goelet,p.等人的方法使用了标记过的终止子和与在多态性位点3'端的序列互补的引物的混合物。由此通过存在于所评价靶分子的多态性位点中的核苷酸来确定合并的标记过的终止子并且该终止子与存在于所评价靶分子的多态性位点中的核苷酸互补。与cohen等人(法国专利2,650,840;pct申请第wo91/02087号)的方法相比,goelet,p.等人的方法可以是非均相测定,其中引物或靶分子被固定于固相。
154.已描述数种引物引导的用于测定dna中的多态性位点的核苷酸并入程序(komher,j.s.等人,nucl.acids.res.17:7779
‑
7784(1989);sokolov,b.p.,nucl.acids res.18:3671(1990);syvanen,a.
‑
c.等人,genomics 8:684
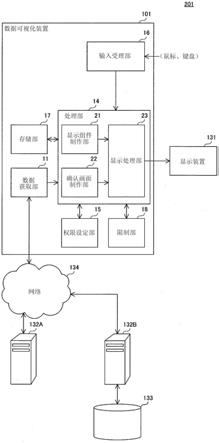
‑
692(1990);kuppuswamy,m.n.等人,proc.natl.acad.sci.(u.s.a.)88:1143
‑
1147(1991);prezant,t.r.等人,hum.mutat.1:159
‑
164(1992);ugozzoli,l.等人,gata 9:107
‑
112(1992);nyren,p.等人,anal.biochem.208:171
‑
175(1993))。这些方法与gba的不同之处在于,它们利用并入经过标记的脱氧核苷酸来区别多态性位点处的碱基。在此类形式中,由于信号与并入的脱氧核苷酸的数量成比例,故在同一核苷酸的操作中出现的多态现象可以产生与该操作的长度成比例的信号(syvanen,a.
‑
c.等人,amer.j.hum.genet.52:46
‑
59(1993))。
155.许多方案直接从数百万个独立dna或rna分子中并行获得序列信息。实时单分子边合成边测序技术依赖于荧光核苷酸的检测,因为这些核苷酸被并入与测序模板互补的新生dna链中。在一种方法中,将30
‑
50个碱基长度的寡核苷酸以5'端共价锚定至玻璃盖玻片上。这些锚定链执行两种功能。首先,如果模板被配置成具有与表面结合的寡核苷酸互补的捕捉尾部,则其充当靶模板链的捕捉位点。这些锚定链还充当模板引导的引物延伸的引物,形成序列读取的基础。捕捉引物用作固定位点以便使用多个合成、检测以及染料
‑
连接子化学裂解以移除染料的循环进行序列测定。每个循环由添加聚合酶/标记过得核苷酸混合物、冲洗、成像及染料裂解组成。在一种替代方法中,聚合酶被修饰成具有荧光供体分子并且被固
定于玻璃载片上,而各核苷酸用附接至γ
‑
磷酸的受体萤光部分进行颜色编码。当核苷酸被并入从头合成的链中时,该系统检测荧光标记的聚合酶与荧光修饰的核苷酸之间的相互作用。还存在其它边合成边测序技术。
156.任何适合的边合成边测序平台都可以用于鉴别突变。如上文所描述,目前有四个主要的边合成边测序平台:来自roche/454life sciences的基因组测序仪、来自illumina/solexa的1g分析仪、来自applied biosystems的solid系统以及来自helicos biosciences的heliscope系统。pacific biosciences和visigen biotechnologies也描述过边合成边测序平台。在一些实施方案中,使所测序的多个核酸分子结合至支撑物(例如固体支撑物)上。为了将核酸固定于支撑物上,可以在模板的3'和/或5'端添加捕捉序列/通用引发位点。可以通过使捕捉序列与共价附接至支撑物的互补序列杂交而使核酸结合至支撑物。捕捉序列(又称为通用捕捉序列)是与附接至支撑物的序列互补的核酸序列,该序列还可以充当通用引物。
157.作为捕捉序列的替代,可以将偶合对(如抗体/抗原、受体/配体,或抗生物素
‑
生物素对,如例如美国专利申请第2006/0252077号中所述)的一个成员连接至各片段以将其捕捉在涂有该偶合对的相应第二成员的表面上。
158.在捕捉后,可以例如实施例和美国专利第7,283,337号中所描述,通过例如单分子检测/测序,包括模板依赖性边合成边测序对该序列进行分析。在边合成边测序时,使表面结合的分子在聚合酶存在下暴露于多个标记过得核苷酸三磷酸。模板序列由并入正在生长的链的3'端的标记过的核苷酸的顺序决定。这可以实时进行或者可以按分步重复模式进行。对于实时分析,可以将不同光学标记并入各核苷酸并且可以利用多种激光器刺激并入的核苷酸。
159.测序还可以包括其它大规模平行测序或下一代测序(ngs)技术和平台。大规模平行测序技术和平台的其它实例有illumina hiseq或miseq、thermo pgm或proton、pac bio rs ii或sequel、qiagen公司的gene reader及oxford nanopore minion。可以使用当前其它类似的大规模平行测序技术,以及这些技术的改进形式。
160.任何细胞类型或组织都可以用于获得用于本文所描述的方法中的核酸样品。举例来说,dna或rna样品可以从肿瘤或体液,例如利用已知技术(例如静脉穿刺)获得的血液,或唾液获得。或者,可以对干燥样品(例如毛发或皮肤)进行核酸测试。此外,可以从肿瘤获得一份测序样品,并且可以从正常组织获得另一份测序样品,其中正常组织与肿瘤同属相同组织类型。可以从肿瘤获得一份测序样品,并且可以从正常组织获得另一份测序样品,其中正常组织与肿瘤属于不同组织类型。
161.肿瘤可以包括以下一种或多种:肺癌、黑素瘤、乳癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、b细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病和t细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
162.或者,可以使用蛋白质质谱法鉴别或验证结合至肿瘤细胞上的mhc蛋白质的突变肽的存在。肽可以用酸从肿瘤细胞或从自肿瘤免疫沉淀的hla分子洗脱,并且接着使用质谱法鉴别。
163.iv.新抗原
164.新抗原可以包括核苷酸或多肽。举例来说,新抗原可以是编码多肽序列的rna序
列。因此,可用于疫苗中的新抗原包括核苷酸序列或多肽序列。
165.本文公开了包含通过本文所公开的方法鉴别的肿瘤特异性突变的分离的肽、包含已知肿瘤特异性突变的肽,以及通过本文所公开的方法鉴别的突变多肽或其片段。新抗原肽可以在其编码序列背景下描述,其中新抗原包括编码相关多肽序列的核苷酸序列(例如dna或rna)。
166.由新抗原核苷酸序列编码的一个或多个多肽可以包含以下至少一种:以低于1000nm的ic50值的与mhc的结合亲和力;对于长度是8
‑
15个,即8、9、10、11、12、13、14或15个氨基酸的i类mhc肽,在该肽内或附近存在促进蛋白酶体裂解的序列基元;及存在促进tap转运的序列基元。对于长度是6
‑
30个,即6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的ii类mhc肽,在该肽内或附近存在促进通过细胞外或溶酶体蛋白酶(组织蛋白酶)的切割或hla
‑
dm催化的hla结合的序列基元。
167.一个或多个新抗原可以被呈递于肿瘤表面上。
168.一个或多个新抗原可以在患肿瘤的受试者中具有免疫原性,例如能够在该受试者体内引起t细胞应答或b细胞应答。
169.在产生用于患肿瘤的受试者的疫苗的情况下,可以考虑排除在受试者体内诱导自体免疫应答的一个或多个新抗原。
170.至少一个新抗原肽分子的尺寸可以包括但不限于约5个、约6个、约7个、约8个、约9个、约10个、约11个、约12个、约13个、约14个、约15个、约16个、约17个、约18个、约19个、约20个、约21个、约22个、约23个、约24个、约25个、约26个、约27个、约28个、约29个、约30个、约31个、约32个、约33个、约34个、约35个、约36个、约37个、约38个、约39个、约40个、约41个、约42个、约43个、约44个、约45个、约46个、约47个、约48个、约49个、约50个、约60个、约70个、约80个、约90个、约100个、约110个、约120个或更多个氨基分子残基,以及由其中可衍生的任何范围。在特定实施例方案中,新抗原肽分子等于或少于50个氨基酸。
171.新抗原肽和多肽可以:对于i类mhc是15个或更少残基长度并且通常由介于约8个与约11个之间的残基,特别是9个或10个残基组成;对于ii类mhc是6
‑
30个残基(包括端点在内)。
172.必要时,可以通过若干方式设计出更长的肽。在一种情况下,当预测出或已知肽在hla等位基因上呈递的可能性时,较长的肽可以由以下任一种组成:(1)朝各相应基因产物的n端和c端延伸2
‑
5个氨基酸的个别呈递的肽;(2)一些或全部呈递肽与各自的延伸序列的串接。在另一情况下,当测序披露在肿瘤中存在较长的(>10个残基)新表位序列(例如由产生新颖肽序列的移码、通读或包括内含子引起)时,较长的肽将由以下组成:(3)由新颖肿瘤特异性氨基酸组成的整个延伸段,由此绕过了对基于计算或体外测试来选择hla呈递最强的较短肽的需求。在两种情况下,较长链的使用使患者细胞能够进行内源性加工并且可以产生更有效的抗原呈递和t细胞应答的诱导作用。
173.新抗原肽和多肽可以被呈递于hla蛋白质上。在一些方面,新抗原肽和多肽是以高于野生型肽的亲和力呈递于hla蛋白质上。在一些方面,新抗原肽或多肽的ic50值可以是至少低于5000nm、至少低于1000nm、至少低于500nm、至少低于250nm、至少低于200nm、至少低于150nm、至少低于100nm、至少低于50nm或更低。
174.在一些方面,新抗原肽和多肽当施用给受试者时不会诱导自体免疫应答和/或激
发免疫耐受性。
175.还提供了包含至少两个或更多个新抗原肽的组合物。在一些实施方案中,该组合物含有至少两个不同的肽。至少两个不同的肽可以来源于同一多肽。不同的多肽意味着,该肽的长度、氨基酸序列或两者不同。这些肽来源于已知或被发现含有肿瘤特异性突变的任何多肽。可以作为新抗原肽的来源的适合多肽可以见于例如cosmic数据库。cosmic策划了有关人癌症中的体细胞突变的全面信息。肽含有肿瘤特异性突变。在一些方面,肿瘤特异性突变是特定癌症类型的驱动突变。
176.具有所希望的活性或特性的新抗原肽和多肽可以被修饰成用于提供某些所希望的属性,例如改良的药理学特征,同时增加或至少保持未修饰肽的大体上所有生物活性以结合所希望的mhc分子并活化适当t细胞。举例来说,新抗原肽和多肽可以经历各种变化,如保守性或非保守性取代,其中此类变化可能在其使用中提供某些优势,如改良的mhc结合、稳定性及呈递。保守性取代意思指氨基酸残基被在生物上和/或化学上类似的另一氨基酸残基置换,例如一个疏水性残基被另一个置换,或一个极性残基被另一个置换。取代包括如gly、ala;val、ile、leu、met;asp、glu;asn、gln;ser、thr;lys、arg;及phe、tyr等的组合。单氨基酸取代的影响还可以使用d
‑
氨基酸探测。此类修饰可以使用众所周知的肽合成程序进行,如例如merrifield,science 232:341
‑
347(1986),barany&merrifield,the peptides,gross&meienhofer编辑(n.y.,academic press),第1
‑
284页(1979);及stewart和young,solid phase peptide synthesis,(rockford,ill.,pierce),第2版(1984)中所述。
177.用各种氨基酸模拟物或非天然氨基酸修饰肽和多肽特别适用于增加该肽和多肽的体内稳定性。稳定性可以通过多种方式测定。举例来说,使用肽酶和各种生物介质如人血浆和血清测试稳定性。参见例如,verhoef等人,eur.j.drug metab pharmacokin.11:291
‑
302(1986)。肽的半衰期可以使用25%人血清(v/v)测定,按常规方式测定。方案大致如下。在使用前,通过离心使汇集的人血清(ab型,未热灭活)脱脂。接着,用rpmi组织培养基将该血清稀释至25%并用于测试肽稳定性。按预定时间间隔,取出少量反应溶液并添加至6%三氯乙酸水溶液或乙醇中。冷却混浊的反应样品(4℃),保持15分钟,然后离心以使沉淀的血清蛋白聚结。接着,通过反相hplc,使用稳定性特异性色谱条件测定肽的存在。
178.这些肽和多肽可以经过修饰以提供除改良的血清半衰期外的所希望的属性。举例来说,可以通过将这些肽连接至含有至少一个能够诱导t辅助细胞应答的表位的序列来增强其诱导ctl活性的能力。免疫原性肽/t辅助偶联物可以借助于间隔子分子连接。间隔子通常包含在生理条件下大体上不带电荷的相对较小的中性分子,如氨基酸或氨基酸模拟物。这些间隔子通常选自例如ala、gly或由非极性氨基酸或中性极性氨基酸组成的其它中性间隔子。应理解,任选存在的间隔子无需包含相同残基且因此可以是异低聚物或同低聚物。当存在时,间隔子通常是至少一个或二个残基,更通常是三个至六个残基。或者,可以在无间隔子情况下将肽连接至t辅助肽。
179.新抗原肽可以直接地或通过间隔子在肽的氨基或羧基末端连接至t辅助肽。新抗原肽或t辅助肽的氨基末端可以被酰基化。示例性t辅助肽包括破伤风类毒素830
‑
843、流感307
‑
319、疟疾环子孢子382
‑
398和378
‑
389。
180.蛋白质或肽可以通过本领域技术人员已知的任何技术制备,包括通过标准分子生物学技术表达蛋白质、多肽或肽、从天然来源分离蛋白质或肽,或化学合成蛋白质或肽。先
前已公开对应于各种基因的核苷酸和蛋白质、多肽和肽序列,并且可以见于本领域普通技术人员已知的计算机化数据库。一种此类数据库是位于美国国家卫生研究院(national institutes of health)网站的国家生物技术信息中心(national center for biotechnology information)的genbank和genpept数据库。已知基因的编码区可以使用本文所公开或本领域普通技术人员已知的技术扩增和/或表达。或者,本领域技术人员已知蛋白质、多肽和肽的各种市售制剂。
181.在另一方面,新抗原包括了编码新抗原肽或其部分的核酸(例如多核苷酸)。该多核苷酸可以是例如单链和/或双链dna、cdna、pna、can、rna(例如mrna),或多核苷酸的天然或稳定化形式,如例如具有硫代磷酸酯主链的多核苷酸,或其组合,并且该多核苷酸可以含有或可以不含内含子。又另一方面提供了一种能够表达多肽或其部分的表达载体。用于不同细胞类型的表达载体是本领域众所周知的并且可以在无过度实验情况下进行选择。一般来说,将dna以适当取向和正确的表达阅读框插入表达载体,如质粒中。必要时,可以将dna连接至能被所希望的宿主识别的适当转录和翻译调控性控制核苷酸序列,不过此类控制一般在表达载体中可用。接着,通过标准技术将载体插入宿主中。相关指导可见于例如sambrook等人(1989)molecular cloning,a laboratory manual,cold spring harbor laboratory,cold spring harbor,n.y.
182.iv.疫苗组合物
183.本文还公开了一种能够引起特异性免疫应答,例如肿瘤特异性免疫应答的免疫原性组合物,例如疫苗组合物。疫苗组合物通常包含多个例如使用本文所描述的方法选择的新抗原。疫苗组合物又可以称为疫苗。
184.疫苗可以含有个数在1个与30个之间的肽,即2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个不同的肽;6、7、8、9、10 11、12、13或14个不同肽;或12、13或14个不同的肽。肽可以包括翻译后修饰。疫苗可以含有个数在1个与100个之间或更多个核苷酸序列,即2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94,95、96、97、98、99、100或更多个不同的核苷酸序列;6、7、8、9、10 11、12、13或14个不同的核苷酸序列;或12、13或14个不同的核苷酸序列。疫苗可以含有个数在1个与30个之间的新抗原序列,即2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94,95、96、97、98、99、100或更多个不同的新抗原序列;6、7、8、9、10 11、12、13或14个不同的新抗原序列;或12、13或14个不同的新抗原序列。
185.在一个实施方案中,不同肽和/或多肽或编码其的核苷酸序列的选择使得这些肽和/或多肽能够与不同mhc分子,如不同的i类mhc分子和/或不同的ii类mhc分子缔合。在一些方面,一种疫苗组合物包含能够与最常出现的i类mhc分子和/或ii类mhc分子缔合的肽和/或多肽的编码序列。因此,疫苗组合物可以包含能够与至少2个优选的、至少3个优选的
或至少4个优选的i类mhc分子和/或ii类mhc分子缔合的不同片段。
186.该疫苗组合物能够引起特异性细胞毒性t细胞应答和/或特异性辅助t细胞应答。
187.疫苗组合物还可以包含佐剂和/或载剂。有用的佐剂和载剂的实例提供于下文中。组合物可以与载剂缔合,如例如蛋白质或抗原呈递细胞,如能够将肽呈递至t细胞的树突状细胞(dc)。
188.佐剂是混合至疫苗组合物中增加或以其它方式改变针对新抗原的免疫应答的任何物质。载剂可以是能够与新抗原缔合的支架结构,例如多肽或多糖。任选地,佐剂是共价或非共价缀合的。
189.佐剂增加针对抗原的免疫应答的能力通常通过免疫介导的反应的显著或实质上增加,或疾病症状的减少来表现。举例来说,体液免疫的增加通常表现为针对抗原所产生的抗体的效价的显著增加,并且t细胞活性增加通常表现为细胞增殖,或细胞毒性,或细胞因子分泌的增加。佐剂也可以通过例如将主要体液或th反应变成主要细胞或th反应来改变免疫应答。
190.适合的佐剂包括但不限于,1018iss、矾、铝盐、amplivax、as15、bcg、cp
‑
870,893、cpg7909、cyaa、dslim、gm
‑
csf、ic30、ic31、咪喹莫特(imiquimod)、imufact imp321、is patch、iss、iscomatrix、juvimmune、lipovac、mf59、单磷酰脂质a、montanide ims 1312、montanide isa 206、montanide isa 50v、montanide isa
‑
51、ok
‑
432、om
‑
174、om
‑
197
‑
mp
‑
ec、ontak、peptel载体系统、plg微粒、雷西莫特(resiquimod)、srl172、病毒颗粒和其它类病毒颗粒、yf
‑
17d、vegf捕捉剂、r848、β
‑
葡聚糖、pam3cys、aquila的来源于皂素的qs21刺激子(aquila biotech,worcester,mass.,usa)、分枝杆菌提取物和合成细菌细胞壁模拟物,以及其它专用佐剂,如ribi的detox.quil或superfos。佐剂,如不完全弗氏佐剂或gm
‑
csf是有用的。先前已描述若干专用于树突状细胞的免疫佐剂(例如mf59)和其制备方法(dupuis m等人,cell immunol.1998;186(1):18
‑
27;allison a c;dev biol stand.1998;92:3
‑
11)。也可以使用细胞因子。若干细胞因子与以下直接相关:影响树突状细胞向淋巴组织(例如tnf
‑
α)的迁移;加速树突状细胞成熟成为t淋巴细胞的有效抗原呈递细胞(例如gm
‑
csf、il
‑
1及il
‑
4)(美国专利第5,849,589号,特定地以引用的方式整体并入本文中)及充当免疫佐剂(例如il
‑
12)(gabrilovich d i等人,j immunother emphasis tumor immunol.1996(6):414
‑
418)。
191.也已经报导过cpg免疫刺激性寡核苷酸能增强佐剂在疫苗环境中的作用。也可以使用其它tlr结合分子,如rna结合性tlr 7、tlr 8和/或tlr 9。
192.有用佐剂的其它实例包括但不限于,化学修饰的cpg(例如cpr、idera)、聚(i:c)(例如聚i:ci2u)、非cpg细菌dna或rna以及免疫活性小分子和抗体,如环磷酰胺、舒尼替尼(sunitinib)、贝伐单抗(bevacizumab)、西乐葆(celebrex)、ncx
‑
4016、西地那非(sildenafil)、他达那非(tadalafil)、伐地那非(vardenafil)、索拉非尼(sorafinib)、xl
‑
999、cp
‑
547632、帕佐盘尼(pazopanib)、zd2171、azd2171、伊匹单抗(ipilimumab)、曲美单抗(tremelimumab)及sc58175,这些可以起到治疗作用和/或充当佐剂。佐剂和添加剂的量和浓度可以由熟练技术人员容易地确定,无需过度实验。其它佐剂包括集落刺激因子,如粒细胞巨噬细胞集落刺激因子(gm
‑
csf,沙格司亭(sargramostim))。
193.疫苗组合物可以包含超过一种不同的佐剂。此外,治疗组合物可以包含任何佐剂
物质,包括上述任一种或其组合。另外,预期疫苗和佐剂可以一起施用或按任何适当的次序分开施用。
194.载剂(或赋形剂)可以独立于佐剂而存在。载剂的功能可以是例如增加特定突变体的分子量以增加活性或免疫原性;赋予稳定性、增加生物活性或增加血清半衰期。此外,载剂可以帮助将肽呈递至t细胞。载剂可以是本领域技术人员已知的任何适合的载剂,例如蛋白质或抗原呈递细胞。载剂蛋白可以是但不限于匙孔血蓝蛋白、血清蛋白如转铁蛋白、牛血清白蛋白、人血清白蛋白、甲状腺球蛋白或卵白蛋白、免疫球蛋白或激素,如胰岛素或棕榈酸。对于人的免疫,载剂一般是对人生理学上可接受的载剂并且是安全的。不过,破伤风类毒素及/或白喉类毒素是适合的载剂。或者,载剂可以是葡聚糖,例如琼脂糖。
195.细胞毒性t细胞(ctl)识别呈结合至mhc分子的肽形式的抗原,而非整个外来抗原本身。mhc分子本身位于抗原呈递细胞的细胞表面上。因此,如果存在肽抗原、mhc分子和apc的三聚体复合物,则可能活化ctl。相应地,如果该肽不仅用于活化ctl,而且如果另外添加具有相应mhc分子的apc,则其可以增强免疫应答。因此,在一些实施方案中,疫苗组合物另外含有至少一种抗原呈递细胞。
196.新抗原也可以被包括在基于病毒载体的疫苗平台中,如牛痘、禽痘、自复制型α病毒、马拉巴病毒(marabavirus)、腺病毒(参见例如tatsis等人,adenoviruses,molecular therapy(2004)10,616—629)或慢病毒,包括但不限于第二代、第三代和/或混合第二/第三代慢病毒和设计成靶向特定细胞类型或受体的任何一代重组慢病毒(参见例如,hu等人,immunization delivered by lentiviral vectors for cancer and infectious diseases,immunol rev.(2011)239(1):45
‑
61;sakuma等人,lentiviral vectors:basic to translational,biochem j.(2012)443(3):603
‑
18;cooper等人,rescue of splicing
‑
mediated intron loss maximizes expression in lentiviral vectors containing the human ubiquitin c promoter,nucl.acids res.(2015)43(1):682
‑
690;zufferey等人,self
‑
inactivating lentivirus vector for safe and efficient in vivo gene delivery,j.virol.(1998)72(12):9873
‑
9880)。取决于以上提到的基于病毒载体的疫苗平台的包装能力,此方法可以递送编码一个或多个新抗原肽的一个或多个核苷酸序列。这些序列可以侧接非突变序列,可以由连接子分开,或者可以在前面具有一个或多个靶向亚细胞区室的序列(参见例如,gros等人,prospective identification of neoantigen
‑
specific lymphocytes in the peripheral blood of melanoma patients,nat med.(2016)22(4):433
‑
8;stronen等人,targeting of cancer neoantigens with donor
‑
derived t cell receptor repertoires,science.(2016)352(6291):1337
‑
41;lu等人,efficient identification of mutated cancer antigens recognized by t cells associated with durable tumor regressions,clin cancer res.(2014)20(13):3401
‑
10)。在引入宿主中后,受感染的细胞表达新抗原,并由此引起针对肽的宿主免疫(例如ctl)反应。可用于免疫方案的牛痘载体和方法描述于例如美国专利第4,722,848号中。另一载体是卡介苗(bacille calmette guerin,bcg)。bcg载体描述于stover等人(nature 351:456
‑
460(1991))中。根据本文的描述,本领域技术人员将显而易见可用于新抗原的治疗性施用或免疫的多种其它疫苗载体,例如,伤寒沙门氏菌(salmonella typhi)载体。
197.iv.a.有关疫苗设计和制造的其它考虑因素
198.iv.a.1.确定涵盖所有肿瘤亚克隆的肽集合
199.躯干肽(truncal peptide),意思指由所有或大部分肿瘤亚克隆呈递的肽,将优先被包括在疫苗中。
53
任选地,如果不存在预测会以较高机率呈递并具有免疫原性的躯干肽,或者如果预测能够以较高机率呈递并具有免疫原性的躯干肽的数量足够小以致可以在疫苗中包括其它非躯干肽,则可以通过估计肿瘤亚克隆的数量和属性并选择肽以使该疫苗所涵盖的肿瘤亚克隆的数量最大来对其它肽进行优先排序。
54
200.iv.a.2.新抗原优先排序
201.与疫苗技术可以支持的量相比,在应用所有以上新抗原过滤器后,仍有许多候选新抗原可包括在疫苗中。另外,可以保留有关新抗原分析的各个方面的不确定性,并且在候选疫苗新抗原的不同特性之间可能存在折中。因此,可以考虑用整合式多维模型代替在选择过程的每个步骤中的预定过滤器,该多维模型将候选新抗原放入具有至少以下轴的空间中并使用整合方法优化选择。
202.1.自体免疫或耐受的风险(生殖系的风险)(通常优选较低的自体免疫风险)。
203.2.测序伪影的机率(通常优选较低的伪影机率)。
204.3.免疫原性的机率(通常优选较高的免疫原性机率)。
205.4.呈递机率(通常优选较高的呈递机率)。
206.5.基因表达(通常优选较高表达)。
207.6.hla基因的覆盖率(参与呈递新抗原集合的hla分子数量增多可以降低肿瘤通过hla分子下调或突变而逃避免疫攻击的机率)。
208.7.hla类别的覆盖率(同时覆盖hla
‑
i和hla
‑
ii可能会增加治疗反应的几率并降低肿瘤逃逸的几率)。
209.另外,任选地,如果预测新抗原由患者肿瘤的全部或部分中丢失或失活的hla等位基因呈递,则可以从疫苗接种中降低(例如,排除)新抗原的优先级。hla等位基因缺失可由体细胞突变、杂合性缺失或基因座纯合缺失引起。用于检测hla等位基因体细胞突变的方法在本领域是公知的,例如(shukla等人,2015)。检测体细胞loh和纯合性缺失(包括hla基因座的缺失)的方法同样被很好地描述。(carter等人,2012;mcgranahan等人,2017;van loo等人,2010)。
210.v.治疗和制造方法
211.还提供了一种通过向受试者施用一个或多个新抗原,如使用本文所公开的方法鉴别的多个新抗原来诱导受试者的肿瘤特异性免疫应答、针对肿瘤接种疫苗、治疗和或缓解受试者的癌症症状的方法。
212.在一些方面,受试者被诊断患有癌症或有发生癌症的风险。受试者可以是需要肿瘤特异性免疫应答的人、狗、猫、马或任何动物。肿瘤可以是任何实体肿瘤,如乳房肿瘤、卵巢肿瘤、前列腺肿瘤、肺肿瘤、肾肿瘤、胃肿瘤、结肠肿瘤、睾丸肿瘤、头颈部肿瘤、胰腺肿瘤、脑肿瘤、黑素瘤及其它组织器官肿瘤;以及血液肿瘤,如淋巴瘤和白血病,包括急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、t细胞淋巴细胞性白血病及b细胞淋巴瘤。
213.新抗原的施用量应足以诱导ctl反应。
214.新抗原可以单独施用或与其它治疗剂组合施用。治疗剂是例如化学治疗剂、放射
或免疫疗法。针对特定癌症的任何适合的治疗性治疗都可以施用。
215.此外,还可以向受试者施用抗免疫抑制/免疫刺激剂,如检查点抑制剂。举例来说,还可以向受试者施用抗ctla抗体或抗pd
‑
1或抗pd
‑
l1。抗体阻断ctla
‑
4或pd
‑
l1可以增强针对患者体内癌细胞的免疫应答。确切地说,经显示,当遵循疫苗接种方案时,有效阻断ctla
‑
4。
216.可以确定包括在疫苗组合物中的各新抗原的最佳量和最佳剂量方案。举例来说,可以制备供静脉内(i.v.)注射、皮下(s.c.)注射、皮内(i.d.)注射、腹膜内(i.p.)注射、肌肉内(i.m.)注射的新抗原或其变体。注射方法包括皮下(s.c.)、皮内(i.d.)、腹腔(i.p.)、肌内(i.m.)及静脉内。dna或rna注射方法包括皮内、肌内、皮下、腹腔及静脉内。本领域技术人员已知施用疫苗组合物的其它方法。
217.疫苗可以被设计成使得组合物中存在的新抗原的选择、数量和/或量具有组织、癌症和/或患者特异性。举例来说,肽的确切选择可以由给定组织中亲本蛋白质的表达模式来指导。该选择可以取决于癌症的具体类型、疾病状态、先前的治疗方案、患者的免疫状态及当然要考虑的患者的hla单倍型。此外,根据特定患者的个人需要,疫苗还可以含有个性化组分。实例包括根据特定患者体内新抗原的表达来改变新抗原的选择或遵循第一轮治疗方案调整后续治疗。
218.对于打算用作癌症疫苗的组合物,在正常组织中大量表达的具有类似正常自身肽的新抗原应当避免或以少量存在于本文所描述的组合物中。另一方面,如果已知患者的肿瘤大量表达某一新抗原,则用于治疗此癌症的相应药物组合物可以大量存在和/或可以包括超过一种对于此特定新抗原或此新抗原的路径具有特异性的新抗原。
219.可以将包含新抗原的组合物施用给患上癌症的个体。在治疗应用中,组合物是以足以引起针对肿瘤抗原的有效ctl反应及治愈或至少部分停滞症状和/或并发症的量施用给患者。适于实现此目的的量定义为“治疗有效剂量”。有效用于此用途的量将取决于例如组成、施用方式、所治疗的疾病的分期和严重程度、患者的体重和一般健康状态,以及处方医师的判断。应了解,组合物一般可以用于严重疾病状态,也就是说,危及生命或可能危及生命的状况,特别是当癌症已经转移的时候。在此类情况下,考虑到要使外来物质最少以及新抗原的相对无毒性质,治疗医师有可能并且会感觉需要施用大体上过量的这些组合物。
220.对于治疗用途,施用可以在检测到或手术移除肿瘤时开始。这之后是增加剂量,直到至少症状大体上减轻并且之后持续一段时间。
221.用于治疗性治疗的药物组合物(例如疫苗组合物)意图用于肠胃外、表面、鼻、口或局部施用。药物组合物可以通过肠胃外施用,例如静脉内、皮下、皮内或肌肉内施用。这些组合物可以施用到手术切除的部位处以诱导针对肿瘤的局部免疫应答。本文公开了供肠胃外施用的组合物,这些组合物包含新抗原溶液并且疫苗组合物被溶解或悬浮于可接受的载剂,例如水性载剂中。可以使用多种水性载剂,例如水、缓冲水、0.9%生理盐水、0.3%甘氨酸、透明质酸等。这些组合物可以通过众所周知的常规灭菌技术灭菌,或者可以经历无菌过滤。由此得到的水溶液可以被包装起来按原样使用,或者被冻干;冻干的制剂在施用之前与无菌溶液组合。必要时,这些组合物可以含有药学上可接受的辅助物质以接近生理条件,如ph调节剂和缓冲剂、张力调节剂、润湿剂等,例如乙酸钠、乳酸钠、氯化钠、氯化钾、氯化钙、脱水山梨糖醇单月桂酸酯、三乙醇胺油酸酯等。
lymphocytes in the peripheral blood of melanoma patients,nat med.(2016)22(4):433
‑
8;stronen等人,targeting of cancer neoantigens with donor
‑
derived t cell receptor repertoires,science.(2016)352(6291):1337
‑
41;lu等人,efficient identification of mutated cancer antigens recognized by t cells associated with durable tumor regressions,clin cancer res.(2014)20(13):3401
‑
10)。在引入宿主中后,受感染的细胞表达新抗原,并由此引起针对肽的宿主免疫(例如ctl)反应。可用于免疫方案的牛痘载体和方法描述于例如美国专利第4,722,848号中。另一载体是卡介苗(bcg)。bcg载体描述于stover等人(nature 351:456
‑
460(1991))中。根据本文的描述,本领域技术人员将显而易见可用于新抗原的治疗性施用或免疫的多种其它疫苗载体。
227.施用核酸的方式使用了编码一个或多个表位的微型基因构建体。为了产生用于在人细胞中表达的编码所选ctl表位的dna序列(微型基因),对这些表位的氨基酸序列进行逆翻译。使用人密码子用法表指导各氨基酸的密码子选择。将这些表位编码dna序列直接邻接,产生连续多肽序列。为了优化表达和/或免疫原性,可以将另外的元件并入微型基因设计中。可以被逆翻译并且包括在微型基因序列中的氨基酸序列的实例包括:辅助t淋巴细胞、表位、前导(信号)序列及内质网滞留信号。此外,通过邻近ctl表位包括合成(例如聚丙氨酸)或天然存在的侧接序列可以改善ctl表位的mhc呈递。通过组装编码微型基因正链和负链的寡核苷酸,将微型基因序列转化成dna。使用众所周知的技术,在适当条件下合成、磷酸化、纯化重叠寡核苷酸(30
‑
100个碱基长)并使其退火。使用t4 dna连接酶接合寡核苷酸的末端。接着,可以将这一编码ctl表位多肽的合成微型基因克隆至所希望的表达载体中。
228.可以使用多种配制物制备注射用纯化质粒dna。这些方法中最简单的方法是在无菌磷酸盐缓冲生理盐水(pbs)中使冻干的dna复水。多种方法已有描述,并且新技术也可以使用。如上文所述,核酸宜用阳离子性脂质配制。此外,还可以使统称为保护性、相互作用性、非缩合性(pinc)的糖酯、促融脂质体、肽和化合物与纯化的质粒dna形成复合物以影响各种变量,如稳定性、肌肉内分散或向特定器官或细胞类型的运输。
229.还公开了一种制造肿瘤疫苗的方法,该方法包括执行本文所公开的方法的各个步骤;及产生包含多个新抗原或该多个新抗原的子集的肿瘤疫苗。
230.本文所公开的新抗原可以使用本领域中已知的方法制造。举例来说,本文所公开的产生新抗原或载体(例如包括至少一个编码一个或多个新抗原的序列的载体)的方法可以包括在适于表达该新抗原或载体的条件下培养宿主细胞,其中该宿主细胞包含至少一个编码该新抗原或载体的多核苷酸;及纯化该新抗原或载体。标准纯化方法包括色谱技术、电泳技术、免疫技术、沉淀、透析、过滤、浓缩及等电聚焦技术。
231.宿主细胞可以包括中国仓鼠卵巢(cho)细胞、ns0细胞、酵母或hek293细胞。宿主细胞可以用一个或多个多核苷酸转化,该一个或多个多核苷酸包含至少一个编码本文所公开的新抗原或载体的核酸序列,任选地其中分离的多核苷酸另外包含可操作地连接到该至少一个编码新抗原或载体的核酸序列的启动子序列。在某些实施方案中,该分离的多核苷酸可以是cdna。
232.vi.新抗原鉴别
233.vi.a.新抗原候选物的鉴别。
234.有关以ngs分析肿瘤和正常外显子组和转录组的研究方法已有描述且被应用于新
抗原鉴别邻域中。
6,14,15
以下实施例考虑了在临床环境中对于新抗原鉴别具有较高灵敏度和特异性的某些优化措施。这些优化措施可以分为两个领域,即与实验室方法有关的优化和与ngs数据分析有关的优化。
235.vi.a.1.实验室方法优化
236.此处提出的方法改进通过将所开发的有关可靠地评估靶癌症组中的癌症驱动基因的概念
16
扩展至新抗原鉴别所需的全外显子组和全转录组环境,解决了从肿瘤含量较低并且体积较小的临床试样中高准确性发现新抗原的难题。确切地说,这些改进包括:
237.1.靶向整个肿瘤外显子组的深度(>500
×
)独特平均覆盖率,以检测由于肿瘤含量低或处于亚克隆状态而以低突变等位基因频率存在的突变。
238.2.靶向整个肿瘤外显子组的均匀覆盖率,其中在<100
×
下覆盖<5%的碱基,由此通过例如以下方式使遗漏新抗原的可能性最低:
239.a.采用基于dna的捕捉探针和个别探针qc
17
240.b.包括针对覆盖较少的区域的额外诱饵
241.3.靶向整个正常外显子组的均匀覆盖率,其中在<20
×
下覆盖<5%的碱基,由此对于体细胞/生殖系状态可能有最少的新抗原未被分类(并因此不能用作tsna)
242.4.为了使需要测序的总量减到最少,序列捕捉探针应被设计成仅针对基因编码区,因为非编码rna不会产生新抗原。其它优化包括:
243.a.针对hla基因的补充探针,这些基因富含gc并且通过标准外显子组测序很难捕捉
18
244.b.排除由于如表达水平不足、蛋白酶体消化欠佳或不常见的序列特征等因素而被预测产生极少或不产生候选新抗原的基因。
245.5.肿瘤rna将通常同样在高深度(>100m个读段)下测序,以便能够进行变体检测、基因和剪接变体(“同功型”)表达水平的定量,及融合物检测。来自ffpe样品的rna将使用基于探针的富集方法
19
,使用与捕捉dna中的外显子组相同或类似的探针进行提取。
246.vi.a.2.ngs数据分析优化
247.分析方法的改进解决了常用研究突变调用方法灵敏度和特异性欠佳的问题,并且特别考虑到了在临床环境中与新抗原鉴别相关的定制。这些包括:
248.1.使用hg38参考人基因组或后续版本进行比对,因为相对于先前的基因组版本,该基因组含有多个mhc区域组装体,较佳地反映了群体多态性。
249.2.通过合并由不同程序得到的结果5,克服单个变体调用程序的局限性
20
250.a.利用一套工具,检测肿瘤dna、肿瘤rna及正常dna中的单核苷酸变体和插入缺失,该套工具包括:基于肿瘤与正常dna的比较的程序,如strelka
21
和mutect
22
;和并入了肿瘤dna、肿瘤rna及正常dna的程序,如unceqr,特别适用于低纯度样品
23
。
251.b.插入缺失将利用执行局部再组装的程序测定,如strelka和abra
24
。
252.c.结构重排将使用专用工具测定,如pindel
25
或breakseq
26
。
253.3.为了检测并防止样品调换,将在选定的多态性位点数量下,比较来自同一患者的样品中的变体调用。
254.4.针对伪调用的广泛过滤将例如通过以下方式进行:
255.a.移除在正常dna中发现的变体,在低覆盖率下可能使用不严格的检测参数,并且
在插入缺失情况下使用容许的接近标准。
256.b.移除由低定位质量或低碱基质量引起的变体
27
。
257.c.移除来源于反复出现的测序伪影的变体,即使在相应的正常情况下未观察到
27
。实例包括主要在一条链上检测到的变体。
258.d.移除不相关的对照物集合中检测到的变体
27
259.5.使用seq2hla
28
、athlates
29
或optitype之一,从正常外显子组中准确地调用hla,并且还将外显子组与rna测序数据组合
28
。其它可能的优化包括采用专用于hla分型的分析,如长读段dna测序
30
,或调适用于接合rna片段的方法以保持连续性
31
。
260.6.针对由肿瘤特异性剪接变体产生的neo
‑
orf的稳健检测将通过使用class
32
、bayesembler
33
、stringtie
34
或类似程序以其参考引导的模式,根据rna
‑
seq数据组装转录物来进行(即,使用已知的转录物结构而非尝试在每个实验中重新构建整个转录物)。尽管cufflinks
35
通常被用于此目的,但它常常会不合情理地产生大量剪接变体,其中有许多比全长基因要短得多,并且无法回收简单的阳性对照。编码序列及无义介导的衰变可能性将通过如splicer
36
和mamba
37
等工具,利用重新引入的突变序列测定。基因表达将利用如cufflinks
35
或express(roberts和pachter,2013)等工具测定。野生型和突变体特异性表达计数和/或相对水平将利用开发用于这些目的的工具,如ase
38
或htseq
39
测定。可能的过滤步骤包括:
261.a.移除被认为表达不足的候选neo
‑
orf。
262.b.移除被预测会触发无义介导的衰变(nmd)的候选neo
‑
orf。
263.7.仅在rna中观察到的无法直接验证为肿瘤特异性抗原的候选新抗原(例如neoorf)将根据额外参数,例如通过考虑以下因素而归类为可能是肿瘤特异性的:
264.a.存在仅支持肿瘤dna的顺式作用移码或剪接位点突变
265.b.在剪接因子中存在仅证实肿瘤dna的反式作用突变。举例来说,在利用r625突变型sf3b1进行的三个独立公布的实验中,尽管一个实验检查到葡萄膜黑素瘤患者
40
,第二个实验检查到葡萄膜黑素瘤细胞系
41
,而第三个实验检查到乳癌患者
42
,但展现最大剪接差异的基因是一致的。
266.c.对于新剪接同功型,在rnaseq数据中存在确证的“新”剪接
‑
接合读段。
267.d.对于新重排,有确证在肿瘤dna中存在而在正常dna中不存在的近似外显子读段
268.e.基因表达概略中缺乏,如gtex
43
(即,使得不太可能为生殖系起源)
269.8.通过直接比较组装的dna肿瘤与正常读段(或来自这些读段的k
‑
mer)来补充基于参考基因组比对的分析以避免基于比对和注释的错误和伪影。(例如对于在生殖系变体或重复序列插入缺失附近出现的体细胞变体)
270.在具有聚腺苷酸化rna的样品中,rna
‑
seq数据中病毒和微生物rna的存在将使用rna compass
44
或类似方法评估,以鉴别可以预测患者响应的其它因素。
271.vi.b.hla肽的分离和检测
272.hla
‑
肽分子的分离在溶胞和溶解组织样品之后,使用经典免疫沉淀(ip)方法进行
55
‑
58
。使用澄清的溶解产物进行hla特异性ip。
273.免疫沉淀是使用偶合至珠粒的抗体进行,其中该抗体对hla分子具有特异性。对于全i类hla免疫沉淀,使用全i类cr抗体,对于ii类hla
–
dr,使用hla
–
dr抗体。在过夜培育期
间,将抗体共价连接至nhs
‑
琼脂糖珠粒。在共价连接后,洗涤珠粒并等分试样用于ip。
59,60
免疫沉淀也可以使用未共价结合至磁珠的抗体进行。通常,使用包被有蛋白a和/或蛋白g的琼脂糖或磁珠将抗体固定在色谱柱上来完成此操作。下面列出了一些可用于选择性富集mhc/肽复合物的抗体。
274.抗体名称特异性w6/32i类hla
‑
a,b,cl243ii类
–
hla
‑
drtu36ii类
–
hla
‑
drln3ii类
–
hla
‑
drtu39ii类
–
hla
‑
dr,dp,dq
275.将澄清的组织溶解产物添加至抗体珠粒中进行免疫沉淀。免疫沉淀后,从溶解产物移除珠粒,并储存溶解产物用于另外的实验,包括另外的ip。洗涤ip珠粒以移除非特异性结合并使用标准技术,从珠粒洗脱下hla/肽复合物。使用分子量旋转柱或c18分级分离,从肽移除蛋白质组分。通过speedvac蒸发使所得肽变干并且在一些情形中在
‑
20c下储存以待ms分析。
276.干燥的肽在适于反相色谱法的hplc缓冲液中复水并装载至c
‑
18微毛细管hplc柱上以在fusion lumos质谱仪(thermo)中进行梯度洗脱。在orbitrap检测器中在高分辨率下收集肽质/荷比(m/z)的ms1谱,随后在所选离子经历hcd片段化后,在离子阱检测器中收集ms2低分辨率扫描谱。另外,可以使用cid或etd片段化方法,或三种技术的任何组合获得ms2谱,以达到该肽的较高氨基酸覆盖率。还可以在orbitrap检测器中用高分辨率质量精度测量ms2谱。
277.使用comet
61,62
,针对蛋白质数据库搜索由各分析得到的ms2谱并使用percolator
63
‑
65
对肽鉴别进行评分。可以使用peaks studio(bioinformatics solutions inc.)进行另外的测序,并且可以使用其它搜索引擎或其它测序方法,包括光谱匹配和从头测序
75
。
278.vi.b.1.支持全面hla肽测序的ms检测限研究。
279.使用肽yvyvadvaak,利用装载至lc柱上的不同量的肽确定检测限。测试肽的量是1pmol、100fmol、10fmol、1fmol及100amol。(表1)结果显示于图1f中。这些结果表明,最低检测限(lod)是埃摩尔(attomol)范围(10
‑
18
),动态范围跨五个数量级,并且信噪比看来足以在低飞摩尔(femtomol)范围(10
‑
15
)内进行测序。
280.肽m/z装载于柱上在1e9个细胞中的拷贝数/细胞566.8301pmol600562.823100fmol60559.81610fmol6556.8101fmol0.6553.802100amol0.06
281.vii.呈递模型
282.vii.a.系统综述
283.图2a是根据一个实施方案,用于鉴别患者体内肽呈递的可能性的环境100的概述。
环境100提供背景以便引入呈递鉴别系统160,该系统本身包括呈递信息存储器165。
284.呈递鉴别系统160是一个或多个在如以下关于图21所论述的计算系统中体现的计算机模型,其接收与mhc等位基因集合有关的肽序列并测定这些肽序列将被该相关mhc等位基因集合中的一个或多个mhc等位基因呈递的可能性。呈递鉴别系统160可以应用于i类和ii类mhc等位基因两者。这在多种情形中都适用。呈递鉴别系统160的一个具体使用情形是,它能够接收与来自患者110的肿瘤细胞的mhc等位基因集合有关的候选新抗原的核苷酸序列,并测定这些候选新抗原将被该肿瘤的相关mhc等位基因中的一个或多个呈递和/或在患者110的免疫系统中诱导免疫原性反应的可能性。可以选出被系统160测定具有高可能性的候选新抗原用于包括在疫苗118中,此类抗肿瘤免疫应答可以由提供肿瘤细胞的患者110的免疫系统引发。另外,可以产生具有tcr的t细胞以用于t细胞疗法,所述t细胞对具有高呈递可能性的候选新抗原具有响应,从而还引起来自患者110的免疫系统的抗肿瘤免疫应答。
285.呈递鉴别系统160通过一个或多个呈递模型测定呈递可能性。确切地说,呈递模型生成给定肽序列是否将由相关mhc等位基因集合呈递的可能性,并且这是基于存储在存储器165中的呈递信息生成的。举例来说,呈递模型可以生成肽序列“yvyvadvaak”是否将由等位基因hla
‑
a*02:01、hla
‑
a*03:01、hla
‑
b*07:02、hla
‑
b*08:03、hla
‑
c*01:04的集合呈递于样品的细胞表面上的可能性。呈递信息165含有关于肽是否结合至不同类型的mhc等位基因以使得这些肽被mhc等位基因呈递的信息,该信息在模型中是根据肽序列中氨基酸的位置确定。呈递模型可以基于呈递信息165预测未被识别的肽序列的呈递是否会与相关mhc等位基因集合相关联。如前所述,呈递模型可以应用于i类和ii类mhc等位基因两者。
286.vii.b.呈递信息
287.图2示出了根据一个实施方案的获得呈递信息的方法。呈递信息165包括两个通用信息类别:等位基因相互作用信息和等位基因非相互作用信息。等位基因相互作用信息包括影响与mhc等位基因的类型相关的肽序列的呈递的信息。等位基因非相互作用信息包括影响与mhc等位基因的类型无关的肽序列的呈递的信息。
288.vii.b.1.等位基因相互作用信息
289.等位基因相互作用信息主要包括经过鉴别的肽序列,已知这些肽序列已经被来自人、小鼠等的一个或多个经过鉴别的mhc分子呈递。值得注意的是,这可能包括或可能不包括从肿瘤样品获得的数据。可以从表达单个mhc等位基因的细胞鉴别出所呈递的肽序列。在这一情形中,所呈递的肽序列一般是从单个等位基因细胞系收集,这些细胞系被工程改造成表达预定mhc等位基因并且随后暴露于合成蛋白质。在mhc等位基因上呈递的肽是通过如酸洗脱等技术分离并通过质谱法鉴别。图2b示出了这一情形的一个实施例,其中分离出在预定mhc等位基因hla
‑
drb1*12:01上呈递的示例肽yemfndksqrapddkmf并通过质谱法鉴别。由于在此情况下,肽是通过被工程改造成表达单一预定mhc蛋白质的细胞鉴别,故呈递的肽与其所结合的mhc蛋白质之间的直接关联是确定已知的。
290.也可以从表达多个mhc等位基因的细胞收集所呈递的肽序列。通常,在人体中,一种细胞表达6种不同类型的mhc
‑
i和至多12种不同类型的mhc
‑
ii分子。如此呈递的肽序列可以从被工程改造成表达多个预定mhc等位基因的多等位基因细胞系鉴别到。还可以从组织样品,如正常组织样品或肿瘤组织样品鉴别如此呈递的肽序列。特别就这一情形来说,mhc分子可以从正常或肿瘤组织免疫沉淀。在多个mhc等位基因上呈递的肽可类似地通过如酸
洗脱等技术分离并通过质谱法鉴别。图2c示出了此种情形的一个实施例,其中将六个示例肽yemfndksf、hroeifshdfj、fjiejfoess、neioreirei、jfksifemmsjdssuiflksjfieifj及knflenfiesofi呈递于所鉴别的i类mhc等位基因hla
‑
a*01:01、hla
‑
a*02:01、hla
‑
b*07:02、hla
‑
b*08:01及ii类mhc等位基因hla
‑
drb1*10:01、hla
‑
drb1:11:01并且分离,并通过质谱法鉴别。相对于单等位基因细胞系,呈递的肽与其所结合的mhc蛋白质之间的直接关联可能是未知的,因为结合肽是在鉴别之前与mhc分子分离。
291.等位基因相互作用信息还可以包括质谱离子流,其取决于肽
‑
mhc分子复合物的浓度和肽电离效率。电离效率以序列依赖性方式随肽而变化。一般来说,电离效率随肽而在约两个数量级内变化,而肽
‑
mhc复合物的浓度在比其更大的范围内变化。
292.等位基因相互作用信息还可以包括给定mhc等位基因与给定肽之间结合亲和力的测量或预测。(72,73,74)一个或多个亲和力模型可以生成此类预测。举例来说,再看回图1d中所示的实施例,呈递信息165可以包括肽yemfndksf与等位基因i类hla
‑
a*01:01之间的1000nm的结合亲和力预测值。ic50>1000nm的肽很少被mhc呈递,且较低的ic50值使呈递机率增加。呈递信息165可以包括肽knflenfiesofi和ii类等位基因hla
‑
drb1:11:01之间的结合亲和力预测。
293.等位基因相互作用信息也可以包括该mhc复合物稳定性的测量或预测。一个或多个稳定性模型可以生成此类预测。较稳定的肽
‑
mhc复合物(即,半衰期较长的复合物)比较可能在肿瘤细胞上及在遭遇疫苗抗原的抗原呈递细胞上以高拷贝数呈递。举例来说,再看回图2c中所示的实施例,呈递信息165可以包括i类分子hla
‑
a*01:01的半衰期是1小时的稳定性预测值。呈递信息165可以包括ii类分子hla
‑
drb1:11:01的半衰期的稳定性预测值。
294.等位基因相互作用信息也可以包括测量或预测的肽
‑
mhc复合物的形成反应速率。以较高速率形成的复合物比较可能以高浓度呈递于细胞表面上。
295.等位基因相互作用信息还可以包括肽的序列和长度。i类mhc分子通常偏好呈递长度介于8与15个肽之间的肽。所呈递的肽中有60
‑
80%的长度是9个。ii类mhc分子通常更优先呈递介于6到30个肽之间的肽。
296.等位基因相互作用信息还可以包括新抗原编码肽上激酶序列基元的存在,以及新抗原编码肽上特定翻译后修饰的不存在或存在。激酶基元的存在会影响翻译后修饰的机率,该翻译后修饰可能增强或干扰mhc结合。
297.等位基因相互作用信息还可以包括翻译后修饰过程中所涉及的蛋白质,例如激酶的表达水平或活性水平(如由rna seq、质谱法或其它方法所测量或预测)。
298.等位基因相互作用信息还可以包括来自表达特定mhc等位基因的其它个体的细胞中具有相似序列的肽的呈递机率,这可通过质谱蛋白组学或其它手段评估。
299.等位基因相互作用信息还可以包括所讨论的个体中特定mhc等位基因的表达水平(例如,如通过rna
‑
seq或质谱法测量)。相较于最强地结合至以低水平表达的mhc等位基因的肽,最强地结合至以高水平表达的mhc等位基因的肽比较可能被呈递。
300.等位基因相互作用信息还可以包括不依赖于总体新抗原编码肽序列而在表达特定mhc等位基因的其它个体中由特定mhc等位基因呈递的机率。
301.等位基因相互作用信息还可以包括不依赖于总体肽序列而在其它个体中由同一家族分子(例如hla
‑
a、hla
‑
b、hla
‑
c、hla
‑
dq、hla
‑
dr、hla
‑
dp)中的mhc等位基因呈递的机
率。举例来说,hla
‑
c分子的表达水平通常低于hla
‑
a或hla
‑
b分子,且由此可推断,由hla
‑
c呈递肽的机率低于由hla
‑
a或hla
‑
b呈递的机率。再举一个例子,hla
‑
dp的表达水平通常低于hla
‑
dr或hla
‑
dq,且由此可推断,由hla
‑
dp呈递肽的机率低于由hla
‑
dr或hla
‑
dq呈递的机率。
302.等位基因相互作用信息还可以包括特定mhc等位基因的蛋白质序列。
303.以下部分中所列的任何mhc等位基因非相互作用信息也可以按mhc等位基因相互作用信息的方式进行建模。
304.vii.b.2.等位基因非相互作用信息
305.等位基因非相互作用信息可以包括在源蛋白质序列内侧接新抗原编码肽的c端序列。对于mhc
‑
i,c端侧接序列可能影响肽的蛋白酶体加工。不过,c端侧接序列是在肽转运至内质网并遇到细胞表面上的mhc等位基因之前,在蛋白酶体作用下自该肽裂解得到。因此,mhc分子接收不到有关c端侧接序列的信息,且由此,c端侧接序列的影响不会随mhc等位基因类型而变化。举例来说,再参看图2c中所示的实施例,呈递信息165可以包括从肽的源蛋白鉴别到的呈递肽fjiejfoess的c端侧接序列foeifndksldkfji。
306.等位基因非相互作用信息也可以包括mrna定量测量。举例来说,可以获得与提供质谱训练数据相同的样品的mrna定量数据。如稍后参照图14g所描述,rna表达水平被鉴别为肽呈递的强预测因子。在一个实施方案中,mrna定量测量值是由软件工具rsem鉴别得到。有关rsem软件工具的详细实施方式可见于bo li及colin n.dewey.rsem:accurate transcript quantification from rna
‑
seq data with or without a reference genome.bmc bioinformatics,12:323,2011年8月。在一个实施方案中,mrna定量是以每一百万条定位读段数中每千碱基转录物的片段数(fpkm)为单位度量。
307.等位基因非相互作用信息还可以包括在源蛋白质序列内侧接所述肽的n端序列。
308.等位基因非相互作用信息还可以包括肽序列的源基因。可以将源基因定义为肽序列的ensembl蛋白家族。在另一些例子中,源基因可以被定义为肽序列的源dna或源rna。可以例如将源基因表示为编码蛋白质的一串核苷酸,或者基于已知编码特定蛋白质的已知dna或rna序列的命名集合将更直接地表示。在另一个例子中,等位基因非相互作用信息还可以包括从数据库如ensembl或refseq中提取的肽序列的源转录本或同工型或潜在的源转录本或同工型的集合。
309.等位基因非相互作用信息还可以包括肽序列来源的细胞的组织类型、细胞类型或肿瘤类型。
310.等位基因非相互作用信息还可以包括在该肽中蛋白酶裂解基元的存在,任选地根据肿瘤细胞中相应蛋白酶的表达(如通过rna
‑
seq或质谱法测量)加权。含有蛋白酶裂解基元的肽不太可能被呈递,因为这些肽比较容易被蛋白酶降解,并因此在细胞内不太稳定。
311.等位基因非相互作用信息还可以包括如在适当细胞类型中测量的源蛋白的转换率。转换率较快(即,半衰期较短)会增加呈递机率;不过,如果在不相似的细胞类型中测量,则此特征的预测能力较低。
312.等位基因非相互作用信息还可以包括如通过rna
‑
seq或蛋白质组质谱法所测量,或如根据在dna或rna序列数据中检测到的生殖系或体细胞剪接突变的注释所预测的源蛋白的长度,任选地考虑在肿瘤细胞中表达水平最高的特定剪接变体(“同功型”)。
313.等位基因非相互作用信息还可以包括肿瘤细胞中蛋白酶体、免疫蛋白酶体、胸腺蛋白酶体或其它蛋白酶的表达水平(可以通过rna
‑
seq、蛋白质组质谱法或免疫组织化学分析测量)。不同的蛋白酶体具有不同的裂解位点偏好。与表达水平成比例的各类型蛋白酶体的裂解偏好将被给予较大权重。
314.等位基因非相互作用信息还可以包括肽的源基因的表达水平(例如通过rna
‑
seq或质谱法测量)。可能的优化措施包括调整表达水平测量值以说明肿瘤样品内基质细胞和肿瘤浸润淋巴细胞的存在。来自表达水平较高的基因的肽比较可能被呈递。来自表达水平不可检测的基因的肽可以不予考虑。
315.等位基因非相互作用信息还可以包括如由无义介导的衰变模型,例如来自rivas等人,science 2015的模型所预测的新抗原编码肽的源mrna将经历无义介导的衰变的机率。
316.等位基因非相互作用信息还可以包括在各种细胞周期阶段期间肽的源基因的典型肿瘤特异性表达水平。以总体较低水平表达(如通过rna
‑
seq或质朴蛋白质组学所测量)但已知在特定细胞周期阶段期间高水平表达的基因所产生的呈递肽可能多于以极低水平稳定表达的基因。
317.等位基因非相互作用信息还可以包括例如uniprot或pdb http://www.rcsb.org/pdb/home/home.do中提供的源蛋白特征的综合目录。这些特征尤其可以包括:蛋白质的二级和三级结构、亚细胞定位11、基因本体(gene ontology,go)项。确切地说,这一信息可以含有在蛋白质水平上起作用的注释,例如5’utr长度;及在特定残基水平上起作用的注释,例如在残基300与310之间的螺旋基元。这些特征还可以包括转角基元、折叠基元和无序残基。
318.等位基因非相互作用信息还可以包括描述含有该肽的源蛋白的结构域的特性的特征,例如:二级或三级结构(例如α螺旋对比β折叠);选择性剪接。
319.等位基因非相互作用信息还可以包括描述在该肽的源蛋白中该肽的位置处存在或不存在呈递热点的特征。
320.等位基因非相互作用信息还可以包括其它个体中来自相关肽的源蛋白的肽的呈递机率(在调整这些个体中源蛋白的表达水平及这些个体的不同hla类型的影响之后)。
321.等位基因非相互作用信息还可以包括由于技术偏差而无法通过质谱法检测到或过量表示该肽的机率。
322.通过基因表达测定如rnaseq、微阵列、靶向组如nanostring所测量的各种基因模块/路径的表达,或通过如rt
‑
pcr等测定(无需含有该肽的源蛋白)所测量的基因模块的单基因/多基因代表提供了有关肿瘤细胞、基质或肿瘤浸润淋巴细胞(til)的状态的信息。
323.等位基因非相互作用信息还可以包括肿瘤细胞中肽的源基因的拷贝数。举例来说,在肿瘤细胞中经历纯合子缺失的基因的肽可以指定为呈递机率是零。
324.等位基因非相互作用信息还可以包括肽结合至tap的机率或肽与tap的结合亲和力测量值或预测值。比较可能结合至tap的肽,或以较高亲和力结合tap的肽比较可能被mhc
‑
i呈递。
325.等位基因非相互作用信息还可以包括肿瘤细胞中tap的表达水平(可以通过rna
‑
seq、蛋白质组质谱法、免疫组织化学分析测量)。对于mhc
‑
i,较高的tap表达水平会增加所
有肽的呈递机率。
326.等位基因非相互作用信息还可以包括肿瘤突变的存在或不存在,包括但不限于:
327.i.已知癌症驱动基因,如egfr、kras、alk、ret、ros1、tp53、cdkn2a、cdkn2b、ntrk1、ntrk2、ntrk3中的驱动突变
328.ii.编码抗原呈递机器中所涉及的蛋白质的基因(例如b2m、hla
‑
a、hla
‑
b、hla
‑
c、tap
‑
1、tap
‑
2、tapbp、calr、cnx、erp57、hla
‑
dm、hla
‑
dma、hla
‑
dmb、hla
‑
do、hla
‑
doa、hla
‑
dobhla
‑
dp、hla
‑
dpa1、hla
‑
dpb1、hla
‑
dq、hla
‑
dqa1、hla
‑
dqa2、hla
‑
dqb1、hla
‑
dqb2、hla
‑
dr、hla
‑
dra、hla
‑
drb1、hla
‑
drb3、hla
‑
drb4、hla
‑
drb5或编码蛋白酶体或免疫蛋白酶体的组分的任何基因)中的突变。呈递依赖于肿瘤中经历功能丧失性突变的抗原呈递机器组分的肽具有降低的呈递机率。
329.存在或不存在功能性生殖系多态现象,包括但不限于:
330.i.编码抗原呈递机器中所涉及的蛋白质的基因(例如b2m、hla
‑
a、hla
‑
b、hla
‑
c、tap
‑
1、tap
‑
2、tapbp、calr、cnx、erp57、hla
‑
dm、hla
‑
dma、hla
‑
dmb、hla
‑
do、hla
‑
doa、hla
‑
dobhla
‑
dp、hla
‑
dpa1、hla
‑
dpb1、hla
‑
dq、hla
‑
dqa1、hla
‑
dqa2、hla
‑
dqb1、hla
‑
dqb2、hla
‑
dr、hla
‑
dra、hla
‑
drb1、hla
‑
drb3、hla
‑
drb4、hla
‑
drb5或编码蛋白酶体或免疫蛋白酶体的组分的任何基因)中的功能性生殖系多态现象
331.等位基因非相互作用信息还可以包括肿瘤类型(例如nsclc、黑素瘤)。
332.等位基因非相互作用信息还可以包括hla等位基因的已知功能,如由例如hla等位基因的后缀所反映。举例来说,等位基因名称hla
‑
a*24:09n中的n后缀指示未表达并因此不可能呈递表位的无效等位基因;完整hla等位基因后缀命名法描述于https://www.ebi.ac.uk/ipd/imgt/hla/nomenclature/suffixes.html。
333.等位基因非相互作用信息还可以包括临床肿瘤亚型(例如鳞状肺癌对比非鳞状肺癌)。
334.等位基因非相互作用信息也可以包括吸烟史。
335.等位基因非相互作用信息还可以包括晒伤史、太阳曝晒史或暴露于其它诱变剂的历史。
336.等位基因非相互作用信息还可以包括肽的源基因在相关肿瘤类型或临床亚型中的典型表达,任选地利用驱动基因突变分层。通常在相关肿瘤类型中高水平表达的基因比较可能被呈递。
337.等位基因非相互作用信息还可以包括所有肿瘤中,或同一类型肿瘤中,或来自具有至少一个共有mhc等位基因的个体的肿瘤中,或具有至少一个共有mhc等位基因的个体体内的同一类型肿瘤中的突变频率。
338.就突变的肿瘤特异性肽而言,用于预测呈递机率的特征清单也可以包括突变注释(例如错义、通读、移码突变、融合等)或预测该突变是否会引起无义介导的衰变(nmd)。举例来说,来自因纯合子早期终止突变而在肿瘤细胞中不翻译的蛋白质区段的肽可以指定为呈递机率是零。nmd使mrna翻译减少,由此降低呈递机率。
339.vii.c.呈递鉴别系统
340.图3是一个高级框图,示出了根据一个实施方案的呈递鉴别系统160的计算机逻辑组件。在本示例实施方案中,呈递鉴别系统160包括数据管理模块312、编码模块314、训练模
块316及预测模块320。呈递鉴别系统160还包括训练数据存储器170和呈递模型存储器175。该模型管理系统160的一些实施方案具有与此处所描述不同的模块。类似地,这些模块的功能分布可能不同于此处描述的模块。
341.vii.c.1.数据管理模块
342.数据管理模块312根据呈递信息165生成数组训练数据170。每组训练数据含有多个数据实例,其中每个数据实例i含有一组自变量z
i
,这些自变量包括至少一个呈递或不呈递肽序列p
i
、一个或多个与该肽序列p
i
相关联的相关mhc等位基因a
i
;和一个因变量y
i
,该因变量表示呈递鉴别系统160有意预测自变量的新值的信息。
343.在本说明书其余部分通篇提到的一个特定的实施方式中,因变量y
i
是一种二元标记,指示肽p
i
是否被该一个或多个相关mhc等位基因a
i
呈递。不过,应理解,在其它实施方式中,取决于自变量z
i
,因变量y
i
可以表示呈递鉴别系统160有意进行预测的任何其它类别的信息。举例来说,在另一实施方案中,因变量y
i
还可以是指示所鉴别的数据实例的质谱离子电流的数值。
344.数据实例i的肽序列p
i
是具有k
i
个氨基酸的序列,其中k
i
可以在随数据实例i而在一定范围内变化。举例来说,该范围对于i类mhc可以是8
‑
15,或对于ii类mhc是6
‑
30。在系统160的一个具体实施方案中,一个训练数据集中的所有肽序列p
i
可以具有相同长度,例如9。肽序列中氨基酸的数量可以取决于mhc等位基因的类型(例如人体中的mhc等位基因等)而变化。数据实例i的mhc等位基因a
i
指示存在的与相应肽序列p
i
相关的mhc等位基因。
345.数据管理模块312还可以包括另外的等位基因相互作用变量,如与训练数据170中所包含的肽序列p
i
和相关mhc等位基因a
i
有关的结合亲和力b
i
和稳定性预测值s
i
。举例来说,训练数据170可以含有肽p
i
与以a
i
指示的各相关mhc分子之间的结合亲和力预测值b
i
。在另一个实施例中,训练数据170可以含有以a
i
指示的各mhc等位基因的稳定性预测值s
i
。
346.数据管理模块312还可以包括等位基因非相互作用变量w
i
,如与肽序列p
i
有关的c端侧接序列和mrna定量测量值。
347.数据管理模块312还鉴别不被mhc等位基因呈递的肽序列,以生成训练数据170。一般来说,这涉及在呈递之前,鉴别包括呈递肽序列在内的源蛋白的“较长”序列。当呈递信息含有工程改造的细胞系时,数据管理模块312鉴别这些细胞所暴露的合成蛋白质中未呈递于细胞的mhc等位基因上的一系列肽序列。当呈递信息含有组织样品时,数据管理模块312鉴别作为呈递肽序列的来源的源蛋白,并且鉴别源蛋白中未呈递于组织样品细胞的mhc等位基因上的一系列肽序列。
348.数据管理模块312还可以利用随机氨基酸序列人工产生肽,并将所产生的序列鉴别为不呈递于mhc等位基因上的肽。这可以通过随机产生肽序列实现,使得数据管理模块312能够容易地生成大量有关不呈递于mhc等位基因上的肽的合成数据。由于实际上,只有少量肽序列被mhc等位基因呈递,故合成产生的肽序列很有可能不会被mhc等位基因呈递,即使这些序列被包括在细胞加工的蛋白质中。
349.图4示出了根据一个实施方案的一组示例性训练数据170a。确切地说,训练数据170a中的前3个数据实例指示由包含等位基因hla
‑
c*01:03以及3个肽序列qceiowareflkeigj、fieuhfwi及fewrhrjtrujr的单等位基因细胞系得到的肽呈递信息。训练数据170a中的第四个数据实例指示由包含等位基因hla
‑
b*07:02、hla
‑
c*01:03、hla
‑
a*
01:01和一个肽序列qiejoeije的多等位基因细胞系得到的肽信息。第一个数据实例指示,肽序列qceioware不被等位基因hla
‑
drb3:01:01呈递。如前两段所论述,阴性标记的肽序列可以由数据管理模块312随机产生,或从呈递肽的源蛋白鉴别得到。训练数据170a还包括肽序列
‑
等位基因对的1000nm的结合亲和力预测值以及半衰期是1小时的稳定性预测值。训练数据170a还包括等位基因非相互作用变量,如肽fjelfisbosjfie的c端侧接序列,以及102tpm的mrna定量测量值。第四个数据实例指示,肽序列qiejoeije被等位基因hla
‑
b*07:02、hla
‑
c*01:03或hla
‑
a*01:01之一呈递。训练数据170a还包括各等位基因的结合亲和力预测值和稳定性预测值,以及该肽的c端侧接序列和该肽的mrna定量测量值。
350.vii.c.2.编码模块
351.编码模块314将训练数据170中所包含的信息编码成可以用于产生一个或多个呈递模型的数字表示。在一个实施方案中,编码模块314在预定的20字母氨基酸字母表内独热编码序列(例如肽序列或c端侧接序列)。确切地说,具有k
i
个氨基酸的肽序列p
i
表示为具有20
·
k
i
个元素的行向量,其中p
i20
·
(j
‑
1) 1
,p
i20
·
(j
‑
1) 2
,
…
,p
i20
·
j
当中对应于字母表中在肽序列第j位的氨基酸的单一元素的值是1。另外,其余元素的值是0。举个例子,对于给定的字母表{a,c,d,e,f,g,h,i,k,l,m,n,p,q,r,s,t,v,w,y},数据实例i的具有3个氨基酸的肽序列eaf可以由具有60个元素的行向量表示p
i
=[0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0]。c端侧接序列c
i
,以及mhc等位基因的蛋白质序列d
h
和呈递信息中的其它序列数据都可以按与上文所描述类似的方式编码。
[0352]
当训练数据170含有氨基酸长度不同的序列时,编码模块314也可以通过添加pad字符以扩充预定字母表,将肽编码成相等长度的向量。举例来说,这可以通过用pad字符对肽序列进行左侧填充,直到该肽序列的长度达到训练数据170中具有最大长度的肽序列来进行。因此,当具有最大长度的肽序列具有k
max
个氨基酸时,编码模块314将各序列以数字方式表示为具有(20 1)k
max
个元素的行向量。举个例子,对于扩充的字母表{pad,a,c,d,e,f,g,h,i,k,l,m,n,p,q,r,s,t,v,w,y}和k
max
=5的最大氨基酸长度,该具有3个氨基酸的相同示例肽序列eaf可以由具有105个元素的行向量表示:p
i
=[1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0]。c端侧接序列c
i
或其它序列数据可以按与上文描述类似的方式编码。因此,肽序列p
i
或c
i
的每个自变量或每一列表示在该序列特定位置处特定氨基酸的存在。
[0353]
尽管以上编码序列数据的方法是参照具有氨基酸序列的序列描述,但该方法可以类似地扩展至其它类型的序列数据,如dna或rna序列数据等。
[0354]
编码模块314还将数据实例i的一个或多个mhc等位基因a
i
编码成具有m个元素的行向量,其中每个元素h=1,2,
…
,m对应于唯一鉴别的mhc等位基因。对应于所鉴别的数据实例i的mhc等位基因的元素的值是1。另外,其余元素的值是0。举个例子,m=4种唯一鉴别的mhc等位基因类型{hla
‑
a*01:01,hla
‑
c*01:08,hla
‑
b*07:02,hla
‑
drb1*10:01}当中对应于多等位基因细胞系的数据实例i的等位基因hla
‑
b*07:02和hla
‑
drb1*10:01可以由具有4个元素的行向量表示:a
i
=[0 0 1 1],其中a
3i
=1和a
4i
=1。尽管本文中用4种鉴别的mhc等
位基因类型描述该实施例,但mhc等位基因类型的数量实际上可以是数百或数千。如先前所论述,每个数据实例i通常含有最多6种不同的与肽序列p
i
相关的i类mhc等位基因类型,和/或最多4种不同的与肽序列p
i
相关的ii类mhc dr等位基因类,和/或最多12种不同的与肽序列p
i
相关的ii类mhc等位基因类型。
[0355]
编码模块314还将每个数据实例i的标记y
i
编码为具有来自集合{0,1}的值的二元变量,其中值1指示肽x
i
由相关的mhc等位基因a
i
中的一个呈递,而值0指示肽x
i
不被任何相关的mhc等位基因a
i
呈递。当因变量y
i
表示质谱离子电流时,编码模块314可以另外使用各种函数,如对[0,∞)之间的离子电流具有(
‑
∞,∞)范围的对数函数等缩放这些值。
[0356]
编码模块314可以将有关肽p
i
和相关mhc等位基因h的一对等位基因相互作用变量x
hi
表示为行向量,其中等位基因相互作用变量的数字表示相继地串接。举例来说,编码模块314可以将x
hi
表示为等于[p
i
]、[p
i b
hi
]、[p
i s
hi
]或[p
i b
hi s
hi
]的行向量,其中b
hi
是肽p
i
和相关mhc等位基因h的结合亲和力预测值,并且类似地s
hi
是关于稳定性。或者,等位基因相互作用变量的一个或多个组合可以个别地存储(例如以个别向量或矩阵形式)。
[0357]
在一个实例中,编码模块314通过将结合亲和力的测量值或预测值并入等位基因相互作用变量x
hi
中来表示结合亲和力信息。
[0358]
在一个实例中,编码模块314通过将结合稳定性的测量值或预测值并入等位基因相互作用变量x
hi
中来表示结合稳定性信息。
[0359]
在一个实例中,编码模块314通过将结合缔合速率的测量值或预测值并入等位基因相互作用变量x
hi
中来表示结合缔合速率信息。
[0360]
在一个实例中,对于由i类mhc分子呈递的肽,编码模块314将肽长度表示为向量在一个实例中,对于由i类mhc分子呈递的肽,编码模块314将肽长度表示为向量其中是指示函数,并且l
k
表示肽p
k
的长度。向量t
k
可以被包括在等位基因相互作用变量x
hi
中。在另一个实例中,对于由ii类mhc分子呈递的肽,编码模块314将肽长度表示为向量中。在另一个实例中,对于由ii类mhc分子呈递的肽,编码模块314将肽长度表示为向量中。在另一个实例中,对于由ii类mhc分子呈递的肽,编码模块314将肽长度表示为向量其中是指示函数,并且l
k
表示肽p
k
的长度。向量t
k
可以被包括在等位基因相互作用变量x
hi
中。
[0361]
在一个实例中,编码模块314通过将基于rna
‑
seq的mhc等位基因表达水平并入等位基因相互作用变量x
hi
中来表示mhc等位基因的rna表达信息。
[0362]
类似地,编码模块314可以将等位基因非相互作用变量w
i
表示为行向量,其中等位基因非相互作用变量的数字表示相继地串接。举例来说,w
i
可以是等于[c
i
]或[c
i m
i w
i
]的行向量,其中w
i
是除肽p
i
的c端侧接序列和与该肽相关的mrna定量测量值m
i
外,还表示任何其它等位基因非相互作用变量的行向量。或者,等位基因非相互作用变量的一个或多个组合可以个别地存储(例如以个别向量或矩阵形式)。
[0363]
在一实例中,编码模块314通过将转换率或半衰期并入等位基因非相互作用变量w
i
中来表示肽序列的源蛋白的转换率。
[0364]
在一个实例中,编码模块314通过将蛋白质长度并入等位基因非相互作用变量w
i
中来表示源蛋白或同功型的长度。
[0365]
在一个实例中,编码模块314通过将包括β1
i
、β2
i
、β5
i
亚单元在内的免疫蛋白酶体特异性蛋白酶体亚单元的平均表达水平并入等位基因非相互作用变量w
i
中来表示免疫蛋白酶体的活化情况。
[0366]
在一个实例中,编码模块314通过将源蛋白的丰度并入等位基因非相互作用变量w
i
中来表示肽的源蛋白或者肽的基因或转录物的rna
‑
seq丰度(通过如rsem等技术以fpkm、tpm为单位定量)。
[0367]
在一个实例中,编码模块314通过将利用rivas等人,science,2015中的模型估计的肽的源转录物会经历无义介导的衰变(nmd)的机率并入等位基因非相互作用变量w
i
中来表示此机率。
[0368]
在一个实例中,编码模块314例如通过使用例如路径中每个基因的rsem,以tpm为单位定量该路径中基因的表达水平,接着计算该路径中所有基因的概括统计量,例如平均值,以此表示经rna
‑
seq评估的基因模块或路径的活化状态。该平均值可以并入等位基因非相互作用变量w
i
中。
[0369]
在一个实例中,编码模块314通过将拷贝数并入等位基因非相互作用变量w
i
中来表示源基因的拷贝数。
[0370]
在一个实例中,编码模块314通过将测量的或预测的tap结合亲和力例如以纳摩尔浓度为单位)包括在等位基因非相互作用变量w
i
中来表示tap结合亲和力。
[0371]
在一个实例中,编码模块314通过将利用rna
‑
seq测量(并利用例如rsem,以tpm为单位定量)的tap表达水平包括在等位基因非相互作用变量w
i
中来表示tap表达水平。
[0372]
在一个实例中,编码模块314在等位基因非相互作用变量w
i
中将肿瘤突变表示为指示变量的向量(即,如果肽p
k
来自具有kras g12d突变的样品,则d
k
=1,否则是0)。
[0373]
在一个实例中,编码模块314将抗原呈递基因中的生殖系多态性表示为指示变量的向量(即,如果肽p
k
来自在tap中具有物种生殖系多态性的样品,则d
k
=1)。这些指示变量都可以被包括在等位基因非相互作用变量w
i
中。
[0374]
在一个实例中,编码模块314根据肿瘤类型(例如nsclc、黑素瘤、结肠直肠癌等)的字母表将肿瘤类型表示为长度一独热编码的向量。这些独热编码的变量都可以被包括在等位基因非相互作用变量w
i
中。
[0375]
在一个实例中,编码模块314通过用不同后缀处理有4个数字的hla等位基因来表示mhc等位基因后缀。举例来说,出于该模型的目的,hla
‑
a*24:09n被认为是与hla
‑
a*24:09不同的等位基因。或者,由于以n后缀结尾的hla等位基因不表达,故可以将以n为后缀的mhc等位基因对所有肽的呈递机率设置成零。
[0376]
在一个实例中,编码模块314根据肿瘤亚型(例如肺腺癌、肺鳞状细胞癌等)的字母表将肿瘤亚型表示为长度一独热编码的向量。这些独热编码的变量都可以被包括在等位基因非相互作用变量w
i
中。
[0377]
在一个实例中,编码模块314将吸烟史表示为二元指示变量(如果患者有吸烟史,则d
k
=1,否则是0),该变量可以包括在等位基因非相互作用变量w
i
中。或者,可以根据吸烟严重程度的字母表,将吸烟史编码为长度一独热编码的变量。举例来说,吸烟状态可以在1
‑
5级量表上评级,其中1指示非吸烟者,并且5指示当前多量吸烟者。由于吸烟史主要与肺部肿瘤相关,故当训练有关多种肿瘤类型的模型时,此变量也可以在患者有吸烟史时定义为等于1并且肿瘤类型是肺部肿瘤,否则是零。
[0378]
在一个实例中,编码模块314将晒伤史表示为二元指示变量(如果患者有重度晒伤史,则d
k
=1,否则是0),该变量可以包括在等位基因非相互作用变量w
i
中。由于重度晒伤主要与黑素瘤相关,故当训练有关多种肿瘤类型的模型时,此变量也可以在患者有重度晒伤史时定义为等于1并且肿瘤类型是黑素瘤,否则是零。
[0379]
在一个实例中,编码模块314通过使用参考数据库如tcga将有关人基因组中各基因或转录物的特定基因或转录物的表达水平分布表示为表达水平分布的概括统计量(例如平均值、中值)。确切地说,对于肿瘤类型是黑素瘤的样品中的肽p
k
,不仅可以将肽p
k
的源基因或转录物的基因或转录物表达水平测量值包括在等位基因非相互作用变量w
i
中,而且还包括通过tcga测量的黑素瘤中肽p
k
的源基因或转录物的平均和/或中值基因或转录物表达水平。
[0380]
在一个实例中,编码模块314根据突变类型(例如错义突变、移码突变、nmd诱导的突变等)的字母表将突变类型表示为长度一独热编码的变量。这些独热编码的变量都可以被包括在等位基因非相互作用变量w
i
中。
[0381]
在一个实例中,编码模块314在等位基因非相互作用变量w
i
中将蛋白质的蛋白质水平特征表示为源蛋白的注释值(例如5’utr长度)。在另一个实例中,编码模块314通过在等位基因非相互作用变量w
i
中包括指示变量来表示p
i
的残基水平的源蛋白注释,即,如果肽p
i
与螺旋基元重叠则等于1,否则是0,或者如果肽p
i
完全包含在螺旋基元内则等于1。在另一个实例中,表示肽p
i
中包含在螺旋基元注释内的残基的比例的特征可以包括在等位基因非相互作用变量w
i
中。
[0382]
在一个实例中,编码模块314将人蛋白质组中蛋白质或同功型的类型表示为指示向量o
k
,该向量的长度等于人蛋白质组中蛋白质或同功型的数量,并且如果肽p
k
来自蛋白质i,则相应元素o
ki
是1,否则是0。
[0383]
在一个实例中,编码模块314将肽p
i
的源基因g=gene(p
i
)表示为具有l个可能类别的分类变量,其中l表示索引的源基因1、2、...l的数目的上限。
[0384]
在一个实例中,编码模块314将肽p
i
的组织类型、细胞类型、肿瘤类型或肿瘤组织学类型t=tissue(p
i
)表示为具有m个可能类别的分类变量,其中m表示索引类型1、2、...m的数目的上限。组织的类型可以包括,例如,肺组织、心脏组织、肠组织、神经组织等。细胞的类型可以包括树突状细胞、巨噬细胞、cd4 t细胞等。肿瘤类型可以包括肺腺癌、肺鳞状细胞癌、黑素瘤、非霍奇金淋巴瘤等。
[0385]
编码模块314还可以将有关肽p
i
和相关mhc等位基因h的变量z
i
的总体集合表示为行向量,其中等位基因相互作用变量x
i
和等位基因非相互作用变量w
i
的数字表示相继地串接。举例来说,编码模块314可以将z
hi
表示为等于[x
hi w
i
]或[w
i x
hi
]的行向量。
[0386]
viii.训练模块
[0387]
训练模块316构建一个或多个呈递模型,这些模型生成肽序列是否会被与这些肽序列相关的mhc等位基因呈递的可能性。确切地说,给定肽序列p
k
及与肽序列p
k
相关联的一组mhc等位基因a
k
,每个呈递模型生成估计值u
k
,指示肽序列p
k
会被与一个或多个相关mhc等
位基因a
k
呈递的可能性。
[0388]
viii.a.综述
[0389]
训练模块316基于由存储在165中的呈递信息产生的存储于存储器170中的训练数据集来构建一个或多个呈递模型。一般来说,不管呈递模型的具体类型如何,所有呈递模型都捕捉训练数据170中自变量与因变量之间的相关性以使损失函数减到最小。确切地说,损失函数l(y
i∈s
,u
i∈s
;θ)表示训练数据170中一个或多个数据实例s的因变量y
i∈s
与由呈递模型生成的数据实例s的估计可能性u
i∈s
值之间的偏差。在本说明书其余部分通篇所提到的一个特定实施方式中,损失函数(y
i∈s
,u
i∈s
;θ)是由以下等式(1a)提供的负对数可能性函数:
[0390][0391]
不过,实际上,可以使用另一损失函数。举例来说,当对质谱离子电流进行预测时,损失函数是由以下等式1b提供的均方损失:
[0392][0393]
呈递模型可以是一种参数模型,其中一个或多个参数θ在数学上指明自变量与因变量之间的相关性。通常,使损失函数(yi∈s,u
i∈s
;θ)最小的参数型呈递模型的各种参数是通过基于梯度的数值优化算法,如批量梯度算法、随机梯度算法等来确定。或者,呈递模型可以是非参数模型,其中模型结构是由训练数据170决定并且并不严格基于固定参数集合。
[0394]
viii.b.独立等位基因模型
[0395]
训练模块316可以在独立等位基因(per
‑
allele)基础上构建呈递模型以预测肽的呈递可能性。在此情况下,训练模块316可以基于由表达单个mhc等位基因的细胞产生的训练数据170中的数据实例s训练呈递模型。
[0396]
在一种实施方式中,训练模块316通过下式使特定等位基因h对于肽p
k
的估计呈递可能性u
k
建模:
[0397][0398]
其中肽序列x
hk
表示编码的有关肽p
k
和相应mhc等位基因h的等位基因相互作用变量,f(
·
)是任何函数,并且为便于说明,在本文通篇称为变换函数。此外,g
h
(
·
)是任何函数,为便于说明,在本文通篇称为相关性函数(dependency function),并且基于所测定的mhc等位基因h的一组参数θ
h
产生等位基因相互作用变量x
hk
的相关性分数。有关各mhc等位基因h的参数集合θ
h
的值可以通过使关于θ
h
的损失函数减到最小来测定,其中i是由表达单个mhc等位基因h的细胞所产生的训练数据170的子集s中的每个实例。
[0399]
相关性函数g
h
(x
hk
;θ
h
)的输出值表示至少基于等位基因相互作用特征x
hk
,并且确切地说,基于肽p
k
的肽序列中氨基酸的位置的针对mhc等位基因h的相关性分数,其指示mhc等位基因h将呈递相应新抗原。举例来说,如果mhc等位基因h可能呈递肽p
k
,则mhc等位基因h的相关性分数可能具有较高值,而如果不可能呈递,则可能具有较低值。变换函数f(
·
)将输入,并且更确切地说,在此情形中将由g
h
(x
hk
;θ
h
)生成的相关性分数变换成适当值以指示肽p
k
将由mhc等位基因呈递的可能性。
[0400]
在本说明书其余部分通篇提到的一个特定实施方式中,f(
·
)是对于适当域范围
具有在[0,1]内的范围的函数。在一个实施例中,f(
·
)是由下式提供的expit函数:
[0401][0402]
作为另一个实施例,当域z的值等于或大于0时,f(
·
)也可以是由下式提供的双曲正切函数:
[0403]
f(z)=tanh(z)
ꢀꢀꢀ
(5)
[0404]
或者,当质谱离子电流的预测值超出范围[0,1]时,f(
·
)可以是任何函数,如恒等函数、指数函数、对数函数等。
[0405]
因此,可以通过将有关mhc等位基因h的相关性函数g
h
(
·
)应用于肽序列p
k
的编码形式以产生相应相关性分数来产生肽序列p
k
将由mhc等位基因h呈递的独立等位基因可能性。相关性分数可以由变换函数f(
·
)变换以产生肽序列p
k
将由mhc等位基因h呈递的独立等位基因可能性。
[0406]
viii.b.1有关等位基因相互作用变量的相关性函数
[0407]
在本发明通篇提到的一个特定实施方案中,相关性函数g
h
(
·
)是由下式提供的仿射函数:
[0408][0409]
该函数将x
hk
中的每个等位基因相互作用变量与所测定的相关mhc等位基因h的参数集合θ
h
中的相应参数线性地组合。
[0410]
在本说明书通篇提到的另一个特定实施方式中,相关性函数g
h
(
·
)是由下式提供的网络函数:
[0411][0412]
以具有分一层或多层布置的一系列节点的网络模型nn
h
(
·
)表示。一个节点可以通过连接而连接至其它节点,这些连接各自在参数集合θ
h
中具有相关参数。在一个特定节点处的值可以表示为通过与该特定节点相关联的激活函数所映射的相关参数加权的连接至该特定节点的节点值的总和。由于呈递模型可以并入具有不同氨基酸序列长度的非线性和工艺数据,与仿射函数相比,网络模型是有利的。确切地说,通过非线性建模,网络模型可以捕捉在肽序列不同位置处的氨基酸之间的相互作用以及这一相互作用如何影响肽呈递。
[0413]
一般来说,网络模型nn
h
(
·
)可以被构造成前馈网络,如人工神经网络(ann)、卷积神经网络(cnn)、深度神经网络(dnn),和/或循环神经网络(rnn),如长短期记忆网络(lstm)、双向lstm网络、双向循环网络、深度双向循环网络、多层感知器网络(mlp)等。
[0414]
在本说明书其余部分通篇提到的一个实例中,h=1,2,
…
,m中的每个mhc等位基因与独立网络模型相关联,并且nn
h
(
·
)表示来自与mhc等位基因h相关联的网络模型的输出。
[0415]
图5示出了与任意mhc等位基因h=3相关联的示例网络模型nn3(
·
)。如图5中所示,关于mhc等位基因h=3的网络模型nn3(
·
)包括在层l=1处的三个输入节点、在层l=2处的四个节点、在层l=3处的两个节点及在层l=4处的一个输出节点。网络模型nn3(
·
)与一组十个参数θ3(1),θ3(2),
…
,θ3(10)相关。网络模型nn3(
·
)接收关于mhc等位基因h=3的三个等位基因相互作用变量x
3k
(1)、x
3k
(2)及x
3k
(3)的输入值(包括编码的多肽序列数据和所用任何其它训练数据的个别数据实例)并输出值nn3(x
3k
)。网络函数还可以包括一个或多
个网络模型,每个网络模型采用不同的等位基因相互作用变量作为输入。
[0416]
在另一个实施例中,鉴别的mhc等位基因h=1,2,
…
,m与单个网络模型nn
h
(
·
)相关联,并且nn
h
(
·
)表示与mhc等位基因h相关的单个网络模型的一个或多个输出。在此类实例中,参数集合θ
h
可以对应于该单个网络模型的一组参数,并因此,参数集合θ
h
可以是所有mhc等位基因共有的。
[0417]
图6a示出了mhc等位基因h=1,2,
…
,m共享的示例网络模型nn
h
(
·
)。如图6a中所示,网络模型nn
h
(
·
)包括m个输出节点,各自对应于mhc等位基因。网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并输出m值,包括对应于mhc等位基因h=3的值nn3(x
3k
)。
[0418]
在又另一实例中,单个网络模型nn
h
(
·
)可以是在给定mhc等位基因h的等位基因相互作用变量x
hk
和编码的蛋白质序列d
h
情况下,输出相关性分数的网络模型。在此类实例中,参数集合θ
h
也可以对应于该单个网络模型的一组参数,并因此,参数集合θ
h
可以是所有mhc等位基因共有的。因此,在此类实例中,nn
h
(
·
)可以表示在给定该单个网络模型的输入[x
hk d
h
]情况下,该单个网络模型nn
h
(
·
)的输出。由于训练数据中未知的mhc等位基因的肽呈递可能性只能通过鉴别其蛋白质序列进行预测,故此类网络模型是有利的。
[0419]
图6b示出了mhc等位基因共享的示例网络模型nn
h
(
·
)。如图6b中所示,网络模型nn
h
(
·
)接收mhc等位基因h=3的等位基因相互作用变量和蛋白质序列作为输入,并输出对应于mhc等位基因h=3的相关性分数nn3(x
3k
)。
[0420]
在又另一个实施例中,相关性函数g
h
(
·
)可以表示为:
[0421][0422]
其中g’h
(x
hk
;θ’h
)是具有一组参数θ’h
的仿射函数、网络函数等,其中有关mhc等位基因的等位基因相互作用变量的一组参数的偏差参数θ
h0
表示mhc等位基因h的基线呈递机率。
[0423]
在另一种实施方式中,偏差参数θ
h0
可以是mhc等位基因h的基因家族共有的。也就是说,mhc等位基因h的偏差参数θ
h0
可以等于θ
基因(h)0
,其中基因(h)是mhc等位基因h的基因家族。举例来说,i类mhc等位基因hla
‑
a*02:01、hla
‑
a*02:02及hla
‑
a*02:03可以指定给“hla
‑
a”基因家族,并且这些mhc等位基因各自的偏差参数θ
h0
可以是共有的。作为另一个实例,ii类mhc等位基因hla
‑
drb1:10:01、hla
‑
drb1:11:01及hla
‑
drb3:01:01可以指定给“hla
‑
drb”基因家族,并且这些mhc等位基因各自的偏差参数θ
h0
可以是共有的。
[0424]
再回到等式(2),作为一个实施例,在使用仿射相关性函数g
h
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=3呈递的可能性可以由下式得到:
[0425][0426]
其中x
3k
是鉴别的mhc等位基因h=3的等位基因相互作用变量,并且θ3是通过损失函数最小化测定的mhc等位基因h=3的参数集合。
[0427]
作为另一个实施例,在使用独立网络变换函数g
h
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=3呈递的可能性可以由下式得到:
[0428]
[0429]
其中x
3k
是鉴别的mhc等位基因h=3的等位基因相互作用变量,并且θ3是测定的与mhc等位基因h=3相关联的网络模型nn3(
·
)的参数集合。
[0430]
图7示出了使用示例网络模型nn3(
·
)生成与mhc等位基因h=3相关联的肽p
k
的呈递可能性。如图7中所示,网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
)。该输出由函数f(
·
)映射以产生估计的呈递可能性u
k
。
[0431]
viii.b.2.具有等位基因非相互作用变量的独立等位基因
[0432]
在一种实施方式中,训练模块316并入等位基因非相互作用变量并通过下式使肽p
k
的估计呈递可能性u
k
建模:
[0433][0434]
其中w
k
表示肽p
k
的编码的等位基因非相互作用变量,g
w
(
·
)是基于测定的等位基因非相互作用变量的一组参数θ
w
的等位基因非相互作用变量w
k
的函数。确切地说,有关各mhc等位基因h的参数集合θ
h
和有关等位基因非相互作用变量的参数集合θ
w
的值可以通过使关于θ
h
和θ
w
的损失函数减到最小来测定,其中i是由表达单个mhc等位基因的细胞所产生的训练数据170的子集s中的每个实例。
[0435]
相关性函数g
w
(w
k
;θ
w
)的输出表示基于等位基因非相互作用变量的影响的等位基因非相互作用变量的相关性分数,其指示肽p
k
是否会由一个或多个mhc等位基因呈递。举例来说,如果肽p
k
与已知会积极地影响肽p
k
的呈递的c端侧接序列相关,则等位基因非相互作用变量的相关性分数可能具有较高值,并且如果肽p
k
与已知会不利地影响肽p
k
的呈递的c端侧接序列相关,则可能具有较低值。
[0436]
根据等式(8),可以通过将有关mhc等位基因h的函数g
h
(
·
)应用于肽序列p
k
的编码形式以产生等位基因相互作用变量的相应相关性分数来产生肽序列p
k
将由mhc等位基因h呈递的独立等位基因可能性。有关等位基因非相互作用变量的函数g
w
(
·
)也应用于等位基因非相互作用变量的编码形式以产生等位基因非相互作用变量的相关性分数。将两个分数合并,并通过变换函数f(
·
)变换该合并的分数以产生肽序列p
k
将由mhc等位基因h呈递的独立等位基因可能性。
[0437]
或者,训练模块316可以通过将等位基因非相互作用变量w
k
添加至等式(2)中的等位基因非相互作用变量x
hk
中,来将等位基因非相互作用变量w
k
包括在预测值中。因此,呈递可能性可以由下式得到:
[0438][0439]
viii.b.3有关等位基因非相互作用变量的相关性函数
[0440]
与有关等位基因相互作用变量的相关性函数g
h
(
·
)类似,有关等位基因非相互作用变量的相关性函数g
w
(
·
)可以是仿射函数或网络函数,其中独立网络模型与等位基因非相互作用变量w
k
相关联。
[0441]
确切地说,相关性函数g
w
(
·
)是由下式提供的仿射函数:
[0442]
g
w
(w
k
;θ
w
)=w
k
·
θ
w
。
[0443]
该函数将等位基因非相互作用变量w
k
与参数集合θ
w
中的相应参数线性地组合。
[0444]
相关性函数g
w
(
·
)还可以是由下式提供的网络函数:
[0445]
g
h
(w
k
;θ
w
)=nn
w
(ww
k
;θ
w
)。
[0446]
该函数是由具有参数集合θ
w
中的相关参数的网络模型nn
w
(
·
)表示。网络函数可能还包括一个或多个网络模型,每个网络模型采用不同的等位基因非相互作用变量作为输入。
[0447]
在另一个实施例中,有关等位基因非相互作用变量的相关性函数g
w
(
·
)可以由下式提供:
[0448][0449]
其中g’w
(w
k
;θ’w
)是仿射函数,具有等位基因非相互作用参数集合θ’w
的网络函数等,m
k
是肽p
k
的mrna定量测量值,h(
·
)是变换该定量测量值的函数,并且θ
wm
是有关等位基因非相互作用变量的参数集合中的一个参数,该参数与mrna定量测量值组合以生成有关mrna定量测量值的相关性分数。在本说明书其余部分通篇所提到的一个特定实施方案中,h(
·
)是对数函数,不过实际上,h(
·
)可以是多种不同函数中的任一种。
[0450]
在又另一个实例中,有关等位基因非相互作用变量的相关性函数g
w
(
·
)可以由下式提供:
[0451][0452]
其中g’w
(w
k
;θ’w
)是仿射函数、具有等位基因非相互作用参数集合θ’w
的网络函数等,o
k
是部分vii.c.2中描述的表示人蛋白质组中有关肽p
k
的蛋白质和同功型的指示向量,并且θ
wo
是有关等位基因非相互作用变量的参数集合中的一组参数,其与指示向量组合。在一种变化形式中,当o
k
的维度和参数集合θ
wo
明显较高时,可以在测定参数值时将参数正则项,如添加至损失函数中,其中||
·
||表示l1范数、l2范数、组合等。超参数λ的最佳值可以通过适当方法测定。
[0453]
在又另一个实例中,有关等位基因非相互作用变量的相关性函数g
w
(
·
)可以由下式提供:
[0454][0455]
其中g’w
(w
k
;θ’w
)是仿射函数、具有等位基因非相互作用参数集合θ’w
的网络函数等,(基因(p
k
=l))是指示函数,如上文对于等位基因非相互作用变量所述,如果肽p
k
来自源基因l,则其等于1,并且θ
wl
是指示源基因l的“抗原性”的参数。在一种变化形式中,当l显著较高并且因此参数θ
wl=1,2,
…
,l
数量也显著较高时,可以在测定参数值时将参数正则项,如添加至损失函数中,其中||
·
||表示l1范数、l2范数、组合等。可以通过适当的方法来确定超参数λ的最优值。
[0456]
在又另一个实例中,有关等位基因非相互作用变量的相关性函数g
w
(
·
)可以由下式提供:
[0457][0458]
其中g’w
(w
k
;θ’w
)是仿射函数、具有等位基因非相互作用参数集合θ’w
的网络函数
等,(基因(p
k
)=l,组织(p
k
)=m)是指示函数,如上文对于等位基因非相互作用变量所述,如果肽p
k
来自源基因l并且如果肽p
k
来自组织类型m,则其等于1,并且θ
wlm
是指示源基因l和组织类型m的组合的抗原性的参数。具体地,组织类型m的基因l的抗原性可以表示在控制rna表达和肽序列背景之后,组织m的细胞呈递来自基因l的肽的残余倾向。
[0459]
在一种变化形式中,当l或m显著较高并且因此参数θ
wlm=1,2,
…
,lm
数量也显著较高时,可以在测定参数值时将参数正则项,如添加至损失函数中,其中||
·
||表示l1范数、l2范数、组合等。可以通过适当的方法来确定超参数λ的最优值。在另一种变化形式中,可以在测定参数值时将参数正则项添加至损失函数中,使得相同源基因的系数不会在组织类型之间有显著差异。例如,惩罚项如可以惩罚损失函数中不同组织类型之间抗原性的标准偏差,其中是源基因l的组织类型之间的平均抗原性。
[0460]
实际上,等式(10)、(11)、(12a)和(12b)中的任一个的附加项可以组合以产生等位基因非相互作用变量的相关性函数g
w
(
·
)。例如,可以将等式(10)中表示mrna定量测量的项h(
·
)和等式(12)中表示源基因抗原性的项与任何其它仿射或网络函数一起相加,以生成等位基因非相互作用变量的相关性函数。
[0461]
再回到等式(8),作为一个实施例,在使用仿射变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=3呈递的可能性可以由下式产生:
[0462][0463]
其中w
k
是所鉴别的肽p
k
的等位基因非相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0464]
作为另一个实施例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=3呈递的可能性可以由下式得到:
[0465][0466]
其中w
k
是所鉴别的肽p
k
的等位基因相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0467]
图8示出了使用示例网络模型nn3(
·
)和nn
w
(
·
)生成与mhc等位基因h=3相关联的肽p
k
的呈递可能性。如图8中所示,网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
)。网络模型nn
w
(
·
)接收有关肽p
k
的等位基因非相互作用变量w
k
并生成输出nn
w
(w
k
)。将输出合并,并由函数f(
·
)映射以产生估计的呈递可能性u
k
。
[0468]
viii.c.多等位基因模型
[0469]
训练模块316还可以在存在两个或更多个mhc等位基因的多等位基因环境中构建呈递模型以预测肽的呈递可能性。在此情况下,训练模块316可以基于由表达单个mhc等位基因的细胞、表达多个mhc等位基因的细胞或其组合产生的训练数据170中的数据实例s训练呈递模型。
[0470]
viii.c.1.实施例1:独立等位基因模型的最大值
[0471]
在一种实施方式中,训练模块316使与一组多个mhc等位基因h相关联的肽p
k
的估计呈递可能性u
k
随基于表达单等位基因的细胞所测定的集合h中每个mhc等位基因h的呈递可能性的变化建模,如上文结合等式(2)
‑
(11)所描述。确切地说,呈递可能性u
k
可以是的任何函数。在一种实施方式中,如等式(12)中所示,该函数是最大值函数,并且呈递可能性u
k
可以测定为集合h中每个mhc等位基因h的呈递可能性最大值。
[0472][0473]
viii.c.2.实施例2.1:和的函数(funciton
‑
of
‑
sums)模型
[0474]
在一种实施方式中,训练模块316通过下式使肽p
k
的估计呈递可能性u
k
建模:
[0475][0476]
其中元素a
hk
对于与肽序列p
k
相关的多个mhc等位基因h是1,并且x
hk
表示编码的有关肽p
k
和相应mhc等位基因的等位基因相互作用变量。有关各mhc等位基因h的参数集合θ
h
的值可以通过使关于θ
h
的损失函数减到最小来测定,其中i是由表达单个mhc等位基因的细胞和/或表达多个mhc等位基因的细胞所产生的训练数据170的子集s中的每个实例。相关性函数g
h
可以呈以上viii.b.1部分中介绍的相关性函数g
h
中的任一种的形式。
[0477]
根据等式(13),可以通过将相关性函数g
h
(
·
)应用于有关mhc等位基因h中的每一个的肽序列p
k
的编码形式以产生等位基因相互作用变量的相应分数来产生肽序列p
k
将由一个或多个mhc等位基因h呈递的呈递可能性。将每个mhc等位基因h的分数合并,并通过变换函数f(
·
)变换以产生肽序列p
k
将由mhc等位基因集合h呈递的呈递可能性。
[0478]
等式(13)的呈递模型与等式(2)的独立等位基因模型的不同之处在于,每个肽p
k
的相关等位基因的数量可以大于1。换句话说,对于与肽序列p
k
相关的多个mhc等位基因h,a
hk
中超过一个元素值可以是1。
[0479]
举个例子,在使用仿射变换函数g
h
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0480][0481]
其中x
2k
、x
3k
是鉴别的mhc等位基因h=2、h=3的等位基因相互作用变量,并且θ2、θ3是测定的mhc等位基因h=2、h=3的参数的集合。
[0482]
作为另一个实例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0483][0484]
其中nn2(
·
)、nn3(
·
)是鉴别的mhc等位基因h=2、h=3的网络模型,并且θ2、θ3是测定的mhc等位基因h=2、h=3的参数的集合。
[0485]
图9示出了使用示例网络模型nn2(
·
)和nn3(
·
)生成与mhc等位基因h=2、h=3相关联的肽p
k
的呈递可能性。如图9中所示,网络模型nn2(
·
)接收有关mhc等位基因h=2的等位基因相互作用变量x
2k
并生成输出nn2(x
2k
),并且网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
)。将输出合并,并由函数f(
·
)映射以产
生估计的呈递可能性u
k
。
[0486]
viii.c.3.实施例2.2:利用等位基因非相互作用变量的和的函数模型
[0487]
在一种实施方式中,训练模块316并入等位基因非相互作用变量并通过下式使肽p
k
的估计呈递可能性u
k
建模:
[0488][0489]
其中w
k
表示编码的有关肽p
k
的等位基因非相互作用变量。确切地说,有关各mhc等位基因h的参数集合θ
h
和有关等位基因非相互作用变量的参数集合θ
w
的值可以通过使关于θ
h
和θ
w
的损失函数减到最小来测定,其中i是由表达单个mhc等位基因的细胞和/或表达多个mhc等位基因的细胞所产生的训练数据170的子集s中的每个实例。相关性函数g
w
可以呈以上viii.b.3部分中介绍的相关性函数g
w
中的任一种的形式。
[0490]
因此,根据等式(14),可以通过将函数g
h
(
·
)应用于有关mhc等位基因h中的每一个的肽序列p
k
的编码形式以产生有关每个mhc等位基因h的等位基因相互作用变量的相应相关性分数来产生肽序列p
k
将由一个或多个mhc等位基因h呈递的呈递可能性。有关等位基因非相互作用变量的函数g
w
(
·
)也应用于等位基因非相互作用变量的编码形式以产生等位基因非相互作用变量的相关性分数。将分数合并,并通过变换函数f(
·
)变换该合并的分数以产生肽序列p
k
将由mhc等位基因h呈递的呈递可能性。
[0491]
在等式(14)的呈递模型中,每个肽p
k
的相关等位基因的数量可以大于1。换句话说,对于与肽序列p
k
相关的多个mhc等位基因h,a
hk
中超过一个元素的值可以是1。
[0492]
举个例子,在使用仿射变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0493][0494]
其中w
k
是所鉴别的肽p
k
的等位基因非相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0495]
作为另一个实施例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0496][0497]
其中w
k
是所鉴别的肽p
k
的等位基因相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0498]
图10示出了使用示例网络模型nn2(
·
)、nn3(
·
)及nn
w
(
·
)生成与mhc等位基因h=2、h=3相关联的肽p
k
的呈递可能性。如图10中所示,网络模型nn2(
·
)接收有关mhc等位基因h=2的等位基因相互作用变量x
2k
并生成输出nn2(x
2k
)。网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
)。网络模型nn
w
(
·
)接收有关肽p
k
的等位基因非相互作用变量w
k
并生成输出nn
w
(w
k
)。将输出合并,并由函数f(
·
)映射以产生估计的呈递可能性u
k
。
[0499]
或者,训练模块316可以通过将等位基因非相互作用变量w
k
添加至等式(15)的等位基因非相互作用变量x
hk
中,来将等位基因非相互作用变量w
k
包括在预测值中。因此,呈递
可能性可以由下式得到:
[0500][0501]
viii.c.4.实施例3.1:使用隐式独立等位基因可能性的模型
[0502]
在另一种实施方式中,训练模块316通过下式使肽p
k
的估计呈递可能性u
k
建模:
[0503][0504]
其中元素a
hk
对于与肽序列p
k
相关联的多个mhc等位基因h∈h是1,u’kh
是mhc等位基因h的隐式独立等位基因呈递可能性,向量v是其中元素v
h
对应于a
hk
·
u’kh
的向量,s(
·
)是映射元素v的函数,并且r(
·
)是限幅函数(clipping function),其将输入值削减至给定范围中。如以下更详细地描述,s(
·
)可以是求和函数或二阶函数,但应理解在其它实施方案中,s(
·
)可以是任何函数,如最大值函数。有关隐式独立等位基因可能性的参数集合θ的值可以通过使关于θ的损失函数减到最小来测定,其中i是由表达单个mhc等位基因的细胞和/或表达多个mhc等位基因的细胞所产生的训练数据170的子集s中的每个实例。
[0505]
使等式(17)的呈递模型中的呈递可能性随各自对应于肽p
k
将由个别mhc等位基因h呈递的可能性的隐式独立等位基因呈递可能性u’kh
的变化建模。隐式独立等位基因可能性与viii.b部分的独立等位基因呈递可能性的不同之处在于,有关隐式独立等位基因可能性的参数可以从多等位基因环境习得,其中除单等位基因环境外,呈递肽与相应mhc等位基因之间的直接关联也是未知的。因此,在多等位基因环境中,呈递模型不仅可以估计肽p
k
是否会由作为整体的一组mhc等位基因h呈递,而且还可以提供指示最可能呈递肽p
k
的mhc等位基因h的个别可能性其优势在于,呈递模型可以在无有关表达单mhc等位基因的细胞的训练数据存在下产生隐式可能性。
[0506]
在本说明书其余部分通篇提到的一个特定实施方式中,r(
·
)是具有范围[0,1]的函数。举例来说,r(
·
)可以是限幅函数:
[0507]
r(z)=min(max(z,0),1),
[0508]
其中选择z与1之间的最小值作为呈递可能性u
k
。在另一种实施方式中,当域z的值等于或大于0时,r(
·
)是由下式提供的双曲正切函数:
[0509]
r(z)=tanh(z)。
[0510]
viii.c.5.实施例3.2:函数的和(sum
‑
of
‑
functions)模型
[0511]
在一个特定实施方式中,s(
·
)是求和函数,并且呈递可能性是通过对隐式独立等位基因呈递可能性求和得到:
[0512][0513]
在一种实施方式中,mhc等位基因h的隐式独立等位基因呈递可能性是由下式得到:
[0514][0515]
由此通过下式估计出呈递可能性:
[0516][0517]
根据等式(19),可以通过将函数g
h
(
·
)应用于有关mhc等位基因h中的每一个的肽序列p
k
的编码形式以产生等位基因相互作用变量的相应相关性分数来产生肽序列p
k
将由一个或多个mhc等位基因h呈递的呈递可能性。每个相关性分数都先通过函数f(
·
)变换以产生隐式独立等位基因呈递可能性u’kh
。将独立等位基因可能性u’kh
合并,并且可以将限幅函数应用于合并的可能性以将值削减至范围[0,1]中以产生肽序列p
k
将由mhc等位基因集合h呈递的呈递可能性。相关性函数g
h
可以呈以上viii.b.1部分中介绍的相关性函数g
h
中的任一种的形式。
[0518]
举个例子,在使用仿射变换函数g
h
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0519][0520]
其中x
2k
、x
3k
是鉴别的mhc等位基因h=2、h=3的等位基因相互作用变量,并且θ2、θ3是测定的mhc等位基因h=2、h=3的参数的集合。
[0521]
作为另一个实施例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0522][0523]
其中nn2(
·
)、nn3(
·
)是鉴别的mhc等位基因h=2、h=3的网络模型,并且θ2、θ3是测定的mhc等位基因h=2、h=3的参数的集合。
[0524]
图11示出了使用示例网络模型nn2(
·
)和nn3(
·
)生成与mhc等位基因h=2、h=3相关联的肽p
k
的呈递可能性。如图9中所示,网络模型nn2(
·
)接收有关mhc等位基因h=2的等位基因相互作用变量x
2k
并生成输出nn2(x
2k
),并且网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
)。每个输出由函数f(
·
)映射以产生估计的呈递可能性u
k
。
[0525]
在另一种实施方式中,当预测质谱离子电流的对数时,r(
·
)是对数函数并且f(
·
)是指数函数。
[0526]
viii.c.6.实施例3.3:利用等位基因非相互作用变量的函数的和模型
[0527]
在一种实施方式中,mhc等位基因h的隐式独立等位基因呈递可能性是由下式得到:
[0528][0529]
由此通过下式产生呈递可能性:
[0530][0531]
以并入等位基因非相互作用变量对肽呈递的影响。
[0532]
根据等式(21),可以通过将函数g
h
(
·
)应用于有关mhc等位基因h中的每一个的肽
序列p
k
的编码形式以产生有关每个mhc等位基因h的等位基因相互作用变量的相应相关性分数来产生肽序列p
k
将由一个或多个mhc等位基因h呈递的呈递可能性。有关等位基因非相互作用变量的函数g
w
(
·
)也应用于等位基因非相互作用变量的编码形式以产生等位基因非相互作用变量的相关性分数。将等位基因非相互作用变量的分数与等位基因相互作用变量的各个相关性分数合并。每个合并的分数都通过函数f(
·
)变换以产生隐式独立等位基因呈递可能性。将隐式可能性合并,并且可以将限幅函数应用于合并的输出以将值削减至范围[0,1]中以产生肽序列p
k
将由mhc等位基因集合h呈递的呈递可能性。相关性函数g
w
可以呈以上viii.b.3部分中介绍的相关性函数g
w
中的任一种的形式。
[0533]
举个例子,在使用仿射变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0534][0535]
其中w
k
是所鉴别的肽p
k
的等位基因非相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0536]
作为另一个实施例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的mhc等位基因当中,肽p
k
将由mhc等位基因h=2、h=3呈递的可能性可以由下式得到:
[0537][0538]
其中w
k
是所鉴别的肽p
k
的等位基因相互作用变量,并且θ
w
是测定的等位基因非相互作用变量的参数的集合。
[0539]
图12示出了使用示例网络模型nn2(
·
)、nn3(
·
)及nn
w
(
·
)生成与mhc等位基因h=2、h=3相关联的肽p
k
的呈递可能性。如图12中所示,网络模型nn2(
·
)接收有关mhc等位基因h=2的等位基因相互作用变量x
2k
并生成输出nn2(x
2k
)。网络模型nn
w
(
·
)接收有关肽p
k
的等位基因非相互作用变量w
k
并生成输出nn
w
(w
k
)。将输出合并,并且通过函数f(
·
)映射。网络模型nn3(
·
)接收有关mhc等位基因h=3的等位基因相互作用变量x
3k
并生成输出nn3(x
3k
),再次将该输出与同一网络模型nn
w
(
·
)的输出nn
w
(w
k
)合并,并且通过函数f(
·
)映射。将两个输出合并以产生估计的呈递可能性u
k
。
[0540]
在另一种实施方式中,mhc等位基因h的隐式独立等位基因呈递可能性由下式得到:
[0541][0542]
由此通过下式得到呈递可能性:
[0543][0544]
viii.c.7.实施例4:二阶模型
[0545]
在一种实施方式中,s(
·
)是二阶函数,并且肽p
k
的估计呈递可能性u
k
由下式得到:
[0546]
[0547]
其中元素u’kh
是mhc等位基因h的隐式独立等位基因可能性。有关隐式独立等位基因可能性的参数集合θ的值可以通过使关于θ的损失函数减到最小来测定,其中i是由表达单个mhc等位基因的细胞和/或表达多个mhc等位基因的细胞所产生的训练数据170的子集s中的每个实例。隐式独立等位基因呈递可能性可以呈以上描述的等式(18)、(20)及(22)中所示的任何形式。
[0548]
在一方面,等式(23)的模型可以暗示存在肽p
k
将同时由两个mhc等位基因呈递的可能,其中两个hla等位基因的呈递在统计学上是独立的。
[0549]
根据等式(23),肽序列p
k
将由一个或多个mhc等位基因h呈递的呈递可能性可以通过组合隐式独立等位基因呈递可能性并自总和中减去每对mhc等位基因将同时呈递肽p
k
的可能性以产生肽序列p
k
将由mhc等位基因h呈递的呈递可能性来产生。
[0550]
举个例子,在使用仿射变换函数g
h
(
·
)鉴别的m=4种不同的hla等位基因当中,肽p
k
将由hla等位基因h=2、h=3呈递的可能性可以由下式得到:
[0551][0552]
其中x
2k
、x
3k
是鉴别的hla等位基因h=2、h=3的等位基因相互作用变量,并且θ2、θ3是测定的hla等位基因h=2、h=3的参数的集合。
[0553]
作为另一个实施例,在使用网络变换函数g
h
(
·
)、g
w
(
·
)鉴别的m=4种不同的hla等位基因当中,肽p
k
将由hla等位基因h=2、h=3呈递的可能性可以由下式得到:
[0554][0555]
其中nn2(
·
)、nn3(
·
)是鉴别的hla等位基因h=2、h=3的网络模型,并且θ2、θ3是测定的hla等位基因h=2、h=3的参数的集合。
[0556]
ix.实施例5:预测模块
[0557]
预测模块320接收序列数据并使用呈递模型在序列数据中选择候选新抗原。确切地说,序列数据可以是从患者的肿瘤组织细胞中提取的dna序列、rna序列和/或蛋白质序列。预测模块320将序列数据处理成对于mhc
‑
i具有8
‑
15个氨基酸或对于mhc
‑
ii具有6
‑
30个氨基酸的多个肽序列p
k
。举例来说,预测模块320可以将给定序列“iefroeifjef”处理成具有9个氨基酸的三个肽序列“iefroeifj”、“efroeifje”及“froeifjef”。在一个实施方案中,预测模块320可以通过将从患者的正常组织细胞提取的序列数据与从患者的肿瘤组织细胞提取的序列数据相比较以鉴别含有一个或多个突变的部分,由此鉴别出作为突变肽序列的候选新抗原。
[0558]
预测模块320将一个或多个呈递模型应用于处理的肽序列以估计这些肽序列的呈递可能性。确切地说,预测模块320可以通过将呈递模型应用于候选新抗原来选择一个或多个可能被呈递于肿瘤hla分子上的候选新抗原肽序列。在一种实施方式中,预测模块320选出估计呈递可能性超过预定阈值的候选新抗原序列。在另一种实施方式中,呈递模块选出个具有最高估计呈递可能性的候选新抗原序列(其中一般是可以在疫苗中递送的表位的最大数量)。包括选择用于给定患者的候选新抗原的疫苗可以注射到患者体内以诱导免疫应答。
[0559]
x.实施例6:患者选择模块
[0560]
患者选择模块324基于患者是否满足纳入标准来选择用于疫苗治疗和/或t细胞疗
法的患者子集。在一个实施方案中,基于由呈递模块产生的患者新抗原候选物的呈递可能性来确定纳入标准。通过调整纳入标准,患者选择模块324可以基于患者新抗原候选物的呈递可能性来调整将要接受疫苗和/或t细胞疗法的患者数量。具体地,严格的纳入标准导致较少数量的患者将被用疫苗和/或t细胞疗法治疗,但是可以导致较高比例的疫苗和/或t细胞疗法所治疗患者接受有效治疗(例如接受一种或多种肿瘤特异性新抗原(tsna)和/或一种或多种新抗原响应性t细胞)。另一方面,宽松的纳入标准导致较大数量的患者将被用疫苗和/或t细胞疗法治疗,但是可导致较低比例的疫苗和/或t细胞疗法所治疗患者接受有效治疗。患者选择模块324基于将要接受治疗的患者的目标比例与接受有效治疗的患者比例之间的期望平衡来修改纳入标准。
[0561]
在一些实施方案中,用于选择接受疫苗治疗的患者的纳入标准与用于选择接受t细胞疗法的患者的纳入标准相同。然而,在替代实施方案中,用于选择接受疫苗治疗的患者的纳入标准可能不同于用于选择接受t细胞疗法的患者的纳入标准。以下x.a和x.b节分别讨论了用于选择接受疫苗治疗的患者的纳入标准和用于选择接受t细胞疗法的患者的纳入标准。
[0562]
x.a.对于疫苗治疗的患者选择
[0563]
在一个实施方案中,患者与v种新抗原候选物的相应治疗子集相关联,所述v种新抗原候选物可以潜在地包含在具有疫苗容量v的为患者定制疫苗中。在一个实施方案中,患者的治疗子集是通过呈递模型确定的具有最高呈递可能性的新抗原候选物。例如,如果疫苗可以包含v=20种表位,则疫苗可以包含每个患者的通过呈递模型确定的具有最高呈递可能性的治疗子集。但是,应当理解,在另一些实施方案中,可以基于其它方法来确定患者的治疗子集。例如,患者的治疗子集可以从患者的新抗原候选物集合中随机选择,或者可以部分地基于对肽序列的结合亲和力和稳定性建模的当前现有技术模型,或者包括来自呈递模型的呈递可能性和关于这些肽序列的亲和力或稳定性信息的一些因素组合来确定。
[0564]
在一个实施方案中,如果患者的肿瘤突变负荷等于或高于最小突变负荷,则患者选择模块324确定患者满足纳入标准。患者的肿瘤突变负荷(tmb)指示肿瘤外显子组中非同义突变的总数。在一种实施方式中,如果患者的tmb的绝对数量等于或高于预定阈值,则患者选择模块324可以选择患者进行疫苗治疗。在另一种实施方式中,如果患者的tmb在为患者集合确定的tmb中的阈值百分位数之内,则患者选择模块324可以选择患者用于进行疫苗治疗。
[0565]
在另一个实施方案中,如果基于患者治疗子集的患者效用分数等于或高于最小效用分数,则患者选择模块324确定患者满足纳入标准。在一种实施方式中,效用分数是对治疗子集中被呈递的新抗原的估计数目的度量。
[0566]
可以通过将新抗原呈递建模为一种或多种概率分布的随机变量来预测被呈递的新抗原的估计数目。在一种实施方式中,患者i的效用分数是治疗子集中被呈递的新抗原候选物的预期数目,或其一些函数。作为示例,每种新抗原的呈递可以建模为伯努利随机变量,其中呈递(成功)概率由新抗原候选物的呈递可能性给出。具体来说,对于v种新抗原候选物p
i1
、p
i2
、
…
、p
iv
的治疗子集s
i
,每种新抗原候选物具有最高呈递可能性u
i1
、u
i2
、
…
、u
iv
,则新抗原候选物p
ij
的呈递由随机变量a
ij
给出,其中:
[0567]
p(a
ij
=1)=u
ij
,p(a
ij
=0)=1
‑
u
ij
。
ꢀꢀꢀ
(24)
[0568]
被呈递的新抗原的预期数目由每种新抗原候选物的呈递可能性之和给出。换句话说,患者i的效用分数可以表示为:
[0569][0570]
患者选择模块324选择效用分数等于或高于最小效用的患者子集进行疫苗治疗。
[0571]
在另一种实施方式中,患者i的效用分数是至少阈值数目k的新抗原将被呈递的概率。在一个示例中,将新抗原候选物的治疗子集s
i
中被呈递的新抗原的数目建模为泊松二项式随机变量,其中呈递(成功)概率由每种表位的呈递可能性给出。具体来说,患者i的被呈递的新抗原的数目可以通过随机变量n
i
给出,其中:
[0572][0573]
其中pbd(
·
)表示泊松二项式分布。至少阈值数目k的新抗原将被呈递的概率由被呈递的新抗原的数目n
i
等于或大于k的概率的总和给出。换句话说,患者i的效用分数可以表示为:
[0574][0575]
患者选择模块324选择效用分数等于或高于最小效用的患者子集进行疫苗治疗。
[0576]
在另一种实施方式中,患者i的效用分数是新抗原候选物的治疗子集s
i
中具有低于对一种或多种患者hla等位基因的固定阈值(例如500nm)的结合亲和力或预测的结合亲和力的新抗原的数目。在一个实例中,固定阈值在1000nm至10nm的范围。任选地,效用分数可以仅计数通过rna
‑
seq检测到的那些新抗原。
[0577]
在另一种实施方式中,患者i的效用分数是新抗原候选物的治疗子集s
i
中对一种或多种患者hla等位基因的结合亲和力等于或低于随机肽对该hla等位基因的结合亲和力的阈值百分位数的新抗原的数量。在一个实例中,阈值百分位数是从第10个百分位数到第0.1个百分位数的范围。任选地,效用分数可以仅计数通过rna
‑
seq检测到的那些新抗原。
[0578]
应当理解,关于等式(25)和(27)示出的生成效用分数的实例仅是示例性的,并且患者选择模块324可以使用其它统计学或概率分布来生成效用分数。
[0579]
x.b.对于t细胞疗法的患者选择
[0580]
在另一个实施方案中,代替接受疫苗治疗或除了接受疫苗治疗之外,患者可以接受t细胞疗法。像疫苗治疗一样,在患者接受t细胞疗法的实施方案中,患者可以如上所述与v种新抗原候选物的相应治疗子集相关联。v种新抗原候选物的该治疗子集可用于体外鉴别来自患者的对v种新抗原候选物中的一种或多种有响应的t细胞。然后可以扩增鉴别的t细胞并且输注回患者体内以进行定制的t细胞疗法。
[0581]
可以选择患者以在两个不同的时间点接受t细胞疗法。第一个点是在已经使用模型为患者预测了v种新抗原候选物的治疗子集之后,但在体外筛选对v种新抗原候选物的预测的治疗子集具有特异性的t细胞之前。第二个点是在体外筛选对v种新抗原候选物的预测
的治疗子集具有特异性的t细胞之后。
[0582]
首先,可以在已经为患者预测了v种新抗原候选物的治疗子集之后,但在体外鉴别来自患者的对v种新抗原候选物的预测的子集具有特异性的t细胞之前来选择患者以接受t细胞疗法。具体地,由于来自患者的新抗原特异性t细胞的体外筛选可能是昂贵的,所以可能期望的是仅在患者可能具有新抗原特异性t细胞的情况下才选择患者来筛选新抗原特异性t细胞。为了在体外t细胞筛选步骤之前选择患者,可以使用与用于选择对于疫苗治疗的患者相同的标准。具体地,在一些实施方案中,如果如上所述患者的肿瘤突变负荷等于或高于最小突变负荷,则患者选择模块324可选择患者以接受t细胞疗法。在另一个实施方案中,如果如上所述基于患者的v种新抗原候选物治疗子集的患者效用分数等于或高于最小效用分数,则患者选择模块324可选择患者以接受t细胞疗法。
[0583]
第二,除了或代替在体外鉴别来自患者的对v种新抗原候选物的预测的子集具有特异性的t细胞之前来选择患者以接受t细胞疗法,还可在体外鉴别对v种新抗原候选物的预测的治疗子集具有特异性的t细胞之后选择患者以接受t细胞疗法。具体地,如果在患者t细胞的新抗原识别的体外筛选过程中对患者鉴别了至少阈值量的新抗原特异性tcr,则可以选择患者以接受t细胞疗法。例如,仅在对患者鉴别了至少两种新抗原特异性tcr或者仅在针对两种不同新抗原鉴别了新抗原特异性tcr时,才可选择患者以接受t细胞疗法。
[0584]
在另一个实施方案中,仅在患者的v种新抗原候选物的治疗子集中的至少阈值量的新抗原被患者tcr识别时,才可选择患者以接受t细胞疗法。例如,仅在患者的v种新抗原候选物的治疗子集中的至少一种新抗原被患者tcr识别时,才可选择患者以接受t细胞疗法。在另一些实施方案中,仅在患者的至少阈值量的tcr被鉴别为对特定hla限制类别的新抗原肽具有新抗原特异性时,才可选择患者以接受t细胞疗法。例如,仅在患者的至少一种tcr被鉴别为对i类hla限制的新抗原肽具有新抗原特异性时,才可选择患者以接受t细胞疗法。
[0585]
在甚至另一些实施方案中,仅在特定hla限制类别的至少阈值量的新抗原肽被患者tcr识别时,才可选择患者以接受t细胞疗法。例如,仅在至少一种i类hla限制的新抗原肽被患者tcr识别时,才可选择患者接受t细胞疗法。又例如,仅在至少两种ii类hla限制的新抗原肽被患者tcr识别时,才可选择患者以接受t细胞疗法。在体外鉴别对患者的v种新抗原候选物的预测治疗子集具有特异性的t细胞之后,上述标准的任何组合也可以用于选择患者以接受t细胞疗法。
[0586]
xi.实施例7:显示示例患者选择表现的实验结果
[0587]
通过对模拟患者集合进行患者选择来测试在部分x中描述的患者选择方法的有效性,每个模拟患者均与模拟新抗原候选物的测试集相关联,其中已知模拟新抗原的子集存在于质谱数据中。具体地,测试集中的每个模拟新抗原候选物都与指示新抗原是否存在于来自bassani
‑
sternberg数据集(数据集“d1”)(数据可在www.ebi.ac.uk/pride/archive/projects/pxd0000394上找到)的多等位基因jy细胞系hla
‑
a*02:01和la
‑
b*07:02质谱数据集的标记相关联。如以下结合图13a更详细地描述的,基于非小细胞肺癌(nsclc)患者中突变负荷的已知频率分布,从人蛋白质组中取样了模拟患者的许多新抗原候选物。
[0588]
使用训练集训练相同hla等位基因的独立等位基因呈递模型,所述训练集是来自iedb(数据集“d2”)(数据可以在http://www.iedb.org/doc/mhc_ligand_full.zip中找到)
的单等位基因hla
‑
a*02:01和hla
‑
b*07:02质谱数据的子集。具体地,每个等位基因的呈递模型是等式(8)中所示的独立等位基因模型,其并入了n端和c端侧接序列作为等位基因非相互作用变量,以及网络相关性函数g
h
(
·
)和g
w
(
·
),及expit函数f(
·
)。等位基因hla
‑
a*02:01的呈递模型产生了给定肽将在等位基因hla
‑
a*02:01上呈递的呈递概率,给出了肽序列作为等位基因相互作用变量,并且n端和c端侧接序列作为等位基因非相互作用变量。等位基因hla
‑
b*07:02的呈递模型产生了给定肽将在等位基因hla
‑
b*07:02上呈递的呈递概率,给出了肽序列作为等位基因相互作用变量,并且n端和c端侧接序列作为等位基因非相互作用变量。
[0589]
如在以下实施例中并且参照图13a
‑
13e所阐述的,将用于肽结合预测的多种模型例如经训练的呈递模型和当前现有技术模型应用于每个模拟患者的新抗原候选物的测试集,以基于预测来鉴别患者的不同治疗子集。选择满足纳入标准的患者进行疫苗治疗,并将其与包含患者治疗子集中的表位的定制疫苗相关联。治疗子集的大小根据不同疫苗容量而变化。在用于训练呈递模型的训练集和模拟新抗原候选物的测试集之间没有引入重叠。
[0590]
在以下实施例中,分析了具有在疫苗中所包含的表位中的至少一定数量的被呈递新抗原的选定患者的比例。该统计数据指示了模拟疫苗递送将在患者中引发免疫应答的潜在新抗原的有效性。具体地,如果新抗原存在于质谱数据集d2中,则呈递在测试集中的模拟新抗原。高比例的具有被呈递新抗原的患者指示通过诱导免疫应答通过新抗原疫苗成功治疗的潜力。
[0591]
xi.a.实施例7a:nsclc癌症患者的突变负荷的频率分布
[0592]
图13a示出了nsclc患者中突变负荷的样品频率分布。包括nsclc在内的不同肿瘤类型的突变负荷和突变可以在例如癌症基因组图谱(tcga)(https://cancergenome.nih.gov)上找到。x轴代表每个患者中非同义突变的数目,并且y轴代表具有给定数目的非同义突变的样本患者的比例。图13a的样品频率分布显示了一系列3
‑
1786种突变,其中30%的患者具有少于100个突变。尽管未在图13a中示出,研究表明吸烟者的突变负荷比不吸烟者高,而且突变负荷可能是患者中新抗原负荷的强指标。
[0593]
如在以上部分xi开始时所介绍的,许多模拟患者中的每一个都与新抗原候选物的测试集相关联。对于每个患者,通过从图13a中所示的频率分布对突变负m
i
采样来产生每个患者的测试集。对于每个突变,随机选择来自人类蛋白质组的21
‑
mer肽序列以代表模拟突变序列。通过鉴别跨越21
‑
mer中的突变的每种(8、9、10、11)
‑
mer肽序列,为每个患者i产生新抗原候选物序列的测试集。每个新抗原候选物与都指示新抗原候选物序列是否存在于质谱d1数据集中的标记相关联。例如,存在于数据集d1中的新抗原候选物序列可以与标记“1”相关联,而不存在于数据集d1中的序列可以与标记“0”相关联。如以下更详细描述的,图13b至13e示出了基于测试集中患者的被呈递新抗原进行的患者选择的实验结果。
[0594]
xi.b.实施例7b:基于突变负荷纳入标准具有新抗原呈递的选定患者的比例
[0595]
图13b示出了对于基于患者是否满足最小突变负荷的纳入标准选择的患者,模拟疫苗中被呈递的新抗原的数目。确定在相应的测试中具有至少一定数量的被呈递的新抗原的选定患者的比例。
[0596]
在图13b中,x轴表示基于最小突变负荷(如通过标记“最小突变数”指示的)从疫苗治疗中排除的患者的比例。例如,200“最小突变数”处的数据点表示患者选择模块324仅选
择具有至少200个突变的突变负荷的模拟患者的子集。又例如,300“最小突变数”处的数据点表示患者选择模块324选择了具有至少300个突变的较低比例的模拟患者。y轴表示在没有任何疫苗容量v的测试集中与至少一定数量的被呈递的新抗原相关联的选定患者的比例。具体来说,上方的图显示了呈递至少一种新抗原的选定患者的比例,中间的图显示了呈递至少两种新抗原的选定患者的比例,并且底部的图显示了呈递至少三种新抗原的选定患者的比例。
[0597]
如图13b所示,随着更高的突变负荷,具有被呈递的新抗原的选定患者的比例显著增加。这表明突变负荷作为纳入标准可以有效地选择新抗原疫苗更有可能诱导成功免疫应答的患者。
[0598]
xi.c.实施例7c:通过呈递模型对比于现有技术模型鉴别的疫苗的新抗原呈递的比较
[0599]
图13c在与包含基于呈递模型鉴别的治疗子集的疫苗相关的选定患者和与包含通过当前现有技术模型鉴别的治疗子集的疫苗相关的选定患者之间,比较了模拟疫苗中被呈递的新抗原的数目。左图假定有限的疫苗容量v=10,而右图假定有限的疫苗容量v=20。基于效用分数选择患者,所述效用分数指示被呈递新抗原的预期数目。
[0600]
在图13c中,实线表示与包含基于等位基因hla
‑
a*02:01和hla
‑
b*07:02的呈递模型鉴别治疗子集的疫苗相关的患者。通过将每个呈递模型应用于测试集中的序列,并且鉴别具有最高呈递可能性的v个新抗原候选物,来鉴别每个患者的治疗子集。虚线表示与包含基于单等位基因hla
‑
a*02:01的当前现有技术模型netmhcpan鉴别的治疗子集的疫苗相关的患者。netmhcpan的实现细节在http://www.cbs.dtu.dk/services/netmhcpan中详细提供。通过将netmhcpan模型应用于测试集中的序列,并且鉴别具有最高估计结合亲和力的v个新抗原候选物,来鉴别每个患者的治疗子集。两个图的x轴表示基于预期效用分数从疫苗治疗中排除的患者的比例,所述预期效用分数指示基于呈递模型鉴别的治疗子集中被呈递的新抗原的预期数目。预期效用分数的确定如参考部分x中的等式(25)所述。y轴表示呈递疫苗中所包含的至少一定数量的新抗原(1、2或3种新抗原)的选定患者的比例。
[0601]
如图13c中所示,同与包含基于现有技术模型的治疗子集的疫苗相关的患者相比,与包含基于呈递模型的治疗子集的疫苗相关的患者以显著更高的比例接受包含被呈递新抗原的疫苗。例如,如右图所示,与基于呈递模型的疫苗相关的选定患者中有80%接受疫苗中至少一种被呈递的新抗原,相比之下,与基于当前现有技术模型的疫苗相关的选定患者中仅有40%。结果表明,如本文所述的呈递模型对于选择可能引发用于治疗肿瘤的免疫应答的疫苗的新抗原候选物是有效的。
[0602]
xi.d.实施例7d:hla覆盖率对通过呈递模型鉴别的疫苗的新抗原展呈递的影响
[0603]
图13d在与包含基于hla
‑
a*02:01的单独立等位基因呈递模型鉴别的治疗子集的疫苗相关的选定患者和与包含基于hla
‑
a*02:01和hla
‑
b*07:02的双独立等位基因呈递模型鉴别的治疗子集的疫苗相关的选定患者之间,比较了模拟疫苗中被呈递的新抗原的数目。疫苗容量设定为v=20个表位。对于每个实验,根据基于不同治疗子集确定的期望效用分数选择患者。
[0604]
在图13d中,实线表示与包含基于hla等位基因hla
‑
a*02:01和hla
‑
b*07:02的双呈递模型的治疗子集的疫苗相关的患者。通过将每个呈递模型应用于测试集中的序列,并且
鉴别具有最高呈递可能性的v个新抗原候选物,来鉴别每个患者的治疗子集。虚线表示与包含基于hla等位基因hla
‑
a*02:01的单呈递模型的治疗子集的疫苗相关的患者。通过将仅单个hla等位基因的呈递模型应用于测试集中的序列,并且鉴别具有最高呈递可能性的v个新抗原候选物,来鉴别每个患者的治疗子集。对于实线图,x轴表示基于通过双呈递模型鉴别的治疗子集的预期效用分数从疫苗治疗中排除的患者的比例。对于虚线图,x轴表示基于通过单呈递模型鉴别的治疗子集的预期效用分数从疫苗治疗中排除的患者的比例。y轴表示呈递至少一定数量的新抗原(1、2或3种新抗原)的选定患者的比例。
[0605]
如图13d中所示,同与包含通过单呈递模型鉴别的治疗子集的疫苗相关的患者相比,与包含通过双hla等位基因的呈递模型鉴别治疗子集的疫苗相关的患者以显著更高的比例呈递新抗原。结果表明了建立具有高hla等位基因覆盖率的呈递模型的重要性。
[0606]
xi.e.实施例7e:通过突变负荷对比于通过被呈递抗原的预期数目选择的患者的新抗原呈递的比较
[0607]
图13e在基于肿瘤突变负荷选择的患者和通过预期效用分数选择的患者之间,比较了模拟疫苗中被呈递的新抗原的数量。基于通过具有v=20个表位的呈递模型鉴别的治疗子集确定预期效用分数。
[0608]
在图13e中,实线表示与包含通过呈递模型鉴别的治疗子集的疫苗相关的基于预期效用分数选择的患者。通过将呈递模型应用于测试集中的序列,并且鉴别具有最高呈递可能性的v=20个新抗原候选物,来鉴别每个患者的治疗子集。基于根据部分x中的等式(25)的鉴别的治疗子集的呈递可能性来确定预期效用分数。虚线表示与还包含通过呈递模型鉴别的治疗子集的疫苗相关的基于突变负荷选择的患者。实线图的x轴表示基于预期效用分数从疫苗治疗中排除的患者的比例,虚线图的x轴表示基于突变负荷从疫苗治疗中排除的患者的比例。y轴表示接受包含至少一定数量的被呈递的抗原(1、2或3种新抗原)的疫苗的选定患者的比例。
[0609]
如图13e中所示,与基于突变负荷选择的患者相比,基于预期效用分数选择的患者以更高的比率接受包含被呈递新抗原的疫苗。然而,基于突变负荷选择的患者比未选择的患者以更高的比率接受包含被呈递新抗原的疫苗。因此,突变负荷是成功的新抗原疫苗治疗的有效患者选择标准,尽管预期效用分数更有效。
[0610]
xii.实施例8:根据留出ii类mhc质谱数据的经过质谱训练的ii类mhc呈递模型的评估
[0611]
根据测试数据t测试上述各种呈递模型的有效性,所述测试数据t是未用于训练呈递模型的训练数据170的子集,或者是与训练数据170具有相似的变量和数据结构的来自训练数据170的单独数据集。
[0612]
指示呈递模型的性能的相关度量是:
[0613][0614]
,其指示被正确预测在相关的hla等位基因上呈递的肽实例的数量与被预测在hla等位基因上呈递的肽实例的数量的比率。在一个实施方式中,如果相应的可能性估计u
i
大于或等于给定的阈值t,则测试数据t中的肽p
i
被预测为在一个或多个相关hla等位基因上呈递。指示呈递模型的性能的另一个相关度量是:
[0615][0616]
,其指示被正确预测在相关hla等位基因上呈递的肽实例的数量与已知在hla等位基因上呈递的肽实例的数量的比率。指示呈递模型的性能的另一个相关度量是接受者工作特性(roc)的曲线下面积(auc)。所述roc将召回率针对假阳性率(fpr)作图,其由下式给出:
[0617][0618]
xii.a.ii类mhc质谱数据的呈递模型的性能
[0619]
xii.a.1.实施例1
[0620]
图14a是使用质谱法从人肿瘤细胞和肿瘤浸润淋巴细胞(til)上的ii类mhc等位基因洗脱的肽的长度的直方图。具体地,对hla
‑
drb1*12:01纯合子等位基因(“数据集1”)和hla
‑
drb1*12:01、hla
‑
drb1*10:01多等位基因样品(“数据集2”)进行了质谱肽组学分析。结果显示,从ii类mhc等位基因洗脱的肽长度范围为6
‑
30个氨基酸。图14a所示的频率分布类似于使用现有技术水平的质谱技术从ii类mhc等位基因洗脱的肽的长度的频率分布,如参考文献69的图1c中所示的。
[0621]
图14b示出了数据集1和数据集2的mrna定量与每残基呈递的肽之间的依赖性。结果表明,对于ii类mhc等位基因,在mrna表达与肽呈递之间存在很强的依赖性。
[0622]
具体地,图14b中的水平轴表示以log
10
转录物/百万(tpm)箱为单位表示的mrna表达。图14b中的垂直轴以对应于10
‑2<log
10
tpm<10
‑1之间的mrna表达的最低箱的肽呈递的倍数表示每残基的肽呈递。一条实线是使数据集1的mrna定量与肽呈递相关的曲线,另一条实线是针对数据集2的曲线。如图14b所示,在相应基因中,mrna表达与每残基的肽呈递之间有很强的正相关性。具体地,来自在101<log
10
tpm<102的范围内的rna表达的基因的肽被呈递的可能性为底箱的5倍以上。
[0623]
结果表明,呈递模型的性能可通过结合mrna定量测量而大大提高,因为这些测量强烈地预测了肽呈递。
[0624]
图14c比较了使用数据集1和数据集2训练和测试的示例呈递模型的性能结果。对于示例性呈递模型的每组模型特征,图14c描绘了当模型特征集合中的特征被分类为等位基因相互作用特征时,或者当模型特征集合中的特征被分类为等位基因非相互作用特征变量时,10%的召回率下的ppv值。如图14c所示,对于示例呈递模型的每组模型特征,当模型特征组中的特征被分类为等位基因相互作用特征时确定的10%的召回率下的ppv值显示在左侧,并且当模型特征组中的特征被分类为等位基因非相互作用特征时确定的10%的召回率下的ppv值显示在右侧。注意,出于14c的目的,肽序列的特征总是被归类为等位基因相互作用特征。结果表明,呈递模型在10%的召回率下达到了14%直至29%不等的ppv值,这比随机预测的ppv高得多(约500倍)。
[0625]
本实验考虑了长度为9
‑
20的肽序列。数据被分成训练集、验证集和测试集。来自数据集1和数据集2的50个残基块(residue block)的肽块被分配给训练集和测试集。去除蛋白质组中任何地方重复的肽,确保在训练集和测试集中均没有肽序列出现。通过去除非呈递的肽,训练集和测试集中肽呈递的普遍性增加了50倍。这是因为数据集1和数据集2来自
人肿瘤样品,其中只有一小部分细胞是ii类hla等位基因,导致比ii类hla等位基因的纯样品低约10倍的肽产量,由于质谱灵敏度不完善,这仍然是一个低估。训练集包含1,064个已呈递的肽和3,810,070个未呈递的肽。测试集包含314个呈递肽和807,400个非呈递的肽。
[0626]
示例性模型1是使用网络相关性函数g
h
(
·
)、expit函数f(
·
)和恒等函数r(
·
)的等式(22)中的函数的和模型。网络相关性函数g
h
(
·
)被构造为具有256个隐藏节点和已校正的线性单位(relu)激活的多层感知器(mlp)。除了肽序列之外,等位基因相互作用变量w包含独热编码的的c端和n端侧接序列、指示肽p
i
的源基因g=基因(p
i
)的指数的分类变量和指示mrna定量测量的变量。示例性模型2与示例性模型1相同,只是从等位基因相互作用变量中省略了c端和n端侧接序列。示例性模型3与示例性模型1相同,只是从等位基因相互作用变量中省略了源基因的指数。示例性模型4与示例性模型1相同,只是从等位基因相互作用变量中省略了mrna定量测量。
[0627]
示例性模型5是利用等式(12)的网络相关性函数g
h
(
·
)、expit函数f(
·
)、恒等函数r(
·
)和相关性函数g
w
(
·
)的等式(20)中的函数的和模型。相关性函数g
w
(
·
)还包括以mrna定量测量为输入的网络模型(其被构造为具有16个隐藏节点和relu激活的mlp)以及以c
‑
侧接序列为输入的网络模型(其被构造为具有32个隐藏节点和relu激活的mlp)。网络关性函数g
h
(
·
)被构造为具有256个隐藏节点和已校正的线性单位(relu)激活的多层感知器。示例性模型6与示例性模型5相同,只是省略了c端和n端侧接序列的网络模型。示例性模型7与示例性模型5相同,只是从等位基因非交互作用变量中省略了源基因的指数。示例性模型8与示例性模型5相同,只是省略了用于mrna定量测量的网络模型。
[0628]
测试集中呈递的肽的普遍性约为1/2400,因此,随机预测的ppv也约为1/2400=0.00042。如图14c所示,表现最好的呈递模型实现了约29%的ppv值,这比随机预测的ppv值大约好500倍。
[0629]
xii.a.2.实施例2
[0630]
图14d是描绘对总共73个样品中的每个样品使用质谱法测序的肽的数量的直方图,所述样品包含含有ii类hla分子的人肿瘤(nsclc、淋巴瘤和卵巢癌)和细胞系(ebv)。如图14d所示,每个样品平均有900个肽被测序。此外,对于多个样品中的每个样品,图14d所示的直方图描绘了在不同q值阈值下使用质谱法测序的肽的数量。具体地,对于多个样品中的每个样品,图14d描绘了利用小于0.01的q值、利用小于0.05的q值以及利用小于0.2的q值,使用质谱法测序的肽的数量。
[0631]
如上所述,图14d的73个样品中的每个样品包含ii类hla分子。更具体地,图14d的73个样品中的每个样品包含hla
‑
dr分子。hla
‑
dr分子是ii类hla分子的一种类型。更具体地,图14d的73个样品中的每个样品包含hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和/或hla
‑
drb5分子。hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和hla
‑
drb5分子是hla
‑
dr分子的类型。
[0632]
虽然该特定实验是使用包含hla
‑
dr分子,特别是hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和hla
‑
drb5分子的样品进行的,但在替代实施方案中,该实验可使用包含一种或多种任何类型的ii类hla分子的样品来进行。例如,在替代实施方案中,可使用包含hla
‑
dp和/或hla
‑
dq分子的样品进行相同的实验。使用相同的技术对任何一种或多种类型的ii类mhc分子进行建模,并且仍然获得可靠的结果的能力是本领域技术人员所公知的。例
如,jensen,kamilla kjaergaard,等人
76
是最近一篇科学论文的一个实例,所述论文使用相同的方法对针对hla
‑
dr分子以及hla
‑
dq和hla
‑
dp分子的结合亲和力进行建模。因此,本领域技术人员将理解,本文所述的实验和模型用于单独或同时对不仅hla
‑
dr分子,还对任何其它ii类mhc分子进行建模,同时仍能产生可靠的结果。
[0633]
为了对总共73个样品中每个样品的肽进行测序,对每个样品进行质谱法。然后用comet搜索样品的所得质谱,并用percolator进行评分,以对肽进行测序。然后,针对多个不同的percolator q值阈值,确定样品中测序的肽的数量。具体地,对于样品,确定了以小于0.01的percolator q值、以小于0.05的percolator q值和以小于0.2的percolator q值测序的肽的数量。
[0634]
对于73个样品中的每个样品,在每一个不同的percolator q值阈值下测序的肽的数量描绘于图14d中。例如,如图14d中所看到的,对于第一样品,使用质谱法对约4700个q值小于0.2的肽进行测序,使用质谱法对约3600个q值小于0.05的肽进行测序,以及使用质谱法对约3200个q值小于0.01的肽进行测序。
[0635]
总的来说,图14d证明了使用质谱法在低q值下对含有ii类mhc分子的样品中的大量肽进行测序的能力。换句话说,图14d中描绘的数据证明了使用质谱法对可由ii类mhc分子呈递的肽进行可靠测序的能力。
[0636]
图14e是描绘其中特定的ii类mhc分子等位基因被鉴别的样品的数量的直方图。更具体地,对于总共73个包含ii类hla分子的样品,图14e描述了其中鉴别出某些ii类mhc分子等位基因的样品的数量。
[0637]
如上面关于图14d所述,图14d的73个样品中的每个样品包含hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和/或hla
‑
drb5分子。因此,图14e描绘了其中鉴别出hla
‑
drb1、hla
‑
drb3、hla
‑
drb4和hla
‑
drb5分子的某些等位基因的样品的数量。为了鉴别样品中存在的hla等位基因,对样品进行hla ii类dr分型。然后,为了确定其中鉴定出特定的hla等位基因的样品的数量,仅将其中使用hla ii类dr分型鉴别hla等位基因的样品的数量相加。例如,如图14e所描绘的,总共73个样品中的17个样品含有ii类hla分子等位基因hla
‑
drb3*01:01。换句话说,总共73个样品中的17个样品包含hla
‑
drb3分子的等位基因hla
‑
drb3*01:01。总的来说,图14e描绘了从73个包含ii类hla分子的样品中鉴别出多种的ii类hla分子等位基因的能力。
[0638]
图14f是直方图,其描绘了在总共73个样品中,对于一系列肽长度中的每个肽长度,由ii类mhc分子呈递的肽的比例。为了确定总共73个样品中每个样品中每个肽的长度,将每个肽都使用如上面关于图14d所述的质谱法进行测序,然后仅定量测序的肽中的残基数量。
[0639]
如上所述,ii类mhc分子通常呈递长度为9至20个氨基酸的肽。因此,图14f描绘了对于9至20个氨基酸(包括9个和20个氨基酸)的每个肽长度,在73个样品中由ii类mhc分子呈递的肽的比例。例如,如图14f所示,在73个样品中由ii类mhc分子呈递的肽中的约23%的肽包含14个氨基酸的长度。
[0640]
基于图14f中描绘的数据,在73个样品中由ii类mhc分子呈递的肽的模态长度(modal length)被鉴别为长度为14个和15个氨基酸。在73个样品中针对由ii类mhc分子呈递的肽鉴别的这些模态长度与先前的由ii类mhc分子呈递的肽的模态长度的报告一致。另
外,这也与之前的报告一致,图14f的数据表明超过60%的来自73个样品的由ii类mhc分子呈递的肽包含除14个和15个氨基酸外的长度。换句话说,图14f表明,虽然ii类mhc分子呈递的肽最常见的长度是14或15个氨基酸,但ii类mhc分子呈递的大部分肽的长度不是14或15个氨基酸。因此,假设所有长度的肽由ii类mhc分子呈递的概率相等,或者ii类mhc分子仅呈递包含14或15个氨基酸长度的肽,这是一个很差的假设。如下面参考图14l所详细讨论的,这些错误的假设目前被用于许多现有技术水平的模型中,所述模型用于预测ii类mhc分子的肽呈递,因此,这些模型预测的呈递可能性通常是不可靠的。
[0641]
图14g是描绘73个样品中存在的基因的基因表达与ii类mhc分子对基因表达产物的呈递普遍性之间的关系的线形图。更具体地,图14g描绘了基因表达与由基因表达产生的残基比例之间的关系,所述残基形成由ii类mhc分子呈递的肽的n端。为了定量总共73个样品中的每个样品的基因表达,对每个样品中包含的rna进行rna测序。在图14g中,基因表达通过rna测序以每百万转录物(tpm)为单位进行测量。为了确定73个样品中每个样品的基因表达产物的普遍性,对每个样品进行了hla ii类dr肽组学数据的确定。
[0642]
如图14g中所描绘的,对于73个样品,基因表达水平与ii类mhc分子对表达的基因产物的残基的呈递之间有很强的相关性。具体地,如图14g所示,由表达最少的基因产生的肽由ii类mhc分子呈递的可能性比由表达最多的基因产生的肽低100倍以上。简而言之,更高表达的基因的产物更频繁地由ii类mhc分子呈递。
[0643]
图14h至图14i和图14k至图14l是比较各种呈递模型在预测肽测试数据集中的肽将由测试数据集中存在的ii类mhc分子中的至少一种呈递的可能性方面的性能的线形图。如图14h至图14i和图14k至图14l所示,模型在预测肽将由测试数据集中存在的ii类mhc分子中的至少一种呈递的可能性方面的性能通过确定由模型做出的每个预测的真阳性率与假阳性率的比率来确定。针对给定的模型确定的这些比率可在具有定量假阳性率的x轴和定量真阳性率的y轴的线形图中被可视化为roc(接受者工作特性)曲线。曲线下面积(auc)用于定量模型的性能。具体地,相对于具有较小的auc的模型,具有较大auc的模型具有更高的性能(即,更高的精度)。在图14h、图14i和图14l中,斜率为1(即,真阳性率与假阳性率的比率为1)的黑色虚线描绘了随机猜测肽呈递的可能性的预期曲线。该虚线的auc为0.5。关于上述第xii.节的顶部详细论述了roc曲线和auc度量。
[0644]
图14h是线图,其在给定不同组的等位基因相互作用变量和等位基因非相互作用变量的情况下,比较了五个示例性呈递模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能。换句话说,图14h定量了各种等位基因相互作用变量和等位基因非相互作用变量对于预测肽将由ii类mhc分子呈递的可能性的相对重要性。
[0645]
五个用于生成图14h的线形图的roc曲线的示例性呈递模型中的每个示例性呈递模型的模型体系结构包含五个sigmoids的和模型的集合。集合中的每个sigmoids的和模型被配置成对每个样品多达四个独特的hla
‑
dr等位基因的肽呈递进行建模。此外,集合中的每个sigmoids的和模型被配置成基于以下等位基因相互作用变量和等位基因非相互作用变量来预测肽呈递的可能性:肽序列、侧接序列、以tpm为单位的rna表达、基因标识符和样品标识符。集合中每个sigmoids的和模型的等位基因相互作用分量是具有relu激活作为256个隐藏单元的单隐藏层mlp。
[0646]
在使用示例性模型预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性之
前,训练并验证示例性模型。为了训练、验证和最终测试示例性模型,上面针对73个样品所述的数据被分成训练数据集、验证数据集和测试数据集。
[0647]
为了确保在训练数据集、验证数据集和测试数据集中的超过一者中不出现肽,执行了以下程序。首先,将所有来自总共73个样品的在蛋白质组中出现在超过一个位置中的肽去除。然后,将来自总共73个样品的肽划分成10个相邻的肽块。来自总共73个样品的每个肽块被唯一地分配给训练数据集、验证数据集或测试数据集。这样,没有肽出现在训练数据集、验证数据集和测试数据集中的超过一者中。
[0648]
在总共73个样品中的38,035,453个肽中,训练数据集包含来自总共73个样品中的69个的ii类mhc分子呈递的33,570个肽。训练数据集中包含的33,570个肽的长度为9至20个氨基酸,包括9个和20个氨基酸。使用adam优化器和早期停止,在训练数据集上训练用于生成图14h中的roc曲线的示例性模型。
[0649]
验证数据集由来自训练数据集中使用的相同的69个样品的3,925个由ii类mhc分子呈递的肽组成。验证集仅用于早期停止。
[0650]
测试数据集包含由ii类mhc分子提供的肽,所述肽是使用质谱法从肿瘤样品中鉴别的。具体地,测试数据集包含从四个肿瘤样品中鉴别的232种肽。测试数据集中包含的肽不包含在上述训练数据集中。
[0651]
如上所述,图14h定量了各种等位基因相互作用变量和等位基因非相互作用变量对于预测肽将由ii类mhc分子呈递的可能性的相对重要性。同样如上所述,用于生成图14h的线形图的roc曲线的示例性模型被配置成基于以下等位基因相互作用变量和等位基因非相互作用变量来预测肽呈递可能性:肽序列、侧接序列、以tpm为单位的rna表达、基因标识符和样品标识符。为了定量这五个变量中的四个变量(肽序列、侧接序列、rna表达和基因标识符)对于预测肽将由ii类mhc分子呈递的可能性的相对重要性,使用来自测试数据集的数据,利用所述四个变量的不同组合来测试上述五个示例性模型中的每个示例性模型。具体地,对于测试数据集中的每个肽,示例性模型1基于肽序列、侧接序列、基因标识符和样品标识符而不是基于rna表达,生成了肽呈递可能性的预测。类似地,对于测试数据集中的每个肽,示例性模型2基于肽序列、rna表达、基因标识符和样品标识符而不是基于侧接序列,生成了肽呈递可能性的预测。类似地,对于测试数据集中的每个肽,示例性模型3基于侧接序列、rna表达、基因标识符和样品标识符,而不是基于肽序列,生成了肽呈递可能性的预测。类似地,对于测试数据集中的每个肽,示例性模型4基于侧接序列、rna表达、肽序列和样品标识符而不是基于基因标识符,生成了肽呈递可能性的预测。最后,对于测试数据集中的每个肽,示例性模型5基于侧接序列、rna表达、肽序列、样品标识符和基因标识符的所有五个变量生成了肽呈递可能性的预测。
[0652]
这五个示例性模型中的每一个的性能描绘于图14h的线形图中。具体地,五个示例性模型中的每一个都与roc曲线相关联,所述roc曲线描绘了由模型做出的每个预测的真阳性率与假阳性率的比率。例如,图14h描绘了示例性模型1的曲线,所述示例性模型1基于肽序列、侧接序列、基因标识符和样品标识符而不是基于rna表达来生成肽呈递可能性的预测。图14h描绘了示例性模型2的曲线,所述示例性模型2基于肽序列、rna表达、基因标识符和样品标识符而不是基于侧接序列生成了肽呈递可能性的预测。图14h还描绘了示例性模型3的曲线,所述示例性模型3基于侧接序列、rna表达、基因标识符和样品标识符而不是基
于肽序列生成了肽呈递可能性的预测。图14h还描绘了示例性模型4的曲线,所述示例性模型4基于侧接序列、rna表达、肽序列和样品标识符而不是基于基因标识符生成了肽呈递可能性的预测。最后图14h描绘了示例性模型5的曲线,所述示例性模型5基于侧接序列、rna表达、肽序列、样品标识符和基因标识符的所有五个变量生成了肽呈递可能性的预测。
[0653]
如上所述,通过确定roc曲线的auc来定量模型在预测肽将由ii类mhc分子呈递的可能性方面的性能,所述roc曲线描绘了由模型做出的每个预测的真阳性率与假阳性率的比率。与具有较小auc的模型相比,具有较大auc的模型具有更高的性能(即,更高的精度)。如图14h所示,基于侧接序列、rna表达、肽序列、样品标识符和基因标识符的所有五个变量生成肽呈递可能性预测的示例性模型5的曲线,获得了为0.98的最高auc。因此,使用所有五个变量来生成肽呈递预测的示例性模型5实现了最佳性能。基于肽序列、rna表达、基因标识符和样品标识符而不是基于侧接序列生成肽呈递可能性的预测的示例性模型2的曲线,获得了为0.97的第二高auc。因此,侧接序列可被确定为预测肽将由ii类mhc分子呈递的可能性的最不重要的变量。基于侧接序列、rna表达、肽序列和样品标识符而不是基于基因标识符生成肽呈递可能性的预测的示例性模型4的曲线,获得了为0.96的第三高的auc。因此,基因标识符可被确定为预测肽将由ii类mhc分子呈递的可能性的第二最不重要的变量。基于侧接序列、rna表达、基因标识符和样品标识符而不是基于肽序列生成肽呈递可能性的预测的示例性模型3的曲线,获得了为0.88的最低auc。因此,肽序列可被确定为预测肽将由ii类mhc分子呈递的可能性的最重要变量。基于肽序列、侧接序列、基因标识符和样品标识符而不是基于rna表达生成肽呈递可能性的预测的示例性模型1的曲线,获得了为0.95的第二低auc。因此,rna表达可被确定为对于预测肽将由ii类mhc分子呈递的可能性是第二最重要的变量。
[0654]
图14i是比较四种不同呈递模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能的线形图。
[0655]
图14i中测试的第一模型在本文中被称为“结合亲和力”模型。图14i的结合亲和力模型是同类最佳的现有技术模型,即netmhcii 2.3模型,其利用最小netmhcii 2.3预测的结合亲和力作为生成预测的标准。具体地,netmhcii 2.3模型基于ii类mhc分子类型和肽序列生成肽呈递可能性的预测。使用netmhcii 2.3网站(www.cbs.dtu.dk/services/netmhcii/,pmid 29315598)
76
测试了netmhcii 2.3模型。
[0656]
图14i中测试的第二模型在本文中被称为“mlp”模型。mlp(多层感知器)模型是上述呈递模型的一个实施方案,其中等位基因非相互作用变量w
k
和等位基因相互作用变量x
hk
被输入到单独的相关性函数(例如神经网络)中,然后将这些单独的相关性函数的输出相加。具体地,完全非相互作用模型是上述呈递模型的一个实施方案,其中等位基因非相互作用变量w
k
被输入到相关性函数g
w
中,等位基因相互作用变量x
hk
被输入到单独的相关性函数g
h
中,并且将相关性函数g
w
和相关性函数g
h
的输出加在一起。因此,在一些实施方案中,完全非相互作用模型使用如上所示的等式8来确定肽呈递的可能性。此外,关于第viii.b.2.节的顶部、第viii.b.3.节的底部、第viii.c.3.节的顶部和第viii.c.6.节的顶部详细地论述了完全非相互作用模型的实施方案,其中等位基因非相互作用变量w
k
被输入到相关性函数g
w
中,等位基因相互作用变量x
hk
被输入到单独的相关性函数g
h
中,并且将相关性函数g
w
和相关性函数g
h
的输出相加。
[0657]
图14i中测试的第三模型在本文中被称为“rnn”模型。rnn模型包括循环神经网络(recurrent neural network),并且类似于上面描述的完全非相互作用模型。然而,rnn模型的循环神经网络的层不同于mlp模型的神经网络的层。具体地,rnn模型的循环神经网络的输入层接受可变长度的肽串,所述肽串一次模拟一个肽。对肽一次将单个氨基酸输入神经网络节点,所述神经网络节点的输出与序列中的下一个氨基酸一起被输送到节点的输入中,直至整个序列被建模。循环层由于以下两个原因特别适用于ii类mhc肽建模:(1)数据的顺序性质被模型捕获,以及(2)肽的长度可以变化,而不需要人工填充。循环神经网络的下一层是p=0.2的脱落层,最后是具有relu激活的密集64节点层。
[0658]
图14i中测试的第四模型在本文中被称为“bi
‑
lstm”模型。bi
‑
lstm模型包括双向长短期记忆神经网络。除了肽输入层,bi
‑
lstm模型与非相互作用模型相同。bi
‑
lstm模型的输入层接受20
‑
mer肽串,并随后将20
‑
mer肽串嵌入为(n,20,21)张量。bi
‑
lstm模型的双向长短期记忆神经网络的下一层包括具有128个节点的循环长短期记忆层,p=0.2的脱落层以及最后具有relu激活的密集64节点层。在常规lstm模型中,序列数据的顺序被认为是有方向的(例如,从左到右或从右到左读取)。在双向lstm中,将序列数据在两个方向(从左到右和从右到左)上进行处理。肽结合本质上是没有方向的任务,因此在两个方向上对序列建模可以确保来自序列两端的信息在模型预测中占据同样的权重。
[0659]
简单转向图14j,图14j描绘了图14i的bi
‑
lstm模型的示例性实施方案,其被配置成预测hla
‑
drb(ii类mhc基因)的肽呈递。如图14j所示,bi
‑
lstm模型包括接受等位基因非相互作用特征(例如,rna序列、样品id、蛋白质id和侧接序列)的共享神经网络和一组不同的神经网络,每个神经网络与不同的hla
‑
drb等位基因相关联,并被配置成接受编码的肽序列(等位基因相互作用特征)。该组神经网络中的每个不同的神经网络包括bi
‑
lstm神经网络。在图14j的bi
‑
lstm模型的示例性实施方案中,与不同等位基因相关联的不同神经网络的组包括4个不同的神经网络,因为每个患者样品中hla
‑
drb基因最多与4个不同等位基因相关联。然而,在其中bi
‑
lstm模型被配置成预测另一个hla基因的肽呈递的替代实施方案中,不同的神经网络的组包含等于患者样品中给定的hla基因的等位基因的最大可能数量的数量的不同神经网络。神经网络的组中的每个不同的神经网络确定输入到模型中的肽将由与给定的神经网络相关联的hla
‑
dbr等位基因呈递的可能性。然后将这些可能性中的每一种可能性都与共享神经网络的输出组合。最后,将组合的可能性相加,生成肽将由hla
‑
dbr基因呈递的总体可能性。
[0660]
回到图14i,在使用图14i的四个模型中的每一个预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性之前,对模型进行了训练和验证。结合亲和力模型使用其自身的保存在免疫表位数据库(iedb,www.iedb.org)中的基于hla
‑
肽结合亲和力测定的训练和验证数据集进行训练和验证。另外三个模型使用上述69个样品的训练数据集进行训练,并使用上述验证数据集进行验证。在该模型的训练和验证之后,使用来自上述测试数据集的4个留出肿瘤样品来测试四个模型中的每一个。具体地,对于四个模型中的每一个,将来自测试数据集的4个留出肿瘤样品中的每一个肽输入到模型中,并且该模型随后输出该肽的呈递可能性。
[0661]
四个模型中的每一个的性能描绘于图14i的线形图中。具体地,四个模型中的每一个都与roc曲线相关联,所述roc曲线描绘了模型做出的每个预测的真阳性率与假阳性率的
比率。例如,图14i描绘了结合亲和力模型的roc曲线、rnn模型的roc曲线、mlp模型的roc曲线和bi
‑
lstm模型的roc曲线。
[0662]
如上所述,通过确定roc曲线的auc来定量模型在预测肽将由ii类mhc分子呈递的可能性方面的性能,所述roc曲线描绘了由模型做出的每个预测的真阳性率与假阳性率的比率。与具有较小auc的模型相比,具有较大auc的模型具有更高的性能(即,更高的精度)。如图14i中所示,bi
‑
lstm的曲线实现了为0.98的最高auc。因此,bi
‑
lstm模型实现了最佳性能。bi
‑
lstm模型的这种峰值性能部分归因于以下事实,即bi
‑
lstm具有最强的精确预测可变长度的肽、相对较长长度的肽和具有重复氨基酸的肽的能力。mlp和rnn模型的曲线获得了为0.97的第二高auc。因此,mlp和rnn模型获得了第二好的性能。结合亲和力模型的曲线获得了为0.79的最低auc。因此,结合亲和力模型获得了最差的性能。请注意,图14i中测试的bi
‑
lstm,、mlp和rnn模型中的每一个的auc大于0.9。因此,尽管它们之间肽输入层的体系结构不同,但这些模型能够实现相对精确的肽呈递预测,这与具有低得多的auc的结合亲和力模型不同。
[0663]
图14k是描绘了上面关于图14i所论述的“bi
‑
lstm”模型、“mlp”模型、“rnn”模型和“结合亲和力”模型的全精度召回率曲线的线图中。如图14k所示,并且如基于14i所预期的,“bi
‑
lstm”模型以0.23的auc获得了最佳性能,“rnn”模型以0.16的auc获得了第二好的性能,“mlp”模型以0.11的auc获得了第三好的性能,并且“结合亲和力”模型以0.01的auc获得了最差的性能。特别地,用质谱数据训练的bi
‑
lstm模型明显优于结合亲和力模型,auc增加了20倍以上。
[0664]
图14l是比较被给予两个不同标准的两个示例性同类最佳现有技术模型和被给予两个不同组的等位基因相互作用变量和等位基因非相互作用变量的两个示例性呈递模型在预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性方面的性能的线形图。具体地,图14l是线形图,其比较了利用最小netmhcii 2.3预测的结合亲和力作为生成预测的标准的示例性同类最佳现有技术模型(示例性模型1)、利用最小netmhcii 2.3预测的结合排名作为生成预测的标准的示例性同类最佳现有技术模型(示例性模型2)、基于ii类mhc分子类型和肽序列生成肽呈递可能性的预测的示例性呈递模型(示例性模型4)以及基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性的预测的示例呈递模型(示例性模型3)的性能。
[0665]
用作图14l中的示例性模型1和示例性模型2的同类最佳现有技术模型是netmhcii 2.3模型。netmhcii 2.3模型基于ii类mhc分子类型和肽序列生成肽呈递可能性的预测。使用netmhcii 2.3网站(www.cbs.dtu.dk/services/netmhcii/,pmid 29315598)
76
测试了netmhcii 2.3模型。
[0666]
如上所述,根据两个不同的标准测试netmhcii 2.3模型。具体地,示例性模型1模型根据最小netmhcii 2.3预测的结合亲和力生成肽呈递可能性的预测,并且示例性模型2根据最小netmhcii 2.3预测的结合排名生成肽呈递可能性的预测。
[0667]
用作示例性模型3和示例性模型4的呈递模型是使用通过质谱法获得的数据训练的本文公开的呈递模型的实施方案。如上所述,呈递模型基于两个不同组的等位基因相互作用变量和等位基因非相互作用变量生成肽呈递可能性的预测。具体地,示例性模型4基于ii类mhc分子类型和肽序列(由netmhcii 2.3模型使用的相同变量)生成肽呈递可能性的预
测,并且示例性模型3基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性的预测。
[0668]
在使用图14l的示例性模型预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性之前,对模型进行了训练和验证。netmhcii2.3模型(示例性模型1和示例性模型2)使用其自己的保存在免疫表位数据库(iedb,www.iedb.org)中的基于hla
‑
肽结合亲和力测定的训练数据集和验证数据集进行训练和验证。已知用于训练netmhcii 2.3模型的训练数据集几乎只包含15
‑
mer肽。另一方面,示例性模型3和4使用上面关于图14h所述的训练数据集来训练,并使用上面关于图14h所述的验证数据集进行验证。
[0669]
在模型的训练和验证之后,使用测试数据集测试每个模型。如上所述,netmhcii 2.3模型是在几乎只包含15
‑
mer肽的数据集上训练的,这意味着netmhcii 2.3不具有对不同权重的肽给予不同优先级的能力,从而降低了netmhcii 2.3对包含所有长度肽的hla ii类呈递质谱数据的预测性能。因此,为了在不受可变肽长度影响的模型之间提供公平的比较,测试数据集只包括15
‑
mer肽。具体地,测试数据集包含933个15
‑
mer肽。测试数据集中的933个肽中有40个由ii类mhc分子呈递—具体来说由hla
‑
drb1*07:01、hla
‑
drb1*15:01、hla
‑
drb4*01:03和hla
‑
drb5*01:01分子呈递。测试数据集中包含的肽不包含在上述训练数据集中。
[0670]
为了使用测试数据集测试示例性模型,对于每个示例性模型,对于测试数据集中933个肽中的每个肽,该模型生成了肽的呈递可能性的预测。具体地,对于测试数据集中的每个肽,示例性1模型使用ii类mhc分子类型和肽序列,通过利用测试数据集中四个hla ii类dr等位基因的最小netmhcii 2.3预测的结合亲和力对肽进行排名,生成ii类mhc分子对肽的呈递分数。类似地,对于测试数据集中的每个肽,示例性2模型使用ii类mhc分子类型和肽序列,通过利用测试数据集中的四个hla ii类dr等位基因的最小netmhcii 2.3预测的结合排名(即,分位数归一化结合亲和力)对肽进行排名,生成ii类mhc分子对肽的呈递分数。对于测试数据集中的每个肽,示例性4模型基于ii类mhc分子类型和肽序列生成了ii类mhc分子对肽的呈递可能性。类似地,对于测试数据集中的每个肽,示例性模型3基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列,生成了ii类mhc分子对肽的呈递可能性。
[0671]
四个示例性模型中的每一个的性能描绘于图14l中的线形图中。具体地,四个示例性模型中的每一个都与roc曲线相关联,所述roc曲线描绘了由模型做出的每个预测的真阳性率与假阳性率的比率。例如,图14l描绘了利用最小netmhcii 2.3预测的结合亲和力生成预测的示例性1模型的roc曲线、利用最小netmhcii 2.3预测的结合排名生成预测的示例性2模型的roc曲线、基于ii类mhc分子类型和肽序列生成肽呈递可能性的实施例4模型的roc曲线以及基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性的示例性3模型的roc曲线。
[0672]
如上所述,通过确定roc曲线的auc来定量模型在预测肽将由ii类mhc分子呈递的可能性方面的性能,所述roc曲线描绘了由模型做出的每个预测的真阳性率与假阳性率的比率。与具有较小auc的模型相比,具有较大auc的模型具有更高的性能(即,更高的精度)。如图14l所示,基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性的示例性3模型的曲线获得了为0.95的最高auc。因此,基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性的示例性3模型获得了最佳性能。基
于ii类mhc分子类型和肽序列产生肽呈递可能性的示例性4模型的曲线获得了为0.91的第二高auc。因此,基于ii类mhc分子类型和肽序列生成肽呈递可能性的示例性4模型获得了第二好的性能。利用最小netmhcii 2.3预测的结合亲和力来生成预测的示例性1模型的曲线获得了为0.75的最低auc。因此,利用最小netmhcii 2.3预测的结合亲和力来生成预测的示例1模型获得了最差的性能。利用最小netmhcii 2.3预测的结合排名来生成预测的示例性2模型的曲线获得了为0.76的第二低auc。因此,利用最小netmhcii 2.3预测的结合排名来生成预测的示例性2模型获得了第二差的性能。
[0673]
如图14l所示,示例性模型1和2与示例性模型3和4之间的性能差异很大。具体地,netmhcii 2.3模型(其利用最小netmhcii 2.3预测的结合亲和力或最小netmhcii 2.3预测的结合排名的标准)的性能比本文公开的呈递模型(其基于ii类mhc分子类型和肽序列,或基于ii类mhc分子类型、肽序列、rna表达、基因标识符和侧接序列生成肽呈递可能性)的性能低几乎25%。因此,图14l证明了本文公开的呈递模型能够实现比当前同类最佳现有技术模型(即netmhcii 2.3模型)更加精确得多的呈递预测。
[0674]
更进一步,如上所述,在几乎只含15
‑
mer肽的训练数据集上训练netmhcii 2.3模型。因此,netmhcii 2.3模型未被训练成了解哪些肽长度更可能由ii类mhc分子呈递。因此,netmhcii 2.3模型不会根据肽的长度对其对ii类mhc分子的肽呈递可能性的预测进行加权。换句话说,对于长度在15个氨基酸的模式肽长度之外的肽,netmhcii 2.3模型不会修改其对ii类mhc分子的肽呈递可能性的预测。因此,netmhcii 2.3模型过度预测了长度大于或小于15个氨基酸的肽的呈递可能性。
[0675]
另一方面,本文公开的呈递模型使用通过质谱法获得的肽数据进行训练,因此可在包含所有不同长度的肽的训练数据集上进行训练。因此,本文公开的呈递模型能够了解哪些肽长度更有可能由ii类mhc分子呈递。因此,本文公开的呈递模型可以根据肽的长度对ii类mhc分子的肽呈递可能性的预测进行加权。换句话说,本文公开的呈递模型能够修改其对长度在15个氨基酸的模式肽长度之外的肽的ii类mhc分子的肽呈递的可能性的预测。因此,与目前同类最佳现有技术模型(即netmhcii 2.3模型)相比,本文公开的呈递模型能够实现对长度大于或小于15个氨基酸的肽的显著更精确的呈递预测。这是使用本文公开的呈递模型来预测ii类mhc分子的肽呈递可能性的一个有利方面。
[0676]
xii.a.3.实施例3
[0677]
图14m是直方图,其描绘了在q值小于0.1的情况下,对总共230个样品中的每个样品使用质谱法测序的肽的数量,所述样品包含含有ii类hla分子的人肿瘤(nsclc、淋巴瘤和卵巢癌)和细胞系(ebv)。如图14m所示,在q值小于0.1时,对于每个样品平均有1300个肽被测序。
[0678]
如上关于图14d所述,图14m的230个样品中的每一个样品包含ii类hla分子。更具体地,图14m的230个样品中的每一个样品包含hla
‑
dr分子。hla
‑
dr分子是ii类hla分子的一种类型。甚至更具体地,图14m的230个样品中的每一个样品包含hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和/或hla
‑
drb5分子。hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和hla
‑
drb5分子是hla
‑
dr分子的类型。
[0679]
虽然该特定实验是使用包含hla
‑
dr分子,特别是hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和hla
‑
drb5分子的样品进行的,但在替代实施方案中,该实验可使用包含一
种或多种任何类型的ii类hla分子的样品来进行。例如,在替代实施方案中,可使用包含hla
‑
dp和/或hla
‑
dq分子的样品进行相同的实验。使用相同的技术对任何一种或多种类型的ii类mhc分子进行建模,并且仍然获得可靠的结果的能力是本领域技术人员所公知的。例如,jensen,kamilla kjaergaard,等人
76
是最近一篇科学论文的一个实例,所述论文使用相同的方法对针对hla
‑
dr分子以及hla
‑
dq和hla
‑
dp分子的结合亲和力进行建模。因此,本领域技术人员将理解,本文所述的实验和模型用于单独或同时对不仅hla
‑
dr分子,还对任何其它ii类mhc分子进行建模,同时仍能产生可靠的结果。
[0680]
为了对总共230个样品中的每个样品的肽进行测序,对每个样品进行质谱法。然后用comet搜索样品的所得质谱,并用percolator进行评分,以对肽进行测序。然后,针对多个不同的percolator q值阈值,确定样品中测序的肽的数量。具体地,对于样品,确定了以小于0.01的percolator q值、以小于0.05的percolator q值和以小于0.2的percolator q值测序的肽的数量。
[0681]
对于203个样品中的每个样品,在每一个不同的percolator q值阈值下测序的肽的数量描绘于图14m。例如,如图14m中所看到,对于第一样品,使用质谱法对约8000个q值小于0.1的肽进行测序。
[0682]
总的来说,图14m证明了使用质谱法在低q值下对大量来自含有ii类mhc分子的样品的肽进行测序的能力。换句话说,图14m中描绘的数据证明了使用质谱法对可由ii类mhc分子呈递的肽进行可靠测序的能力。
[0683]
图14n是描绘其中特定的ii类mhc分子等位基因被鉴别的样品的数量的直方图。更具体地,对于总共230个包含ii类hla分子的样品,图14n描绘了其中鉴别出某些ii类mhc分子等位基因的样品的数量。
[0684]
如上面关于图14m所论述的,图14m的230个样品中的每一个样品包含hla
‑
drb1分子、hla
‑
drb3分子、hla
‑
drb4分子和/或hla
‑
drb5分子。因此,图14n描述了其中鉴别出hla
‑
drb1、hla
‑
drb3、hla
‑
drb4和hla
‑
drb5分子的某些等位基因的样品的数量。
[0685]
为了确定样品中存在哪些hla
‑
drb1、hla
‑
drb3、hla
‑
drb4和hla
‑
drb5等位基因,对样品进行了hla ii类dr分型。然后,为了鉴别其中鉴别出特定hla等位基因的样品的数量,仅将其中使用hla ii类dr分型鉴别出hla等位基因的样品的数量相加。例如,如图14n所描绘的,总共230个样品中有28个样品含有ii类hla分子等位基因hla
‑
drb3*03:01。换句话说,在总共230个样品中,有28个样品含有hla
‑
drb3分子的等位基因hla
‑
drb3*03:01。总的来说,图14n描绘了从230个包含ii类hla分子的样品中鉴别多种ii类hla分子等位基因的能力。对于人群体,美国高加索人群中hla
‑
drb1等位基因的等位基因频率可以在maiers,m等人
161
中找到。
[0686]
图14o描绘了结合到i类mhc分子的肽和结合到ii类mhc分子的肽。
162
如图14o所示,每个肽包含肽主链和多个氨基酸。每个mhc分子包含结合沟。然而,如下所述,肽在i类mhc分子和ii类mhc分子的结合沟内差异地结合。
[0687]
如在整个本公开中所论述的,被mhc分子呈递的肽的长度可以不同。具体地,被mhc分子呈递的肽的长度可为9至20个氨基酸。当肽结合到mhc分子并被mhc分子呈递时,肽的“结合核心”位于mhc分子的结合沟内。具体地,肽的结合核心是当肽结合到mhc分子并被分子呈递时,位于mhc分子的结合沟内的肽的氨基酸序列。此外,当肽结合到mhc分子并由被
mhc分子呈递时,肽的结合核心的“结合锚”与所述mhc分子的结合沟物理结合。具体地,肽的结合核心的结合锚是结合核心的特定氨基酸,当肽结合到mhc分子并被mhc分子呈递时,所述特定氨基酸结合到mhc分子的结合沟。
[0688]
如图14o所示,由mhc类分子呈递的肽的结合核心包括肽的整个长度。具体地,如图14o所示,由i类mhc分子呈递的整个肽位于i类mhc分子的结合沟内。相反地,对于由ii类mhc分子呈递的肽,肽的结合核心中可能只包含肽的氨基酸子序列。具体地,如图14o所示,由ii类mhc分子呈递的肽的末端不位于ii类mhc分子的结合沟内。由ii类mhc分子呈递的包含肽的结合核心的氨基酸子序列可以是未知的。然而,正如文献中所承认的,ii类mhc呈递的肽的结合核心最常见的长度是9个氨基酸。
[0689]
此外,除了ii类mhc呈递的肽的结合核心是未知的之外,包含肽的结合核心的结合锚的氨基酸的数量和位置也可以是已知的。然而,如文献中所承认的,ii类mhc呈递的肽的结合核心通常包括3
‑
4个结合锚,并且结合锚通常包括位于结合核心末端的氨基酸。
[0690]
由于结合到mhc i类和ii类mhc分子的肽之间的区别,为了确保最佳的肽呈递预测性能,肽呈递预测模型应该被配置成专门预测ii类mhc分子的肽呈递。具体地,因为组成由ii类mhc分子呈递的肽的结合核心和结合核心的结合锚的氨基酸的子序列可能是未知的,所以ii类mhc肽呈递预测模型应该被配置成对这种不确定性建模。特别地,初始模型被开发来对由ii类mhc分子呈递的肽的结合核心和结合锚位置的不确定性进行建模。
[0691]
图14p描绘了图14q的初始模型的初始神经网络的示例性实施方案,所述初始模型被配置成预测ii类mhc分子的肽呈递。初始模型是被设计成鉴别由ii类mhc分子呈递的肽的结合核心和结合锚,以及使用这些鉴别的结合核心和结合锚来预测ii类mhc分子的肽呈递的呈递模型。初始模型包括接受等位基因非相互作用特征(例如,rna序列、样品id、蛋白质id和侧接序列)的共享神经网络和一组不同的接受等位基因相互作用特征(例如,肽序列)的初始神经网络。具体地,不同的初始神经网络的组中的每个不同的初始神经网络与不同的ii类mhc等位基因(例如,hla
‑
drb等位基因)相关联,并被配置成接受编码的肽序列。如上所述,图14p描绘了初始模型的初始神经网络的示例性实施方案。
[0692]
首先,因为由ii类mhc分子呈递的肽在长度上是可变的(例如,9至20个氨基酸),短于20个氨基酸的最大长度的肽被填充为具有20个氨基酸的长度。具体地,如果肽的长度少于20个氨基酸,则在肽的左侧添加特殊的氨基酸z,然后在肽的右侧添加所述氨基酸z。重复这种填充肽的模式,直至肽具有20个氨基酸的长度。通过填充到肽的侧面,肽的结合核心保持完整,而肽长度在所有肽中保持恒定。
[0693]
初始神经网络的输入层接受填充的肽序列。然后对填充肽进行独热编码。如图14p所描绘的,每个初始神经网络包括三个一维cnn层。三个cnn层中的一层有16个大小为8的过滤器。三个cnn层中的一层有16个大小为10的过滤器。三个cnn层中的一层有16个大小为12的过滤器。有意选择这些过滤器大小,以使初始神经网络集中于鉴别约9个氨基酸的结合核心,如上所述,所述大小在文献中被指示为ii类mhc呈递的肽的最常见的结合核心长度。
[0694]
三个cnn层中的每一个的输出被输入到两个一维cnn层中。两个cnn层中的一层有32个大小为1的过滤器。两个cnn层中的一层有32个大小为2的过滤器。有意选择这些过滤器大小,以鉴别ii类mhc呈递的肽的结合核心内的结合锚的位置。
[0695]
这两个cnn层的输出是串联的。然后,每个串联的输出被馈送到bi
‑
lstm层。bi
‑
lstm层的输出是串联的,并且这种串联被发送到多层感知器。多层感知器的输出包括不同的初始神经网络的输出。换句话说,多层感知器的输出包括输入到不同初始神经网络的肽将由与不同初始神经网络相关联的ii类mhc等位基因呈递的可能性。将来自每个不同的初始神经网络的呈递可能性与来自共享神经网络的输出组合。最后,将组合的可能性相加,生成肽将由一个或多个ii类mhc等位基因呈递的总体可能性。
[0696]
图14q是比较“bi
‑
lstm”呈递模型与“初始”呈递模型在预测肽测试数据集中的肽将由测试数据集中存在的ii类mhc分子中的至少一种呈递的可能性方面的性能的线形图。具体地,图14q是描绘“bi
‑
lstm”模型和“初始”模型的全精度召回率曲线的线形图。auc用于定量每个模型的性能。
[0697]
图14q中测试的第一模型是“bi
‑
lstm”模型。bi
‑
lstm模型是上面关于图14i和图14j详细地论述的模型。
[0698]
图14q中测试的第二模型是“初始”模型。初始模型是上面关于图14p详细地论述的模型。
[0699]
在使用模型预测肽测试数据集中的肽将由ii类mhc分子呈递的可能性之前,对示例性模型进行了训练和验证。为了训练、验证和最终测试示例性模型,上面针对230个样品所述的数据被分成训练数据集、验证数据集和测试数据集。
[0700]
为了确保在训练数据集、验证数据集和测试数据集中的超过一者中不出现肽,执行了以下程序。首先,除去所有来自230个样品的在蛋白质组中的超过一个位置上出现的肽。然后,将来自230个样品的其余肽分成10个相邻肽的块。每个相邻肽的块被唯一分配给训练数据集、验证数据集或测试数据集。这样,没有肽出现在训练数据集、验证数据集和测试数据集中的超过一者中。
[0701]
训练数据集包括来自总共230个样品中的226个样品的188,210个由ii类mhc分子呈递的肽。训练数据集中包含的188,210个肽的长度为9至20个氨基酸,包括9个和20个氨基酸。bi
‑
lstm模型和初始模型各自使用adam优化器和早期停止在训练数据集上进行训练。
[0702]
验证数据集包括来自训练数据集中使用的相同的226个样品的21,764个由ii类mhc分子呈递的肽。验证数据集仅用于早期停止。
[0703]
测试数据集包括由ii类mhc分子呈递的肽,所述肽是使用质谱法从肿瘤样品中鉴别的。具体地,测试数据集包含从四个肿瘤样品中鉴别的232个肽。测试数据集中包含的肽不包含在如上所述的训练数据集中。
[0704]
在分别使用训练数据集和验证数据集对bi
‑
lstm模型和初始模型进行训练和验证之后,使用测试数据集对模型进行测试。bi
‑
lstm模型和初始模型在测试数据集上的性能在图14q中被描绘为全精度召回率曲线和auc分数。如图14q所示,初始模型的表现优于bi
‑
lstm模型,并且auc达到0.347。bi
‑
lstm模型实现了0.238的auc。
[0705]
xii.a.4.实施例4
[0706]
为了进一步评估本文公开的预测模型是否可应用于ii类hla肽呈递,获得了两个细胞系的公开的ii类质谱数据,每个细胞系表达单个i类hla等位基因。一个细胞系表达hla
‑
drb1*15:01,并且另一个表达hla
‑
drb5*01:01
150
。将这两个细胞系用于训练数据。对于测试数据,从表达hla
‑
drb1*15:01和hla
‑
drb5*01:01两者的单独细胞系获得ii类质谱数据。
151
rna测序数据在训练或测试细胞系中均不可用,因此用来自不同的b细胞系b721.221
92
的rna测序数据代替。
[0707]
使用与i类hla数据相同的程序将肽集分为训练、验证和测试集,不同之处在于ii类数据包括了长度在9至20之间的肽。训练数据包括由hla
‑
drb1*15:01呈递的330种肽和由hla
‑
drb5*01:01呈递的103种肽。测试数据集包括由hla
‑
drb1*15:01或hla
‑
drb5*01:01呈递的223种肽,以及4708种未呈递的肽。
[0708]
我们在训练数据集上训练了10个模型的集合,以预测ii类hla肽呈递。这些模型的架构和训练过程与用于预测i类呈递的那些相同,不同之处在于ii类模型将独热编码且零填充至长度20而不是11的序列作为输入肽序列。
[0709]
图15比较了“ms模型”、“netmhciipan排名”:netmhciipan3.1
152
(取hla
‑
drb1*15:01和hla
‑
drb5*01:01中的最低netmhciipan百分位数排名)和“netmhciipan nm”:netmhciipan3.1(取hla
‑
drb1*15:01和hla
‑
drb5*01:01中的最强亲和力(单位为nm))在对hla
‑
drb1*15:01/hla
‑
drb5*01:01测试数据集中肽的排名中的预测性能。“ms模型”是本文公开的ii类mhc呈递预测模型。
[0710]
具体地,图15描绘了这些排名方法的接受者工作特性(roc)曲线以及roc曲线下面积auc(图a)和auc
0.1
(图b)统计。auc
0.1
是介于0和0.1fpr*10之间的auc,通常在表位预测字段中被考虑
19
。netmhciipan nm和排名方法的表现相似。ms模型表现最佳,显著超过了比较器方法的性能,特别是在roc曲线的关键高特异性区域(auc
01
0.41相比于0.27)。
[0711]
xii.b.为ii类mhc等位基因确定的呈递模型参数的实例
[0712]
下面显示了为多等位基因呈递模型(等式(16))的变型确定的一组参数(所述参数为ii类mhc等位基因hla
‑
drb1*12:01和hla
‑
drb1*10:01生成隐式独立等位基因呈递可能性):
[0713]
u=expit(relu(x
·
w1 b1),w2 b2),
[0714]
其中relu(
·
)是已校正的线性单位(relu)函数,w1、b1、w2和b2是为模型确定的参数集θ。等位基因相互作用变量x包含在1x 399)矩阵中,所述矩阵由1行独热编码的和中间填充的肽序列/输入肽组成。w1的维度为(399x 256),b1的维度为(1x 256),w2的维度为(256x2),并且b2为(1x2)。输出的第一列表示由等位基因hla
‑
drb1*12:01呈递肽序列的隐式独立等位基因概率,并且输出的第二列表示由等位基因hla
‑
drb1*10:01呈递的肽序列的隐式独立等位基因。出于演示目的,附录a中列出了b1、b2、w1和w2的值
[0715]
xiii.实施例9:t细胞数据的ii类mhc呈递模型评估
[0716]
为了评估对ii类mhc等位基因的肽呈递的准确预测是否转化为鉴别人肿瘤cd4 t细胞表位(即,免疫疗法靶点)的能力,从免疫表位数据库(immune epitope database)(iedb)
88
下载了已发表的cd4 t细胞多聚体/四聚体测定数据。这些数据由来自人样品的3,470个长度为9
‑
20个残基的肽组成,所述样品具有18个不同的hla
‑
drb等位基因,包括14个hla
‑
drb1等位基因、2个hla
‑
drb3等位基因、1个hla
‑
drb4等位基因和1个hla
‑
drb5等位基因。平均而言,每个等位基因有33个包含该等位基因的样品。完全ii类mhc ms模型(与上文第xii.a.2节中描述的模型相同)与结合亲和力预测因子netmhcii 2.3进行了比较。在18个等位基因中,完全ii类mhc ms模型的曲线下平均roc面积(roc auc)为0.81,标准偏差为0.08,而netmhcii 2.3模型的roc auc仅为0.65,较大的标准偏差为0.13。这些结果证明了完全ii类mhc ms模型预测cd4 t细胞表位的卓越能力。基于独立等位基因,对于一些更常见
的等位基因,如hla
‑
drb1*01:01,两个模型之间的roc auc更相似。例如,对于hla
‑
drb1*01:01等位基因,完全ii类mhc ms模型的roc auc为0.83,而netmhcii 2.3模型的roc auc为0.81。然而,大多数等位基因在两个模型之间具有广泛得多的性能分布。在18个独立等位基因测试中,完全ii类mhc ms模型在17个等位基因上优于netmhcii 2.3模型。仅在一个等位基因hla
‑
drb1*15:02中,netmhcii 2.3的性能优于完全mhcii类ms模型。然而,该等位基因在完全ii类mhc ms模型的训练数据中未得很好的体现,该模型仅包括一个含有该等位基因的样品。
[0717]
xiv.实施例10:回顾性新抗原t细胞数据的ii类mhc呈递模型评估
[0718]
本实施例还评估了对ii类mhc分子的肽呈递的准确预测是否转化为鉴别人肿瘤cd4 t细胞表位的能力。为了进行该评估,对由ii类mhc呈递模型预测的肽的cd4 免疫原性进行排名。
[0719]
用于该评估的合适测试数据集包括被肿瘤细胞表面上的ii类mhc分子呈递并被t细胞识别的肽。另外,正式的性能评估不仅需要阳性标记的(即,t细胞识别的)肽,而且还需要足够数量的阴性标记的(即,经测试但未被t识别的)肽。质谱数据解决肿瘤呈递,但不能解决t细胞识别;相反地,引发和疫苗接种后的t细胞测定解决了t细胞前体的存在和t细胞识别,但是不能解决肿瘤呈递。例如,其源基因在肿瘤中低水平表达的强hla结合肽可引起免疫后的强cd4 t细胞应答,由于该肽未由肿瘤呈递,因此在治疗上是无用的。
[0720]
为了获得用于该评估的合适的测试数据集,从最近的研究中收集了已发表的数据。
163
收集的测试数据集包括45名患者中的69个对til具有反应性的阳性标记的单核苷酸变体(snv)突变cd4 。如上所述,收集的测试数据集也包括阴性标记的snv突变。具体地,每个患者平均有104个阴性标记的snv突变,中位数为106个。
[0721]
测试数据集中的每个snv突变被表示为25个氨基酸的序列,其中snv突变位于序列中间,在氨基酸位置13处。对于每个25个氨基酸的序列,然后生成包含snv突变的长度为9至20个氨基酸的所有可能的肽。每个25个氨基酸的序列产生118个可能的肽。对于每种可能的肽,在肽的左侧和右侧添加5个氨基酸的侧接序列。
[0722]
为了模拟用于个性化免疫疗法的抗原的选择,使用本文公开的初始模型和netmhciipan 3.2结合亲和力模型,利用tpm=1的基因表达阈值,按照患者的ii类mhc等位基因的呈递可能性的顺序对测试数据集中每个患者的snv突变进行排名。使用的初始模型被训练成预测32个不同的ii类mhc等位基因的肽呈递,所述32个不同的ii类mhc等位基因覆盖了测试数据集中患者中存在的30个ii类mhc等位基因中的25个。
[0723]
对于初始模型,为了计算每个患者的每个snv突变的出现可能性,使用初始模型确定了患者的已鉴别的mhcii类等位基因中的每一个对118个可能的肽中的每一个的snv突变的呈递分数。然后,鉴别出通过初始模型为每个患者的ii类mhc等位基因确定的最高的呈递分数。最后,将这些每一个患者的ii类mhc等位基因的最高呈递分数相加,以确定患者的snv突变呈递的总体可能性。
[0724]
对于netmhciipan 3.2模型,为了计算每个患者的每个snv突变的呈递可能性,使用netmhciipan 3.2模型确定了每个患者的已鉴别的ii类mhc等位基因中的每一个对118个可能的肽中的每一个的snv突变的结合亲和力。然后,鉴别出通过netmhciipan 3.2为每个患者的ii类mhc等位基因确定的最高反向结合亲和力。注意,最高反向结合亲和力被确定,
因为较低的结合亲和力表明更大的呈递可能性。最后,将每个患者的ii类mhc等位基因的最高反向结合亲和力相加,以确定患者的snv突变的总体呈递可能性。
[0725]
接下来,按照如通过初始模型和netmhciipan 3.2模型两者所测定的患者的ii类mhc等位基因的呈递可能性的顺序对每个患者的snv突变进行排名。由于抗原特异性免疫疗法在所靶向的ii类mhc特异性数量上受到技术限制(例如,目前的个性化疫苗编码约10
‑
20种体细胞突变
80
–
82
,其中约10个可以是ii类mhc特异性的),因此对每个患者的前1、2、3、4、5和10名snv突变进行了排名。
[0726]
另外,作为对照,对于每名患者,对源自tpm>=1的基因的患者的snv突变中的每一个进行随机排名。具体地,对于每名患者,在100次试验中对源自tpm>=1的基因的患者的snv突变中的每一个进行随机排名,以确定每名患者的每个snv突变的总体排名。
[0727]
在对snv突变进行排名后,通过计数每名具有至少一个预先存在的t细胞应答的患者的前1、2、3、4、5和10名snv突变中预先存在的t细胞应答的数量来比较预测模型。然后,比较由不同模型为每名具有至少一个预先存在的t细胞应答的患者鉴别的前1、2、3、4、5和10名snv突变的由t细胞识别(例如,预先存在的t细胞应答)的snv突变的比例。具体地,下表2描绘了在前1、2、3、4、5和10名预测中阳性标记的snv突变占由给定模型预测的总共69个阳性标记的snv突变的百分比。如表2所示,与netmhciipan 3.2模型和随机预测相比,初始模型更有可能准确预测cd4 免疫原性ii类mhc呈递的肽。
[0728]
模型前1名前2名前3名前4名前5名前10名开始9%17%17%19%20%32%netmhciipan 3.29%12%16%18%19%29%随机1%3%5%6%8%16%
[0729]
表2
[0730]
因此,该评估建立了初始模型不仅可以鉴别能够如先前文献
81,82,97
中所述启动t细胞的新抗原,而且更严格地说,还可以鉴别由肿瘤呈递给t细胞的新抗原的卓越能力。
[0731]
xvi.实施例11:在癌症患者中前瞻性鉴定新抗原反应性t细胞
[0732]
该前瞻性实施例将证明改进的预测可以从常规患者样品中鉴别新抗原。为此,将分析接受抗pd(l)1疗法的转移性nsclc患者的存档ffpe肿瘤活检和5
‑
30ml外周血。使用肿瘤全外显子组测序、肿瘤转录组测序和匹配的正常外显子组测序将鉴别每位患者的体细胞突变(snv和短插入缺失)。将应用ii类mhc完全ms模型对每位患者确定20个新表位的优先级,以针对预先存在的抗肿瘤t细胞应答进行测试。为了将分析重点放在可能的cd4应答上,将优先肽合成为8
‑
11
‑
mer最小表位(方法),然后将在短的体外刺激(ivs)培养中用合成的肽培养外周血单核细胞(pbmc)以扩增新抗原反应性t细胞。两周后,将使用ifn
‑
γelispot针对优先的新表位评估了抗原特异性t细胞的存在。在有足够pbmc可用的患者中,还将进行了单独的实验以对所识别的特异性抗原进行完全或部分去卷积。
[0733]
首先,将对每一位患者进行针对患者特异性新抗原肽库的t细胞应答的检测。对于每位患者,各自根据模型排名和任何序列同源性,预测的新抗原将被组合成肽的2个库(同源肽将被分到不同的库中)。然后,对于每位患者,将在ifn
‑
γelispot中用2个患者特异性新抗原肽库刺激该患者的体外扩增pbmc。还将进行dmso阴性对照和pha阳性对照以分别检测背景和t细胞活力。值比背景增加>2倍的样品将被认为是阳性的响应性患者。此外,为了
验证体外培养条件仅扩增预先存在的体内引发的记忆t细胞,而不能够在体外从头引发,将在hla匹配的健康供体中用新抗原进行了一系列对照实验。预期将使用ifn
‑
γelispot在用患者特异性肽库测试的大多数患者中鉴别出预先存在的新抗原反应性t细胞。另外,预期大多数患者将对至少一种所测的新抗原肽有响应。
[0734]
xv.a.肽
[0735]
将购买定制的重组冻干肽购,并以10
‑
50mm的浓度在无菌dmso中重构,等分并储存在
‑
80℃。
[0736]
xv.b.人外周血单个核细胞(pbmc)
[0737]
将购买来自健康供体的冷冻保存的hla型pbmc(其已被确认为hiv、hcv和hbv血清阴性)并将其储存在液氮中直到使用。还将购买新鲜血液样品和leukopaks,并且在冷冻保存之前通过ficoll
‑
paque密度梯度法分离pbmc。将根据当地临床标准操作程序(sop)和irb批准的方案,在当地临床处理中心对患者pbmc进行了处理。批准irb将包括quorum review irb、comitato etico interaziendale a.o.u.、san luigi gonzaga di orbassano和comit
éꢀ
de la investigaci
ó
n del grupo hospitalario quir
ó
n en barcelona。
[0738]
将通过密度梯度离心分离pbmc,洗涤,计数并以5x106个细胞/ml的密度冷冻保存在cryostor cs10中。冷冻保存的细胞将在cryoport中运输,并且在到达后转移到ln2中储存。冷冻保存的细胞将解冻,并在含有benzonase的optmizer t细胞扩增基础培养基中洗涤两次,并在无benzonase的情况下洗涤一次。将使用guavaviacount试剂和guava easycyte ht细胞计数器(emd millipore)上的模块评估细胞计数和活力。随后细胞将以适合进行测定的浓度和培养基进行重悬(参见下一节)。
[0739]
xv.c.体外刺激(ivs)培养
[0740]
将以与ott等人
81
相似的方法,在同源肽和il
‑
2存在下扩增来自健康供体或患者样品的预先存在的t细胞。简单地说,将把解冻的pbmc放置过夜,并且在24孔组织培养板中在含10iu/ml rhil
‑
2的immunocult
tm
‑
xf t细胞扩增培养基中在肽库(每种肽10μm)的存在下刺激14天。将以2x106个细胞/孔接种细胞,并且每2
‑
3天通过更换2/3的培养基来补料。
[0741]
xv.d.ifnγ酶联免疫斑点(elispot)测定
[0742]
将通过elispot测定
142
进行产生ifnγ的t细胞的检测。简单地说,将收获pbmc(离体或体外扩增),在无血清rpmi中洗涤,并在用抗人ifnγ捕获抗体包被的elispot multiscreen板中在optmizer t细胞扩增基础培养基(离体)或immunocult
tm
‑
xf t细胞扩增培养基(扩增培养物)中在对照或同源肽的存在下培养。在5%co2、37℃的潮湿培养箱中孵育18小时后,细胞将从板中移出,并将使用抗人ifnγ检测抗体、vectastain avidin过氧化物酶复合物和aec底物检测膜结合的ifnγ。将使elispot板干燥,避光保存,然后送走以进行标准化评估
143
。
[0743]
xv.e.颗粒酶b elisa和msd多重测定
[0744]
将使用3重测定msd u
‑
plex biomarker测定(目录号k15067l
‑
2)进行elispot上清液中分泌的il
‑
2、il
‑
5和tnf
‑
α的检测。将根据制造商的说明进行测定。对于每种细胞因子,将使用已知标准品的系列稀释液计算分析物浓度(pg/ml)。将根据制造商的说明,使用granzyme belis进行elispot上清液中颗粒酶b的检测。简单地说,将使elispot上清液在样品稀释液中以1:4稀释,并与颗粒酶b标准品的系列稀释液一起运行以计算浓度
(pg/ml)。
[0745]
xv.f.ivs测定的阴性对照实验
–
在健康供体中测试的来自肿瘤细胞系的新抗原
[0746]
将进行用于在健康供体中测试的来自肿瘤细胞系的新抗原的ivs测定的阴性对照实验。在此类实验中,将在ivs培养中,用含有阳性对照肽(先前暴露于感染性疾病)、源自肿瘤细胞系的hla匹配的新抗原(未暴露)和源自所述供体为血清阴性的病原体的肽的肽库刺激健康供体pbmc。将在用dmso(阴性对照)、pha和常见感染性疾病多肽(阳性对照)、新抗原(未暴露)或hiv和hcv肽(供体将被确认为是血清阴性的)刺激之后,随后通过ifnγelispot(105个细胞/孔)分析扩增的细胞。
[0747]
xv.g.ivs测定的阴性对照实验
–
在健康供体中测试的来自患者的新抗原
[0748]
将进行用于在健康供体中测试反应性的来自患者的新抗原的ivs测定的阴性对照实验。具体地,将进行健康供体中对hla匹配的新抗原肽库的t细胞应答的评估。将在离体ifn
‑
γelispot中用对照(dmso、cef和pha)或hla匹配的患者来源的新抗原肽刺激健康供体pbmc。另外,将在ifn
‑
γelispot中用对照(dmso、cef和pha)或hla匹配的患者来源的新抗原肽库刺激在新抗原库或cef库的存在下扩增的ivs培养后的健康供体pbmc。
[0749]
xvi.实施例8
‑
11的方法
[0750]
以下实施例8
‑
11的方法用将来时论述,因为它们将用于执行未来的、预期的实施例10
‑
11。然而,尽管使用将来时来描述下面的方法,但这些方法在过去也曾用于实施例8和9的实行。
[0751]
xvi.a.质谱
[0752]
xvi.a.1.试样
[0753]
用于质谱分析的存档冷冻组织试样将获自商业来源。还将从患者中前瞻性地收集一部分试样。
[0754]
xvi.a.2.hla免疫沉淀
[0755]
将在组织样品裂解和溶解之后,将使用建立的免疫沉淀(ip)方法进行hla
‑
肽分子的分离
87,124
‑
126
。新鲜冷冻组织将被粉碎,将加入裂解缓冲液(1%chaps,20mm tris
‑
hcl,150mm nacl,蛋白酶和磷酸酶抑制剂,ph=8)以溶解组织,并将使所得溶液在4c下离心2小时以沉淀碎屑。将把澄清的裂解物用于hla特异性ip。如先前所描述的将使用抗体w6/32进行免疫沉淀
127
。将把裂解物添加至抗体珠粒,并在4c旋转过夜以进行免疫沉淀。免疫沉淀后,将把珠粒从裂解物中除去。将洗涤ip珠粒以除去非特异性结合,并将用2n乙酸从珠粒上洗脱hla/肽复合物。将使用分子量旋转柱从肽中去除蛋白质组分。将使所得肽通过speedvac蒸发至干,并在ms分析之前保存在
‑
20c。
[0756]
xvi.a.3.肽测序
[0757]
干燥的肽将在hplc缓冲液a中复原,并加载到c
‑
18微毛细管hplc柱上,以梯度洗脱到质谱仪中。将使用180分钟内0
‑
40%b(溶剂a
–
0.1%甲酸,溶剂b
‑
80%乙腈的0.1%甲酸)的梯度将肽洗脱到fusion lumos质谱仪中。在所选离子的hcd裂解后,将在orbitrap检测器中以120,000的分辨率收集肽质量/电荷(m/z)的ms1图谱,然后在orbitrap或离子阱检测器中收集了20个ms2低分辨率扫描。ms2离子的选择将使用依赖数据的采集模式进行,并且将在离子的ms2选择之后30秒进行动态排除。将把ms1扫描的自动增益控制(agc)设置为4x105,并且将把ms2扫描的设置为1x104。对于测序hla肽,可以选择 1、 2和 3电荷状态用
于ms2片段化。
[0758]
将使用comet
128,129
针对蛋白质数据库搜索每个分析的ms2图谱,并将使用percolator
130
‑
132
对肽鉴别进行评分。
[0759]
xvi.b.机器学习
[0760]
xvi.b.1.数据编码
[0761]
对于每个样品,训练数据点将是来自参考蛋白质组的所有8
‑
11
‑
mer(含)肽,这些肽正确映射到样品中表达的一个基因。将通过将每个训练样品的训练数据集连接形成整体训练数据集。将选择长度8
‑
11,因为该长度范围捕获了所有i类hla呈递肽的约95%;但是,可以使用相同的方法来为模型增加长度12
‑
15,但要以适度增加计算需求为代价。将使用独热编码方案将肽和侧接序列向量化。将通过使用填充字符扩展氨基酸字母并将所有肽填充到最大长度11来将多种长度(8
‑
11)的肽表示为固定长度的向量。将把训练肽的源蛋白的rna丰度表示为从rsem
133
获得的同工型水平每百万转录物(tpm)估计值的对数。对于每个肽,将把独立肽的tpm计算为对于包含肽的每个同工型的独立同工型tpm估计值的总和。将从训练数据中排除来自以0 tpm表达的基因的肽,并且在测试时,为未表达基因的肽的分配0的呈递概率。最后,将为每个肽分配ensembl蛋白质家族id,并且每个唯一ensembl蛋白家族id将对应于独立基因呈递倾向截距(参见下一节)
[0762]
xvi.b.2.模型架构的说明
[0763]
完整呈递模型具有以下功能形式:
[0764][0765]
其中k索引数据集中的hla等位基因,范围从1到m,并且是指示变量,如果肽i来源的样品中存在等位基因k,则其值为1,否则为0。注意,对于给定的肽i,所有但最多6个(6对应于肽i来源的样品中的hla类型)将为零。概率之和将被固定为1
‑
∈,例如∈=10
‑6。
[0766]
独立等位基因呈递概率将建模如下:
[0767]
pr(由等位基因α呈递的肽i)=sigmoid{nn
a
(肽i)
nn
侧接(侧接
i
) nn
rna
(log(tpm
i
)) α
样品(i)
β
蛋白(i)
},
[0768]
其中变量具有以下含义:sigmoid是sigmoid(又称expit)函数,肽
i
是肽i的独热编码的中间填充氨基酸序列,nn
α
是具有线性最后一层激活的神经网络,其模拟了肽序列对呈递概率的贡献,侧接
i
是其源蛋白中肽i的独热编码的侧接序列,nn
侧接
是具有线性最后一层激活的神经网络,其模拟了侧接序列对呈递概率的贡献,tpm
i
是肽i的源mrna的以tpm为单位的表达,样品(i)是肽i来源的样品(即患者),α
样品(i)
是每样品截距,蛋白(i)是肽i的源蛋白,并且β
蛋白(i)
是每蛋白质截距(也就是每基因的呈递倾向)。
[0769]
模型的组件神经网络将具有以下架构:
[0770]
·
每个nn
α
是单隐藏层多层感知器(mlp)的一个输出节点,具有输入维度231(11个残基x每个残基21种可能的字符(包括填充字符)),宽度256,隐藏层中的已校正的线性单位(relu)激活,以及训练数据集中每hla等位基因α的一个输出节点。
[0771]
·
nn
侧接
是单隐藏层mlp,具有输入维度210(n端侧接序列的5个残基 c端侧接序列的5个残基x每个残基21种可能的字符(包括填充字符)),宽度32,隐藏层中的已校正的线性
单位(relu)激活和输出层中的线性激活。
[0772]
·
nn
rna
是单隐藏层mlp,具有输入维度1,宽度16,隐藏层中的已校正的线性单位(relu)激活和输出层中的线性激活。
[0773]
应注意,模型的一些组件(例如nn
α
)取决于特定的hla等位基因,但是许多组件(nn
侧接
、nn
rna
、α
样品(i)
、β
蛋白(i)
)不是。前者称为“等位基因相互作用”,后者称为“等位基因非相互作用”。将根据生物学现有技术知识选择建模为等位基因相互作用或非相互作用的特征:hla等位基因能识别肽,因此肽序列将被建模为等位基因相互作用,但是没有关于源蛋白、rna表达或侧接序列的信息被传递至hla分子(因为肽在其在内质网中遇到hla时已从同源蛋白分离),因此这些特征将被建模为等位基因非相互作用。该模型将在keras v2.0.4
134
和theano v0.9.0
135
中实现。
[0774]
肽ms模型将使用与完全ms模型相同的去卷积程序(等式1),但是将使用仅考虑肽序列和hla等位基因的简化的等位基因模型生成了独立等位基因呈递概率:
[0775]
pr(由等位基因α呈递的肽i)=sigmoid{nn
a
(肽
i
)}。
[0776]
肽ms模型将使用与结合亲和力预测相同的特征,但是模型的权重将是在不同的数据类型上进行训练的(即质谱数据相比于hla肽结合亲和力数据)。因此,肽ms模型和完全ms模型的预测性能的比较将揭示了非肽特征(即rna丰度、侧接序列、基因id)对总体预测性能的贡献,并且肽ms模型和结合亲和力模型的预测性能的比较将揭示了改进肽序列建模对整体预测性能的重要性。
[0777]
xvi.b.3.训练/验证/测试分组
[0778]
通过使用以下程序,没有肽将出现在多于一个训练/验证/测试集中:首先将从参考蛋白质组中去除出现在多于一种蛋白质中的所有肽,然后将蛋白质组划分为10个相邻的肽块。每个块都将被唯一分配到训练、验证或测试集。这样,将没有肽出现在多于一个训练、验证或测试集中。验证集将仅用于提前停止。来自单等位基因样品的肽将被包括在训练数据中,但是并入到训练和验证集中的肽集(呈递的和非呈递的)将不与用作测试数据的肽集相交。
[0779]
xvi.b.4.模型训练
[0780]
对于模型训练,所有肽将被建模为独立的,其中每肽损失是负的伯努利对数似然损失函数(又称对数损失)。形式上,肽i对总损失的贡献为
[0781]
损失(i)=
‑
log(伯努利(y
i
|pr(呈递的肽i))),
[0782]
其中y
i
是肽i的标记;即,如果肽i被呈递,则y
i
=1,否则为0,并且伯努利(y|p)表示考虑i.i.d.二进制观测向量y的参数p∈[0,1]的伯努利似然性。将通过使损失函数最小化来训练模型。
[0783]
为了减少训练时间,将通过随机去除90%的负标记训练数据来调整类平衡。模型权重将使用glorot统一程序61来初始化,并在nvidia maxwell titan x gpu上使用具有标准参数的adam62随机优化器进行训练。由总数据的10%组成的验证集将用于早期停止。将在每个四分之一周期对验证集进行模型评估,并将在验证损失(即验证集上的负伯努利对数似然)未能降低时在第一个四分之一周期后停止模型训练。
[0784]
完全呈递模型将是10个模型副本的集合,每个副本在相同训练数据的混洗副本上独立训练,其中集合中每个模型的模型权重都有不同的随机初始化。在测试时,将通过取模
型副本输出的概率平均值来生成预测。
[0785]
xvi.b.5.基序徽标
[0786]
将使用weblogolib python api v3.5.0
138
产生基序徽标。为了产生结合亲和力徽标,将从免疫表位数据库(iedb
88
)下载了mhc_ligand_full.csv文件,并将保留了符合以下标准的肽:以纳摩尔(nm)为单位的测量,2000年后的参考日期,对象类型等于“线性肽”并且肽中的所有残基均来自规范的20个字母的氨基酸字母表。将使用具有低于500nm的常规结合阈值的测量的结合亲和力的经过滤肽的子集产生徽标。对于在iedb中具有太少结合剂的等位基因对,将不产生徽标。为了产生代表学习的呈递模型的徽标,将针对每个等位基因和每个肽长度预测2,000,000个随机肽的模型预测。对于每个等位基因和每个长度,将通过学习的呈递模型使用排名前1%(即前20,000)的肽产生徽标。重要的是,来自iedb的这种结合亲和力数据将不用于模型训练或测试,而仅用于比较学习的基序。
[0787]
xvi.b.6.结合亲和力预测
[0788]
我们将使用来自netmhcii 2.3的仅结合亲和力预测器预测了肽
‑
mhc结合亲和力,netmhcii 2.3是一种开源、gpu兼容的i类hla结合亲和力预测器。为组合多个hla等位基因中的单个肽的结合亲和力预测,将选择最小结合亲和力。为了组合多个肽的结合亲和力(即,为了对被多个突变肽所跨越的突变进行排名),将选择肽中的最小结合亲和力。对于t细胞数据集上的rna表达阈值,将使用从tcga到tpm>1阈值的肿瘤类型匹配rna
‑
seq数据。在原始出版物中,所有原始t细胞数据集均将以tpm>0进行过滤,因此将不使用要以tpm>0进行过滤的tcga rna
‑
seq数据。
[0789]
xvi.b.7.呈递预测
[0790]
为了组合多个hla等位基因的单个肽的呈递概率,将如等式1中所示鉴别了概率的总和。为了组合多个肽的呈递概率(即,为了对被多个突变肽所跨越的突变进行排名),将鉴别呈递概率的总和。概率上,如果肽的呈递被认为是i.i.d.伯努利随机变量,则概率的总和对应于所呈递的突变肽的预期数目:
[0791][0792]
其中pr[被呈递的表位j]是通过将经训练的呈递模型应用于表位j获得的,n
i
表示跨越突变i的突变表位的数目。例如,对于远离其源基因末端的snv i,有8个跨8
‑
mer、9个跨9
‑
mer、10个跨10
‑
mer和11个跨11
‑
mer,总共n
i
=38个跨越突变的表位。
[0793]
xvi.c.下一代测序
[0794]
xvi.c.1.样本
[0795]
对于对冷冻切除的肿瘤的转录组分析,将从用于ms分析的相同组织样本(肿瘤或邻近的正常组织)中获得rna。对于进行抗pd1疗法的患者中的新抗原外显子组和转录组分析,将从存档ffpe肿瘤活检中获得dna和rna。将使用邻近的正常、相配的血液或pbmc获得用于正常外显子组和hla分型的正常dna。
[0796]
xvi.c.2.核酸提取与文库构建
[0797]
将使用qiagen dneasy柱按照制造商推荐的程序分离来自血液的正常/生殖细胞dna。将使用qiagen allprep dna/rna分离试剂盒按照制造商推荐的程序分离来自组织样
本的dna和rna。将分别通过picogreen和ribogreen荧光(molecular probes)对dna和rna进行定量。将产量>50ng的样本进行文库构建。将按照制造商推荐的方案,通过声学剪切和随后的dna ultra ii文库制备试剂盒产生dna测序文库。将通过热裂解和利用rna ultra ii的文库构建来产生肿瘤rna测序文库。将通过picogreen(molecular probes)定量得到的文库。
[0798]
xvi.c.3.全外显子组捕获
[0799]
将使用xgen whole exome panel对dna和rna测序文库进行外显子富集。1至1.5μg正常dna或肿瘤dna或rna来源的文库将用作输入,并使其杂交超过12小时,然后进行链霉亲和素纯化。将使捕获的文库通过pcr进行最少扩增,并通过nebnext文库定量试剂盒进行定量。将使捕获的文库以等摩尔浓度合并,并使用c
‑
bot进行聚类,并在hiseq4000上以75个碱基配对的末端进行测序,以达到靶标独特的平均覆盖率>500x肿瘤外显子组,>100x正常外显子组和>100m读段肿瘤转录组。
[0800]
xvi.c.4.分析
[0801]
将使用bwa
‑
mem
144
(v.0.7.13
‑
r1126)使外显子组读段(ffpe肿瘤和匹配的正常)与参考人基因组(hg38)进行比对。将使用star(v.2.5.1b)使rna
‑
seq读段(ffpe和冷冻的肿瘤组织样品)与基因组和gencode转录本(v.25)进行比对。将使用rsem
133
(v.1.2.31)和相同的参考转录本对rna表达进行定量。picard(v.2.7.1)将用于标记重复的比对并计算比对量度。对于用gatk
145
(v.3.5
‑
0)对碱基质量评分进行重新校准后的ffpe肿瘤样品,将利用freebayes
146
(1.0.2)使用配对肿瘤
‑
正常外显子组确定取代和短插入缺失变体。过滤器将包括等位基因频率>4%;中值碱基质量>25,支持读段的最小映射质量30和正常中的替代读段计数<=2且获得足够的覆盖率。还将在两条链上都检测到变体。将排除发生在重复区域的体细胞变体。将使用refseq转录本用snpeff
147
(v.4.2)进行翻译和注释。将在肿瘤rna比对中鉴别的非同义、非终止变体进入新抗原预测。optitype
148 1.3.1将用于产生hla类型。
[0802]
xvi.c.5.用于ivs控制实验的肿瘤细胞系和匹配的正常细胞系
[0803]
将全部购买肿瘤细胞系及其正常供体匹配的对照细胞系,并根据销售者的说明使其生长至10
83
‑
10
84
个细胞,然后速冻用于核酸提取和测序。ngs程序将基本上如上所述执行,只是mutect
149
(3.1
‑
0)将仅用于取代突变检测。
[0804]
xvii.实施例12:对来自nsclc患者的外周血的新抗原特异性记忆t细胞的tcr进行前瞻性测序
[0805]
将对来自nsclc患者的外周血的新抗原特异性记忆t细胞的tcr进行测序。在elispot孵育后,将收集来自nsclc患者的外周血单核细胞(pbmc)。具体地,将用患者特异性个体新抗原肽、患者特异性新抗原肽库和dmso阴性对照在ifn
‑
γelispot中刺激来自患者的体外扩增的pbmc。在孵育之后并且添加检测抗体之前,将把pbmc转移至新的培养板中,并在完成elispot测定期间保持在孵育箱中。将根据elispot结果鉴别阳性(响应性)孔。将合并来自阳性孔和阴性对照(dmso)孔的细胞,并用磁性标记的抗体对cd137进行染色,以用于使用miltenyi磁性分离柱进行富集。
[0806]
将使用10x genomics单细胞分辨率配对免疫tcr分析方法对如上所述分离和扩增的富含cd137和耗尽cd137的t细胞级分进行测序。具体来说,将把活t细胞分配到单细胞乳剂中,以用于随后的单细胞cdna产生和全长tcr分析(5’utr至恒定区——确保α和β配对)。
一种方法使用在转录物的5’端的分子条形码化模板转换寡核苷酸,第二种方法使用在3’端的分子条形码化恒定区寡核苷酸,并且第三种方法是将rna聚合酶启动子与tcr的5’端或3’端偶联。所有这些方法能够在单细胞水平上进行α和βtcr对的鉴别和去卷积。所得条形码化的cdna转录物将经历优化的酶和文库构建工作流程,以减少偏差并确保细胞库内克隆型的准确表示。将在illumina的miseq或hiseq4000仪器(配对末端150个循环)上对文库进行测序,目标测序深度为每个细胞约五千至五万个读段。tcra和tcrb链的存在将通过基于正交锚定pcr的tcr测序方法(archer)证实。与基于10x genomics的tcr测序相比,此特定方法的优势在于使用有限的细胞数作为输入,并且酶操作较少。
[0807]
将使用10x软件和定制生物信息学管线分析测序输出,以鉴别t细胞受体(tcr)α和β链对,克隆型将被定义为独特的cdr3氨基酸序列的α、β链对。将针对以大于2个细胞的频率出现的单α和单β链对,对克隆型进行过滤,以产生患者中每个靶标肽的克隆型的最终列表。
[0808]
总而言之,使用上述方法,将鉴别来自患者的外周血的记忆cd4 t细胞,所述细胞对于如上文第xv节中关于实施例11所讨论而鉴别的患者的肿瘤新抗原是新抗原特异性的。将对这些鉴别的新抗原特异性t细胞的tcr进行测序。此外,还将鉴别对于通过上述呈递模型鉴别的患者的肿瘤新抗原是新抗原特异性的经测序tcr。
[0809]
xviii.实施例13:新抗原特异性记忆t细胞在t细胞疗法中的用途
[0810]
在鉴别出对患者肿瘤呈递的新抗原具有新抗原特异性的t细胞和/或tcr之后,这些鉴别出的新抗原特异性t细胞和/或tcr可用于患者的t细胞疗法。具体地,这些鉴别出的新抗原特异性t细胞和/或tcr可用于产生治疗量的用于在t细胞疗法期间输注到患者体内的新抗原特异性t细胞。在本文第xviii.a.和xviii.b.节中讨论了两种用于产生治疗量的用于患者中的t细胞疗法的新抗原特异性t细胞的方法。第一种方法包括从患者样品中扩增鉴别出的新抗原特异性t细胞(第xviii.a.节)。第二种方法包括对已鉴别的新抗原特异性t细胞的tcr进行测序,并将经测序tcr克隆到新的t细胞中(第xviii.b.节)。本文未明确提及的用于产生用于t细胞疗法的新抗原特异性t细胞的替代方法也可以用于产生治疗量的用于t细胞疗法的新抗原特异性t细胞。一旦通过一种或多种这些方法获得了新抗原特异性t细胞,就可以将这些新抗原特异性t细胞输注到患者体内以用于t细胞疗法。
[0811]
xviii.a.从患者样品中鉴别和扩增新抗原特异性记忆t细胞以用于t细胞疗法
[0812]
产生治疗量的用于患者中的t细胞疗法的新抗原特异性t细胞的第一种方法包括扩增从患者样品中鉴别出的新抗原特异性t细胞。
[0813]
具体地,为了将新抗原特异性t细胞扩增至用于患者的t细胞疗法中的治疗量,使用上述呈递模型鉴别最有可能由患者癌细胞呈递的新抗原肽集。另外,从患者获得包含t细胞的患者样品。患者样品可能包含患者的外周血、肿瘤浸润淋巴细胞(til)或淋巴结细胞。
[0814]
在其中患者样品包含患者外周血的实施方案中,可以使用以下方法将新抗原特异性t细胞扩增至治疗量。在一个实施方案中,可以进行引发。在另一个实施方案中,可以使用一种或多种上述方法鉴别已激活t细胞。在另一个实施方案中,可以进行引发和已激活t细胞的鉴别二者。引发和鉴别已激活t细胞二者的优点是使所代表的特异性的数目最大化。引发和鉴别已激或t细胞二者的缺点是这种方法是困难的和费时的。在另一个实施方案中,可以分离不一定被激活的新抗原特异性细胞。在这样的实施方案中,也可以进行这些新抗原特异性细胞的抗原特异性或非特异性扩增。在收集这些引发的t细胞之后,可以对引发的t
细胞进行快速扩增方案。例如,在一些实施方案中,可以对引发的t细胞进行rosenberg快速扩增方案
[0815]
(https://www.ncbi.plm,nih.gov/pmc/articles/pmc2978753/,
[0816]
https://www.ncbi.nlm.nih.gov/pmc/articles/pmc2305721/)
153,154
。
[0817]
在其中患者样品包含患者的til的实施方案中,可以使用以下方法将新抗原特异性t细胞扩增至治疗量。在一个实施方案中,新抗原特异性til可以在体外进行四聚体/多聚体分选,然后可以对分选的til进行如上所述的快速扩增方案。在另一个实施方案中,可以进行til的新抗原非特异性扩增,然后可以对新抗原特异性til进行四聚体分选,然后可以对分选的til进行如上所述的快速扩增方案。在另一个实施方案中,可以在使til经历快速扩增方案之前进行抗原特异性培养。
[0818]
(https://www.ncbi.nlm.nih.gov/pmc/articles/pmc4607110/,
[0819]
https://onlinelibary.wilev.com/doi/pdf/10.1002/cji.201545849)
155,156
。
[0820]
在一些实施例中,可以修改rosenberg快速扩增方案。例如,可以将抗pd1和/或抗41bb添加到til培养物中以模拟更快速的扩增。(https://jitc.biomedcentral.com/articles/10.1186/s40425
‑
016
‑
0164
‑
7)
157
。
[0821]
xviii.b.鉴别新抗原特异性t细胞,对鉴别的新抗原特异性t细胞的tcr进行测序并且将经测序的tcr克隆到新的t细胞中
[0822]
用于产生治疗量的用于患者中的t细胞疗法的新抗原特异性t细胞的第二种方法包括从患者样品中鉴别新抗原特异性t细胞,对鉴别的新抗原特异性t细胞的tcr进行测序,并且将经测序的tcr克隆到新的t细胞中。
[0823]
首先,从患者样品中鉴别新抗原特异性t细胞,并对鉴别的新抗原特异性t细胞的tcr进行测序。可以从其分离出t细胞的患者样品可以包含血液、淋巴结或肿瘤中的一种或多种。更具体地,可以从其分离t细胞的患者样品可以包含外周血单核细胞(pbmc)、肿瘤浸润细胞(til)、离体肿瘤细胞(dtc)、体外引发的t细胞和/或分离自淋巴结的细胞中的一种或多种。这些细胞可以是新鲜的和/或冷冻的。pbmc和体外引发的t细胞可获自癌症患者和/或健康受试者。
[0824]
在获得患者样品之后,可以扩增和/或引发样品。可以实施各种方法来扩增和引发患者样品。在一个实施方案中,可以在肽或串联小基因的存在下模拟新鲜和/或冷冻的pbmc。在另一个实施方案中,可以在肽或串联小基因的存在下用抗原呈递细胞(apc)模拟和引发新鲜和/或冷冻的分离的t细胞。apc的实例包括b细胞、单核细胞、树突状细胞、巨噬细胞或人工抗原呈递细胞(例如呈递相关hla和共刺激分子的细胞或珠粒,在https://www.ncbi.nlm.nih.gov/pmc/articles/pmc2929753中有综述)。在另一个实施方案中,可以在细胞因子(例如il
‑
2、il
‑
7和/或il
‑
15)存在下刺激pbmc、til和/或分离的t细胞。在另一个实施方案中,可以在最大刺激物、细胞因子和/或饲养细胞的存在下刺激til和/或分离的t细胞。在这样的实施方案中,可以通过激活标志物和/或多聚体(例如,四聚体)分离t细胞。在另一个实施方案中,可以用刺激性和/或共刺激性标志物(例如,cd3抗体、cd28抗体和/或珠粒(例如,dynabeads)刺激til和/或分离的t细胞。在另一个实施方案中,可以在富培养基中以高剂量il
‑
2在饲养细胞上使用快速扩增方案扩增dtc。
[0825]
然后,鉴别和分离新抗原特异性t细胞。在一些实施方案中,从患者离体样品分离t
细胞,而无需事先扩增。在一个实施方案中,以上关于第xvii节描述的方法可用于从患者样品中鉴别新抗原特异性t细胞。在另一个实施方案中,通过阳性选择富集特定细胞群或通过阴性选择耗尽特定细胞群来进行分离。在一些实施方案中,通过将细胞与一种或多种抗体或其它结合剂孵育来实现阳性或阴性选择,所述抗体或其它结合剂与分别在阳性或阴性选择的细胞上表达(标志物 )或以相对高的水平表达(标志物
高
)的一种或多种表面标志物特异性结合。
[0826]
在一些实施方案中,通过在非t细胞(例如b细胞、单核细胞或其它白细胞)上表达的标志物(例如cd14)的阴性选择从pbmc样品分离t细胞。在一些方面,cd4 或cd8 选择步骤用于分离cd4 辅助细胞和cd8 细胞毒性t细胞。可以通过对在一种或多种天然、记忆和/或效应t细胞亚群上表达或以相对较高的程度表达的标志物的阳性或阴性选择将这样的cd4 和cd8 种群进一步分选为亚群。
[0827]
在一些实施方案中,例如通过基于与各个亚群相关的表面抗原的阳性或阴性选择进一步富集或耗尽cd4 和cd8 细胞的天然、中枢记忆、效应记忆和/或中枢记忆干细胞。在一些实施方案中,进行中枢记忆t(tcm)细胞的富集以提高效力,例如改善施用后的长期存活、扩增和/或植入,其在一些方面在这样的亚群中特别强效。参见terakura等人(2012)blood.1:72
‑
82;wang等人(2012)j immunother.35(9):689
‑
701。在一些实施方案中,组合富含tcm的cd8 t细胞和cd4 t细胞进一步增强了功效。
[0828]
在一些实施方案中,记忆t细胞存在于cd8 外周血淋巴细胞的cd62l 和cd62l
‑
亚群二者中。可以富集或耗尽pbmc的cd62l
‑
cd8 和/或cd62l cd8 级分,例如使用抗cd8和抗cd62l抗体。
[0829]
在一些实施方案中,中枢记忆t(tcm)细胞的富集是基于cd45ro、cd62l、ccr7、cd28、cd3和/或cd 127的阳性或高表面表达;在一些方面,其基于对表达或高表达cd45ra和/或颗粒酶b的细胞的阴性选择。在一些方面,通过表达cd4、cd14、cd45ra的细胞的耗尽以及表达cd62l的细胞的阳性选择或富集来进行富集tcm细胞的cd8 群的分离。在一方面,从基于cd4表达选择的细胞的阴性级分开始,对其进行基于cd14和cd45ra表达的阴性选择以及基于cd62l的阳性选择来进行中枢记忆t(tcm)细胞的富集。在一些方面,这样的选择同时进行,而在另一些方面,以任一顺序依次进行。在一些方面,用于制备cd8 细胞群或亚群的相同的基于cd4表达的选择步骤也用于产生cd4 细胞群或亚群,使得任选地在一个或多个阳性或阴性选择步骤之后,保留来自基于cd4
‑
的分离的阳性和阴性级分二者并且用于方法的后续步骤。
[0830]
在特定的实例中,对pbmc样品或其它白细胞样品进行cd4 细胞的选择,其中保留阴性级分和阳性级分二者。然后,对阴性级分进行基于cd14和cd45ra或ror1的表达的阴性选择,以及基于中枢记忆t细胞的标志物特征(例如cd62l或ccr7)的阳性选择,其中阳性和阴性选择以任一顺序进行。
[0831]
通过鉴别具有细胞表面抗原的细胞群,将cd4 t辅助细胞分选为天然、中枢记忆和效应细胞。cd4 淋巴细胞可以通过标准方法获得。在一些实施方案中,天然cd4 t淋巴细胞是cd45ro
‑
、cd45ra 、cd62l 、cd4 t细胞。在一些实施方案中,中枢记忆cd4 细胞是cd62l 和cd45ro 。在一些实施方案中,效应cd4 细胞是cd62l
‑
和cd45ro
‑
。
[0832]
在一个实施方案中,为了通过阴性选择富集cd4 细胞,单克隆抗体混合物通常包
括针对cd14、cd20、cd11b、cd16、hla
‑
dr和cd8的抗体。在一些实施方案中,抗体或结合配偶体结合至固体支持物或基质,例如磁珠或顺磁珠,以允许分离细胞以用于阳性和/或阴性选择。例如,在一些实施方案中,使用免疫
‑
磁性(或亲和
‑
磁性)分离技术分离或分隔细胞和细胞群(综述于methods in molecular medicine,第58卷:metastasis research protocols,第2卷:cell behavior in vitro and in vivo,第17
‑
25页,编辑:s.a.brooks and u.schumacher humana press inc.,totowa,n.j.)。
[0833]
在一些方面,将待分离的样品或细胞组合物与小的可磁化或磁响应的材料,例如磁响应颗粒或微粒,例如顺磁珠(例如,dynabeads或macs珠粒)一起孵育。磁响应材料(例如颗粒)通常直接或间接附着至结合配偶体(例如抗体),该结合配偶体特异性结合存在于期望分离(例如期望阴性或阳性选择)的细胞、多个细胞或细胞群上的分子(例如表面标志物)。
[0834]
在一些实施方案中,磁性颗粒或珠粒包含结合至特异性结合成员(例如抗体或其它结合配偶体)的磁响应材料。存在许多用于磁分离方法中的众所周知的磁响应材料。合适的磁性颗粒包括在molday的美国专利第4,452,773号以及欧洲专利说明书ep 452342b中描述的那些,所述专利通过引用并入本文。胶体大小的颗粒,例如在owen的美国专利第4,795,698号和liberti等人的美国专利第5,200,084号中描述的那些是其它示例。
[0835]
孵育通常在一定的条件下进行,所述条件使得附着于磁性颗粒或磁珠的抗体或结合配偶体或者与这样的抗体或结合配偶体特异性结合的分子(例如二抗或其它试剂)特异性地结合细胞表面分子(如果存在于样品中的细胞上的话)。
[0836]
在一些方面,将样品放置在磁场中,并且具有附着于其上的磁响应或可磁化颗粒的那些细胞将被磁体吸引并与未标记的细胞分离。对于阳性选择,保留被磁体吸引的细胞。对于阴性选择,保留未被吸引的细胞(未标记的细胞)。在一些方面,在同一选择步骤期间进行阳性选择和阴性选择的组合,其中保留阳性和阴性级分并进一步处理或经受进一步的分离步骤。
[0837]
在某些实施方案中,将磁响应颗粒包被在一抗或其它结合配偶体、二抗、凝集素、酶或链霉亲和素中。在某些实施方案中,磁性颗粒通过对一种或多种标志物具有特异性的一抗涂层附着到细胞上。在某些实施方案中,用一抗或结合配偶体标记细胞而不是珠粒,然后添加细胞类型特异性的二抗或其它结合配偶体(例如链霉亲和素)包被的磁性颗粒。在某些实施方案中,链霉亲和素包被的磁性颗粒与生物素化的一抗或二抗结合使用。
[0838]
在一些实施方案中,使磁响应颗粒附着于待随后孵育、培养和/或工程改造的细胞;在一些方面,使颗粒附着于用于向患者施用的细胞。在一些实施方案中,从细胞中去除可磁化或磁响应颗粒。用于从细胞中去除可磁化颗粒的方法是已知的,并且包括例如使用竞争性未标记的抗体、与可切割接头缀合的可磁化颗粒或抗体等。在一些实施方案中,可磁化颗粒是可生物降解的。
[0839]
在一些实施方案中,基于亲和力的选择是通过磁激活细胞分选(macs)(miltenyi biotech,auburn,calif.)进行的。磁激活细胞分选(macs)系统能够高纯度选择附着有磁化颗粒的细胞。在某些实施方案中,macs以其中在施加外部磁场之后非靶物质和靶物质顺序洗脱的模式操作。即,附着于磁化颗粒上的细胞被保持在原位,而未附着的物质被洗脱。然后,在完成该第一洗脱步骤后,以某种方式将捕获在磁场中并被阻止洗脱的物质释放出来,
以便可以将其洗脱并回收。在某些实施方案中,非靶t细胞被标记并从异质细胞群中耗尽。
[0840]
在某些实施方案中,使用执行所述方法的分离、细胞制备、分隔、加工、孵育、培养和/或配制步骤中的一个或多个的系统、装置或设备来执行所述分离或分隔。在一些方面,系统用于在封闭或无菌环境中执行这些步骤中的每一个,例如以使错误、用户操作和/或污染最小化。在一个实例中,系统是如国际专利申请公开号wo2009/072003或us 20110003380 a1中所述的系统。
[0841]
在一些实施例中,系统或设备在集成或自包含系统中和/或以自动化或可编程方式执行分离、加工、工程改造和配制步骤中的一个或多个,例如全部。在一些方面,系统或设备包括与该系统或设备通信的计算机和/或计算机程序,其允许用户对加工、分离、工程改造和配制步骤进行编程、控制、评估结果和/或调整多个方面。
[0842]
在一些方面,使用clinimacs系统(miltenyi biotic)进行分离和/或其它步骤,例如,用于在封闭和无菌系统中在临床规模水平上自动分离细胞。组件可以包括集成的微型计算机、磁分离单元、蠕动泵和各种夹管阀。在一些方面,集成计算机控制仪器的所有组件,并指示系统以标准化顺序执行重复程序。在一些方面,磁分离单元包括可移动的永磁体和用于选择柱的保持器。蠕动泵控制整个管组的流速,并与夹管阀一起确保缓冲液通过系统的受控流动和细胞的连续悬浮。
[0843]
在一些方面,clinimacs系统使用提供在无菌、无热原溶液中的抗体偶联的可磁化颗粒。在一些实施方案中,在用磁性颗粒标记细胞后,洗涤细胞以除去过量的颗粒。然后将细胞制备袋连接到管组,该管组又连接到包含缓冲液的袋和细胞收集袋。管组由预组装的无菌管道组成,包括前置柱和分离柱,并且仅供一次性使用。启动分离程序后,系统会自动将细胞样品上样到分离柱上。标记的细胞保留在柱内,而未标记的细胞通过一系列洗涤步骤除去。在一些实施方案中,用于本文所述方法的细胞群体是未标记的并且不保留在柱中。在一些实施方案中,用于本文所述方法的细胞群体是标记的并保留在柱中。在一些实施方案中,在除去磁场后从柱中洗脱用于本文所述方法的细胞群体,并收集在细胞收集袋中。
[0844]
在某些实施方案中,使用clinimacs prodigy系统(miltenyi biotec)进行分离和/或其它步骤。在一些方面,clinimacs prodigy系统配备了细胞处理单元,该单元允许通过离心自动洗涤和分级分离细胞。clinimacs prodigy系统还可以包括机载摄像头和图像识别软件,该软件通过辨别源细胞产品的宏观层来确定最佳的细胞分级分离终点。例如,可以将外周血自动分离为红细胞、白细胞和血浆层。clinimacs prodigy系统还可以包括集成的细胞培养室,该室完成细胞培养方案,例如细胞分化和扩增、抗原加载和长期细胞培养。输入口可以允许无菌去除和补充培养基,并且可以使用集成显微镜监控细胞。参见,例如,klebanoff等人.(2012)j immunother.35(9):651
‑
660,terakura等人.(2012)blood.1:72
‑
82,和wang等人.(2012)j immunother.35(9):689
‑
701。
[0845]
在一些实施方案中,通过流式细胞术收集和富集(或耗尽)本文所述的细胞群,其中针对多种细胞表面标志物染色的细胞被携带在流体流中。在一些实施方案中,通过制备规模(facs)分选收集和富集(或耗尽)本文所述的细胞群。在某些实施方案中,通过使用与基于facs的检测系统组合的微机电系统(mems)芯片来收集和富集(或耗尽)本文所述的细胞群(参见,例如,wo 2010/033140,cho等人(2010)lab chip 10,1567
‑
1573;和godin等人.(2008)j biophoton.1(5):355
‑
376。在两种情况下,可以用多种标志物标记细胞,从而允许
以高纯度分离定义明确的t细胞亚群。
[0846]
在一些实施方案中,用一种或多种可检测的标志物标记抗体或结合配偶体,以促进用于阳性和/或阴性选择的分离。例如,分离可以基于与荧光标记的抗体的结合。在一些实例中,基于对一种或多种细胞表面标志物特异性的抗体或其它结合配偶体的结合的细胞的分离在流体流中进行,例如通过荧光激活细胞分选(facs),包括制备规模(facs)和/或微机电系统(mems)芯片,例如与流式细胞术检测系统组合使用。这样的方法允许同时基于多种标志物进行阳性和阴性选择。
[0847]
在一些实施方式中,制备方法包括在分离、孵育和/或工程改造之前或之后冷冻(例如冷冻保存)细胞的步骤。在一些实施方案中,冷冻和随后的解冻步骤去除了细胞群中的粒细胞,并且在一定程度上去除了单核细胞。在一些实施方案中,例如在洗涤步骤以除去血浆和血小板之后,将细胞悬浮在冷冻溶液中。在一些方面,可以使用多种已知的冷冻溶液和参数中的任何一种。一个实例涉及使用含有20%dmso和8%人血清白蛋白(hsa)或其它合适的细胞冷冻介质的pbs。然后可以用培养基将其1:1稀释,以使dmso和hsa的终浓度分别为10%和4%。其它实例包括ctl
‑
cryo
tm
abc冷冻介质等。然后将细胞以每分钟1度的速率冷冻至
‑
80摄氏度,并存储在液氮储罐的蒸汽相中。
[0848]
在一些实施例中,所提供的方法包括培养、孵育、培养和/或基因工程步骤。例如,在一些实施方案中,提供了用于孵育和/或工程改造耗尽的细胞群体和培养起始组合物的方法。
[0849]
因此,在一些实施方案中,将细胞群体在培养起始组合物中孵育。孵育和/或工程改造可以在培养容器中进行,例如单元、腔室、孔、柱、管、管组、阀、小瓶、培养皿、袋或用于培养或培养细胞的其它容器。
[0850]
在一些实施方案中,在基因工程之前或与基因工程相结合孵育和/或培养细胞。孵育步骤可包括培养、培养、刺激、激活和/或繁殖。在一些实施方案中,将组合物或细胞在刺激条件或刺激剂的存在下孵育。这样的条件包括被设计以诱导群体中细胞的增殖、扩增、活化和/或存活、模拟抗原暴露和/或引发细胞以进行基因工程(例如用于引入重组抗原受体)的那些条件。
[0851]
条件可以包括以下一种或多种:特定培养基、温度、氧含量、二氧化碳含量、时间、剂(例如营养素、氨基酸、抗生素、离子)和/或刺激因子(例如细胞因子、趋化因子、抗原、结合配偶体、融合蛋白、重组可溶性受体)和旨在激活细胞的任何其它剂。
[0852]
在一些实施方案中,刺激条件或剂包括能够激活tcr复合物的细胞内信号传导结构域的一种或多种剂,例如配体。在一些方面,该剂打开或启动t细胞中的tcr/cd3细胞内信号传导级联。这样的剂可以包括抗体,例如对tcr组分和/或共刺激受体具有特异性的抗体,例如抗cd3、抗cd28,其例如与固体支持物如珠粒和/或一种或多种细胞因子结合。任选地,扩增方法可以进一步包括将抗cd3和/或抗cd28抗体添加到培养基中的步骤(例如,以至少约0.5ng/ml的浓度)。在一些实施方案中,刺激剂包括il
‑
2和/或il
‑
15,例如,il
‑
2浓度为至少约10单位/ml。
[0853]
在一些方面,根据例如以下中描述的那些技术进行孵育:riddell等人的美国专利第6,040,177号,klebanoff等人(2012)j immunother.35(9):651
‑
660,terakura等人(2012)blood.1:72
‑
82,和/或wang等人(2012)j immunother.35(9):689
‑
701。
[0854]
在一些实施方案中,通过向培养起始组合物添加饲养细胞,例如非分裂外周血单核细胞(pbmc)(例如,使得对于待扩增的起始群体中的每个t淋巴细胞,所得细胞群体包含至少约5、10、20或40或更多个pbmc饲养细胞);以及孵育培养物(例如足以扩增t细胞的数目的时间)来扩增t细胞。在一些方面,非分裂饲养细胞可以包含γ辐照的pbmc饲养细胞。在一些实施方案中,用约3000至3600拉德范围内的γ射线辐照pbmc以防止细胞分裂。在一些实施方案中,将pbmc饲养细胞用丝裂霉素c灭活。在一些方面,在添加t细胞群体之前将饲养细胞添加至培养基。
[0855]
在一些实施方案中,刺激条件包括适合于人t淋巴细胞生长的温度,例如,至少约25摄氏度,通常至少约30摄氏度,并且通常在或约37摄氏度。任选地,孵育还可以包括添加非分裂的ebv转化的类淋巴母细胞(lcl)作为饲养细胞。lcl可以用约6000至10,000拉德范围内的γ射线辐照。在一些方面,以任何合适的量提供lcl饲养细胞,例如lcl饲养细胞与初始t淋巴细胞的比率为至少约10:1。
[0856]
在一些实施方案中,通过用抗原刺激天然或抗原特异性t淋巴细胞来获得抗原特异性t细胞,例如抗原特异性cd4 t细胞。例如,可以通过从受感染的受试者中分离t细胞并用相同的抗原体外刺激细胞来产生巨细胞病毒抗原的抗原特异性t细胞系或克隆。
[0857]
在一些实施方案中,在用功能测定(例如,elispot)刺激后,鉴别和/或分离新抗原特异性t细胞。在一些实施方案中,通过细胞内细胞因子染色对多功能细胞进行分选来分离新抗原特异性t细胞。在一些实施方案中,使用活化标志物(例如,cd137、cd38、cd38/hla
‑
dr双阳性和/或cd69)鉴别和/或分离新抗原特异性t细胞。在一些实施方案中,使用ii类多聚体和/或活化标志物鉴别和/或分离新抗原特异性cd4 、自然杀伤t细胞和/或记忆t细胞。在一些实施方案中,使用记忆标志物(例如,cd45ra、cd45ro、ccr7、cd27和/或cd62l)鉴别和/或分离新抗原特异性cd4 t细胞。在一些实施方案中,鉴别和/或分离增殖细胞。在一些实施方案中,鉴别和/或分离活化的t细胞。
[0858]
从患者样品中鉴别出新抗原特异性t细胞之后,对鉴别出的新抗原特异性t细胞中的新抗原特异性tcr进行测序。为了对新抗原特异性tcr进行测序,必须首先鉴别tcr。鉴别t细胞的新抗原特异性tcr的一种方法可以包括使t细胞与包含至少一种新抗原的hla
‑
多聚体(例如,四聚体)接触;以及通过hla
‑
多聚体和tcr之间的结合鉴别tcr。鉴别新抗原特异性tcr的另一种方法可以包括获得包含tcr的一种或多种t细胞;用在至少一种抗原呈递细胞(apc)上呈递的至少一种新抗原活化所述一种或多种t细胞;以及通过选择通过与至少一种新抗原相互作用而活化的一种或多种细胞来鉴别tcr。
[0859]
在鉴别出新抗原特异性tcr之后,可以对tcr进行测序。在一个实施方案中,以上关于第xvii节描述的方法可用于对tcr进行测序。在另一个实施方案中,可以对tcr的tcra和tcrb进行批量测序,然后基于频率进行配对。在另一个实施方案中,可以使用howie等人,science translational medicine 2015(doi:10.1126/scitranslmed.aac5624)的方法对tcr进行测序和配对。在另一个实施方案中,可以使用han等人.,nat biotech 2014(pmid 24952902,doi 10.1038/nbt.2938)的方法对tcr进行测序和配对。在另一个实施方案中,可以使用以下中描述的方法获得配对的tcr序列:https://www.biorxiv.org/content/early/2017/05/05/134841和https://patents.google.com/patent/us20160244825a1/.
158,159
[0860]
在另一个实施方案中,可以通过有限稀释产生t细胞的克隆群,然后可以对t细胞的克隆群的tcra和tcrb进行测序。在又一个实施方案中,可以将t细胞分选到具有孔的板上,使得每个孔有一个t细胞,然后可以对每个孔中每个t细胞的tcra和tcrb进行测序和配对。
[0861]
接下来,在从患者样品中鉴别出新抗原特异性t细胞并且对所鉴别的新抗原特异性t细胞的tcr进行测序之后,将经测序的tcr克隆到新的t细胞中。这些经克隆的t细胞含有新抗原特异性受体,例如含有细胞外结构域,包括tcr。还提供了这样的细胞的群体以及包含这样的细胞的组合物。在一些实施方案中,使组合物或群体富集这样的细胞,例如其中表达tcr的细胞占某类型的组合物或细胞(例如t细胞或cd4 细胞)中总细胞的至少1、5、10、20、30、40、50、60、70、80、90、91、92、93、94、95、96、97、98、99或超过99百分比。在一些实施方案中,组合物包含至少一种包含本文公开的tcr的细胞。组合物包括用于施用,例如用于过继细胞疗法的药物组合物和制剂。还提供了用于将细胞和组合物施用给受试者(例如患者)的治疗方法。
[0862]
因此,还提供了表达tcr的基因工程细胞。细胞通常是真核细胞,例如哺乳动物细胞,并且通常是人细胞。在一些实施方案中,细胞源自血液、骨髓、淋巴或淋巴器官,是免疫系统的细胞,例如先天或适应性免疫的细胞,例如髓样或淋巴样细胞,包括淋巴细胞,通常为t细胞和/或nk细胞。其它示例性细胞包括干细胞,例如多能和多潜能干细胞,包括诱导性多能干细胞(ipsc)。细胞通常是原代细胞,例如直接从受试者中分离和/或从受试者中分离并冷冻的细胞。在一些实施方案中,细胞包括t细胞或其它细胞类型的一个或多个子集,例如整个t细胞群体、cd4 细胞、及其亚群,例如由功能、活化状态、成熟度、潜能分化、扩增、再循环、定位和/或持久能力、抗原特异性、抗原受体的类型、在特定器官或区室中的存在、标志物或细胞因子的分泌谱和/或分化程度定义的那些。关于待治疗的受试者,细胞可以是同种异体的和/或自体的。这些方法包括现成的方法。在一些方面,例如对于现成技术,细胞是多能的和/或多潜能的,例如干细胞,例如诱导多能干细胞(ipsc)。在一些实施方案中,该方法包括如本文所述从受试者分离细胞、制备、加工、培养和/或工程改造它们,以及在冷冻保存之前或之后将其重新引入同一患者。
[0863]
t细胞和/或cd8 t细胞的亚型和亚群是天然t(tn)细胞、效应t细胞(teff)、记忆t细胞及其亚型,例如干细胞记忆t(tscm)、中枢记忆t(tcm)、效应记忆t(tem)或终末分化的效应记忆t细胞、肿瘤浸润淋巴细胞(til)、未成熟t细胞、成熟t细胞、辅助t细胞、细胞毒性t细胞、粘膜相关性不变t(malt)细胞、天然和适应性调节性t(treg)细胞、辅助t细胞(例如th1细胞、th2细胞、th3细胞)、th17细胞、th9细胞、th22细胞、滤泡辅助t细胞、α/βt细胞和δ/γt细胞。
[0864]
在一些实施方案中,细胞是自然杀伤(nk)细胞。在一些实施方案中,细胞是单核细胞或粒细胞,例如髓样细胞、巨噬细胞、嗜中性粒细胞、树突状细胞、肥大细胞、嗜酸性粒细胞和/或嗜碱性粒细胞。
[0865]
可以对细胞进行遗传修饰以减少表达或敲除内源tcr。这样的修饰描述在以下中:mol ther nucleic acids.2012dec;1(12):e63;blood.2011aug 11;118(6):1495
‑
503;blood.2012jun 14;119(24):5697
–
5705;torikai,hiroki等人"hla and tcr knockout by zinc finger nucleases:toward“off
‑
the
‑
shelf”allogeneic t
‑
cell therapy for cd19
malignancies.."blood 116.21(2010):3766;blood.2018jan18;131(3):311
‑
322.doi:10.1182/blood
‑
2017
‑
05
‑
787598;和wo2016069283,所述文献通过引用整体并入。
[0866]
可以对细胞进行遗传修饰以促进细胞因子的分泌。这样的修饰描述在以下中:hsu c,hughes ms,zheng z,bray rb,rosenberg sa,morgan ra.primary human t lymphocytes engineered with acodon
‑
optimized il
‑
15gene resist cytokine withdrawal
‑
induced apoptosis and persist long
‑
term in the absence of exogenous cytokine.jimmunol.2005;175:7226
–
34;quintarelli c,vera jf,savoldo b,giordano attianese gm,pule m,foster ae,co
‑
expression of cytokine and suicide genes to enhance the activity and safety of tumor
‑
specific cytotoxic t lymphocytes.blood.2007;110:2793
–
802;和hsu c,jones sa,cohen cj,zheng z,kerstann k,zhou j,cytokine
‑
independent growth and clonal expansion of a primary human cd8 t
‑
cell clone following retroviral transduction with the il
‑
15gene.blood.2007;109:5168
–
77。
[0867]
已显示t细胞上趋化因子受体和肿瘤分泌的趋化因子的错配是造成t细胞向肿瘤微环境的次佳运输的原因。为了提高治疗效果,可以对细胞进行遗传修饰,以提高对肿瘤微环境中趋化因子的识别。这样的修饰描述在以下中:moon,ekcarpenito,csun,jwang,lckapoor,vpredina,j expression of a functional ccr2 receptor enhances tumor localization and tumor eradication by retargeted human t
‑
cells expressing a mesothelin
‑
specific chimeric antibody receptor.clin cancer res.2011;17:4719
‑
4730;和craddock,jalu,abear,apule,mbrenner,mkrooney,cm et al.enhanced tumor trafficking of gd2 chimeric antigen receptor t
‑
cells by expression of the chemokine receptor ccr2b.j immunother.2010;33:780
‑
788。
[0868]
可以对细胞进行遗传修饰以增强共刺激/增强受体(例如cd28和41bb)的表达。
[0869]
t细胞疗法的不良反应可包括细胞因子释放综合征和延长的b细胞耗竭。在受体细胞中引入自杀/安全开关可以改善基于细胞的疗法的安全性谱。因此,可以对细胞进行遗传修饰以包含自杀/安全开关。自杀/安全开关可以是这样的基因,其在表达该基因的细胞上赋予对剂例如药物的敏感性,并且当细胞与该剂接触或暴露于该剂时导致该细胞死亡。示例性的自杀/安全开关描述于protein cell.2017aug;8(8):573
–
589中。自杀/安全开关可以是hsv
‑
tk。自杀/安全开关可以是胞嘧啶脱氨酶、嘌呤核苷磷酸化酶或硝基还原酶。自杀/安全开关可以是美国专利申请公开no.us20170166877a1中描述的rapacide
tm
。自杀/安全开关系统可以是haematologica.2009sep;94(9):1316
–
1320中描述的cd20/利妥昔单抗。这些参考文献通过引用整体并入。
[0870]
tcr可以作为分裂受体(split receptor)引入受体细胞,分裂受体仅在异二聚化小分子的存在下组装。这样的系统描述在science.2015年10月16日;350(6258):aab4077和美国专利第9,587,020号中,所述文献通过引用并入。
[0871]
在一些实施方案中,细胞包含一种或多种核酸,例如编码本文公开的tcr的多核苷酸,其中所述多核苷酸通过基因工程引入,并因此表达本文公开的重组或基因工程tcr。在一些实施方案中,核酸是异源的,即,通常不存在于从所述细胞获得的细胞或样品中,例如是从另一种生物或细胞获得的,例如其通常不在所工程改造的细胞和/或这样的细胞所来
源的生物中发现。在一些实施方案中,核酸不是天然存在的,例如自然界中不存在的核酸,包括包含编码来自多种不同细胞类型的多个结构域的核酸的嵌合组合的核酸。
[0872]
核酸可包括密码子优化的核苷酸序列。不受特定理论或机制的束缚,据信核苷酸序列的密码子优化增加了mrna转录物的翻译效率。核苷酸序列的密码子优化可以包括将天然密码子替换为另一种密码子,所述另一种密码子编码相同氨基酸,但是可以通过在细胞内更容易获得的trna进行翻译,从而提高翻译效率。核苷酸序列的优化还可以减少将会干扰翻译的二级mrna结构,从而提高翻译效率。
[0873]
可使用构建体或载体将tcr引入受体细胞。本文描述了示例性构建体。编码tcr的α和β链的多核苷酸可以在单个构建体中或在分开的构建体中。编码α和β链的多核苷酸可以可操作地连接至启动子,例如异源启动子。异源启动子可以是强启动子,例如ef1α、cmv、pgk1、ubc、β肌动蛋白、cag启动子等。异源启动子可以是弱启动子。异源启动子可以是诱导型启动子。示例性诱导型启动子包括但不限于tre、nfat、gal4、lac等。其它示例性诱导型表达系统描述于美国专利第5,514,578号、第6,245,531号、第7,091,038号和欧洲专利第0517805号中,所述专利通过引用整体本文。
[0874]
用于将tcr引入受体细胞的构建体还可包含编码信号肽的多核苷酸(信号肽元件)。信号肽可以促进引入的tcr的表面运输。示例性信号肽包括但不限于cd4信号肽、免疫球蛋白信号肽,其中具体实例包括gm
‑
csf和iggκ。这样的信号肽在以下中进行了描述:trends biochem sci.2006年10月;31(10):563
‑
71.epub 2006年8月21日;和an,等人“construction of a new anti
‑
cd19 chimeric antigen receptor and the anti
‑
leukemia function study of the transduced t
‑
cells.”oncotarget 7.9(2016):10638
–
10649.pmc.web.2018年8月16日,所述文献通过引用并入本文。
[0875]
在一些情况下,例如,在从单个构建体或开放阅读框表达α和β链的情况,或在该构建体中包含标志基因的情况,该构建体可包含核糖体跳读序列。核糖体跳读序列可以是2a肽,例如p2a或t2a肽。示例性的p2a和t2a肽在scientific reports第7卷,文章编号:2193(2017)中进行了描述,所述文献通过引用整体并入。在一些情况下,在2a元件上游引入了furin/pace切割位点。furin/pace切割位点描述于例如http://www.nuolan.net/substrates.html中。切割肽也可以是因子xa的切割位点。在从单个构建体或开放阅读框表达α和β链的情况下,该构建体可包含内部核糖体进入位点(ires)。
[0876]
构建体可以进一步包含一种或多种标志基因。示例性标志基因包括但不限于gfp、荧光素酶、ha、lacz。如本领域技术人员已知的,标志物可以是可选择的标志物,例如抗生素抗性标志物、重金属抗性标志物或抗生物杀灭剂标志物。标志物可以是用于营养缺陷宿主的互补标志物。示例性的互补标志物和营养缺陷的宿主在gene.2001年1月24日;263(1
‑
2):159
‑
69中进行了描述。这样的标志物可以通过ires、移码序列、2a肽接头、与tcr融合表达,或者由单独的启动子分开表达。
[0877]
用于将tcr引入受体细胞的示例性载体或系统包括但不限于腺相关病毒、腺病毒、腺病毒 修饰的牛痘病毒、安卡拉病毒(mva)、腺病毒 逆转录病毒、腺病毒 仙台病毒、腺病毒 牛痘病毒、甲病毒(vee)复制子疫苗、反义寡核苷酸、长双歧杆菌(bifidobacterium longum)、crispr
‑
cas9、大肠杆菌(e.coli)、黄病毒、基因枪、疱疹病毒、单纯疱疹病毒、乳酸乳球菌、电穿孔、慢病毒、脂质体转染、单核细胞性李斯特菌(listeria monocytogenes)、麻
疹病毒、修饰的牛痘安卡拉病毒(mva)、mrna电穿孔、裸/质粒dna、裸/质粒dna 腺病毒、裸/质粒dna 修饰的牛痘安卡拉病毒(mva)、裸/质粒dna rna转移、裸/质粒dna 牛痘病毒、裸/质粒dna 水泡性口炎病毒、新城疫病毒、非病毒、piggybac
tm
(pb)转座子、基于纳米颗粒的系统、脊髓灰质炎病毒、痘病毒、痘病毒 牛痘病毒、逆转录病毒、rna转移、rna转移 裸/质粒dna、rna病毒、酿酒酵母(saccharomyces cerevisiae)、鼠伤寒沙门氏菌(salmonella typhimurium)、塞姆利基森林病毒(semliki forest virus)、仙台病毒、痢疾志贺氏菌(shigella dysenteriae)、猿猴病毒、sirna、睡美人转座子、变形链球菌(streptococcus mutans)、牛痘病毒、委内瑞拉马脑炎病毒复制子、水泡性口炎病毒和霍乱弧菌(vibrio cholera)。
[0878]
在优选的实施方案中,将tcr通过腺相关病毒(aav)、腺病毒、crispr
‑
cas9、疱疹病毒、慢病毒、脂转染、mrna电穿孔、piggybac
tm
(pb)转座子、逆转录病毒、rna转移或睡美人转座子引入受体细胞。
[0879]
在一些实施方案中,用于将tcr引入受体细胞的载体是病毒载体。病毒载体的实例包括腺病毒载体、腺相关病毒(aav)载体、慢病毒载体、疱疹病毒载体、逆转录病毒载体等。这样的载体在本文中描述。
[0880]
用于将tcr引入受体细胞的tcr构建体的示例性实施方案在图16中示出。在一些实施方案中,tcr构建体在5'
‑
3'方向包含以下多核苷酸序列:启动子序列、信号肽序列、tcrβ可变(tcrβv)序列、tcrβ恒定(tcrβc)序列、切割肽(例如,p2a)、信号肽序列、tcrα可变(tcrαv)序列和tcrα恒定(tcrαc)序列。在一些实施方案中,构建体的tcrβc和tcrαc序列包含一个或多个鼠区域,例如,如本文所述的完整鼠恒定序列或人
→
鼠氨基酸交换。在一些实施方案中,构建体进一步在tcrαc序列的3’包含切割肽序列(例如,t2a)然后是报告基因。在一个实施方案中,该构建体在5'
‑
3'方向包含以下多核苷酸序列:启动子序列、信号肽序列、tcrβ可变(tcrβv)序列、包含一个或多个鼠区域的tcrβ恒定(tcrβc)序列、切割肽(例如p2a)、信号肽序列、tcrα可变(tcrαv)序列和包含一个或多个鼠区域的tcrα恒定(tcrαc)序列、切割肽(例如(t2a)和报告基因。
[0881]
图17描绘了用于将tcr克隆到表达系统中以进行疗法开发的示例性p526构建体骨架核苷酸序列。
[0882]
图18描绘了用于将患者新抗原特异性tcr克隆型1tcr克隆到表达系统中用于疗法开发的示例性构建体序列。
[0883]
图19描绘了用于将患者新抗原特异性tcr克隆型3克隆到表达系统中用于疗法开发的示例性构建体序列。
[0884]
还提供了编码tcr的分离的核酸,包含所述核酸的载体以及包含所述载体和核酸的宿主细胞,以及用于产生tcr的重组技术。
[0885]
核酸可以是重组的。可通过将天然或合成核酸片段连接至可在活细胞中复制的核酸分子或其复制产物来在活细胞外部构建重组核酸。出于本文的目的,复制可以是体外复制或体内复制。
[0886]
为了重组产生tcr,可以分离编码它的核酸并将其插入可复制的载体中以进一步克隆(即,dna的扩增)或表达。在一些方面,核酸可以通过同源重组产生,例如,如美国专利第5,204,244号中所述,其通过引用整体并入本文。
[0887]
许多不同的载体是本领域已知的。载体组分通常包括以下一种或多种:信号序列、复制起点、一种或多种标志基因、增强子元件、启动子和转录终止序列,例如美国专利第5,534,615号中所述,其通过引用并入本文。
[0888]
适用于表达tcr、抗体或其抗原结合片段的示例性载体或构建体包括,例如,puc系列(fermentas life sciences)、pbluescript系列(stratagene,lajolla,ca)、pet系列(novagen,madison,wi),pgex系列(pharmacia biotech,uppsala,sweden)和pex系列(clontech,palo alto,ca)。噬菌体载体,例如agt10、agt11、azapii(stratagene)、aembl4和anm1149也适用于表达本文公开的tcr。
[0889]
xix.治疗概述流程图
[0890]
图20是根据一个实施方案的用于向患者提供定制的新抗原特异性治疗的方法的流程图。在其它实施方案中,该方法可以包括与图20所示的步骤不同的步骤和/或另外的步骤。另外,该方法的步骤可以以与多个实施方案中结合图20描述的顺序不同的顺序执行。
[0891]
如上所述,使用质谱数据训练呈递模型2001。获得患者样品2002。在一些实施方案中,患者样品包含肿瘤活检和/或患者的外周血。对在步骤2002中获得的患者样品进行测序,以鉴别输入到呈递模型中的数据,以预测来自患者样品的肿瘤抗原肽将被呈递的可能性。使用训练的呈递模型来预测在步骤2002中获得的来自患者样品的肿瘤抗原肽的呈递可能性2003。基于预测的呈递可能性为患者鉴别治疗新抗原2004。接下来,获得另一个患者样品2005。该患者样品可以包含患者的外周血、肿瘤浸润淋巴细胞(til)、淋巴、淋巴结细胞和/或任何其它t细胞来源。将步骤2005中获得的患者样品在体内筛选2006新抗原特异性t细胞。
[0892]
在治疗过程中的这一点上,患者可以接受t细胞疗法和/或疫苗治疗。为了接受疫苗治疗,鉴别患者的t细胞对其特异的新抗原2014。然后,产生包含已鉴别的新抗原的疫苗2015。最后,向患者施用疫苗2016。
[0893]
为了接受t细胞疗法,对新抗原特异性t细胞进行扩增和/或对新的新抗原特异性t细胞进行基因工程改造。为了扩增新抗原特异性t细胞以用于t细胞疗法,简单地将细胞扩增2007并输注2008到患者。
[0894]
为了对新的新抗原特异性t细胞进行基因工程改造以用于t细胞疗法,对在体内鉴别出的新抗原特异性t细胞的tcr进行测序2009。接下来,将这些tcr序列克隆到表达载体中2010。然后将表达载体2010转染到新的t细胞中2011。扩增转染的t细胞2012。最后,将扩增的t细胞注输注到患者体内2013。
[0895]
患者可以同时接受t细胞疗法和疫苗疗法。在一个实施方案中,患者首先接受疫苗疗法,然后接受t细胞疗法。这种方法的一个优点是疫苗疗法可以增加肿瘤特异性t细胞的数量和由可检测水平的t细胞识别的新抗原的数量。
[0896]
在另一个实施方案中,患者可以接受t细胞疗法后进行疫苗疗法,其中疫苗中包含的表位集包含由t细胞疗法靶向的一个或多个表位。该方法的一个优点是疫苗的施用可以促进治疗性t细胞的扩增和持久性。
[0897]
xx.示例计算机
[0898]
图21示出了用于实施图1和3中所示实体的示例计算机2100。计算机2100包括耦合至芯片组2104的至少一个处理器2102。芯片组2104包括内存控制器集线器2120和输入/输
出(i/o)控制器集线器2122。内存2106和图形适配器2112耦合至内存控制器集线器2120,并且显示器2118耦合至图形适配器2112。存储装置2108、输入装置2114和网络适配器2116耦合至i/o控制器集线器2122。计算机2100的其它实施方案具有不同的架构。
[0899]
存储装置2108是非暂时性计算机可读存储介质,如硬盘驱动器、致密光盘只读存储器(cd
‑
rom)、dvd或固态内存装置。内存2106保存处理器2102所使用的指令和数据。输入接口2114是触摸屏界面、鼠标、轨迹球或其它类型的指向装置、键盘或其某一组合,并且用于将数据输入计算机2100中。在一些实施方案中,计算机2100可以被配置成通过用户的示意动作从输入接口2114接收输入(例如,命令)。图形适配器2112将图像和其它信息显示于显示器2118上。网络适配器2116将计算机2100耦合至一个或多个计算机网络。
[0900]
计算机2100被调适成执行计算机程序模块以提供本文所述的功能。如本文所使用,术语“模块”是指用于提供指定功能的计算机程序逻辑。因此,模块可以在硬件、固件和/或软件中实施。在一个实施方案中,程序模块被存储于存储装置2108上,装载至内存2106中并由处理器2102执行。
[0901]
图1的实体所使用的计算机2100的类型可以根据实施方案和实体所需的处理能力而变化。举例来说,呈递鉴别系统160可以在单一计算机2100或在通过网络,如在服务器群中彼此通信的多台计算机2100中运行。计算机2100可以缺少以上描述的组件中的一些,如图形适配器2112和显示器2118。参考文献
[0902]
[0903]
[0904]
[0905]
[0906]
[0907]
[0908]
[0909]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。