1.本发明属于药物制备技术领域,具体地,涉及一种罗沙司他关键中间体及罗沙司他中间体的制备方法。
背景技术:
2.贫血是慢性肾脏病(ckd)患者很常见的一种疾病,无论是透析还是非透析ckd患者,发病率和死亡率都非常高。ckd可发病于任何年龄,老年人中更为常见,中国大约有1.2亿ckd患者。中国接受透析的ckd人群超过40万,而且以两位数的增幅快速增长,因此需要抗贫血疗法的患者日益增多。ckd患者当前的抗贫血疗法主要是注射促红细胞生成素,罗沙司他(roxadustat)是一种低氧诱导因子脯氨酰羟化酶(hif-ph)抑制剂,可以提供一种更便捷(口服)、更安全的治疗选择。
3.罗沙司他化学名为[(4-羟基-1-甲基-7-苯氧基异喹啉-3-羰基)氨基]乙酸,商品名艾瑞卓,是由美国菲布罗根(fibrogen)有限公司研发,现已授权日本安斯泰来(astellas)和英国的阿斯利康(astrazeneca),是一种全新的口服低氧诱导因子脯氨酰羟化酶抑制剂,可以诱导红细胞生成,治疗肾性贫血。2018年12月18日,由珐博进(中国)医药技术开发有限公司和阿斯利康投资(中国)有限公司在国内合作开发的一款国产ⅰ类原创新药、全球首个口服低氧诱导因子脯氨酰羟化酶抑制剂获得国家药品监督管理局的上市批准。罗沙司他被批准用于ckd透析患者的贫血治疗,包括血液透析和腹膜透析患者。
[0004]
目前关于罗沙司他的研究,核心在于异喹啉-4号位置羟基的构建和-1号位置甲基的构建,4号位置的羟基化得到4-羟基-7-苯氧基异喹咻-3-甲酸酯或其衍生物,1号位置的甲基化得到中间体1-甲基-7-苯氧基异喹咻-3-甲酸酯或其衍生物。目前国内外主要有以下几条合成路线:
[0005]
原研fibrogen公司的公开号为wo2004108681a1的国际专利公开了以3,4-二氰基硝基苯为起始原料依次经苯酚醚化、氢氧化钾水解、甘氨酸环合、甲醇酯化、金属钠和叔丁醇环合、溴代,得到罗沙司他关键中间体1(化学名称为1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸丁酯),中间体1再经过水解,正丁基锂和碘甲烷甲基化、再经氢氧化钾水解、甘氨酸苄酯酰胺化和钯炭脱苄基等多步反应得到罗沙司他,该合成路线步骤多、收率低,起始原料为2-氰基-4-硝基苯腈,成本很高且没有商业化产品供应,其中间体1分离需要用到色谱柱,分离成本较高,尤其是在甲基化反应使用正丁基锂危险性试剂,限制了其工业化的大规模生产。
[0006][0007]
合成路线一
[0008]
后来,fibrogen公司对该路线进行了改进(专利号wo2014014834a1、cn103539735b):以5-溴苯酞为起始原料,依次经苯酚醚化、二氯亚砜开环氯化、取代氨基酸对接、环合,得到罗沙司他关键中间体1(化学名称为4-羟基-7-苯氧基异喹咻-3-甲酸甲酯),中间体1再经烷基化,取代,酰胺化,pd-c催化还原,得到罗沙司他。该合成路线起始原料5-溴苯酞价格昂贵,没有商业化产品供应,卤代试剂为二氯三苯基正膦,价格昂贵,且该步中间体纯度85%,制备得到中间体1,总收率仅29%,不利于工业化生产。但在甲基化构建过程中,各步产率均大于81%,纯度达到99.5%,烷基化收率和质量都符合工业化需求。
[0009][0010]
合成路线二
[0011]
欧洲专利ep3305769a1公开了以2-溴-4-氟苯甲酸甲酯为起始原料,依次经苯酚醚化反应、cdi参与的噁唑啉化、钯催化偶联反应、酸催化环合,得到罗沙司他关键中间体1(化学名称为4-羟基-1-甲基-7-苯氧基异喹咻-3-甲酸乙酯),同样的,该合成路线起始原料价格较高,偶联反应所用钯催化剂价格也较为昂贵,且偶联反应和环合反应的收率为50%左右,使得该路线收率低、成本高而难以实现工业化生产。
[0012][0013]
合成路线三
[0014]
贝达药业股份有限公司的国际专利wo2013013609a1以3,4-二氰基硝基苯为起始原料,依次经苯酚醚化、氢氧化钾水解、乙酸酐脱水、氰基乙酸甲酯噁唑啉化、盐酸环合、三氯氧磷氯化,得到罗沙司他关键中间体1(化学名称为4-羟基-1-氯-7-苯氧基异喹咻-3-甲酸甲酯);该合成路线需经多步反应制备,起始原料为2-氰基-4-硝基苯腈,成本很高且没有商业化产品供应,环合双键容易断裂,收率较低,仅62%,难以纯化,使得路线成本大幅提升,难以实现大规模工业化生产。
[0015][0016]
合成路线四
[0017]
上海勋和医药科技有限公司的中国专利cn106478504a以间溴苯乙酮为起始原料,依次经苯酚醚化、盐酸羟胺和硼氢化钠还原胺化、2-羰基丙二酸酯成席夫碱、最后经环合得到罗沙司他关键中间体1(化学名称为4-羟基-1-甲基-7-苯氧基-3-异喹啉甲酸酯),该合成路线起始原料不易获得,价格昂贵,且环合反应温度较高,反应条件苛刻,难以环合,同时反应的选择性较差,易产生邻位环合的杂质,因而产品成本高且不易纯化。同时通过实际实验发现按该方法得不到环合产物,可能是c=n双键非常不稳定,易水解,所以不能环合。
[0018]
[0019]
合成路线五
[0020]
苏州明锐医药科技有限公司的中国专利cn104892509b以酪氨酸为起始原料,依次经醇酯化、卤代苯醚化、乙醛环合、碱催化脱氢、双氧水催化羟基化,得到罗沙司他关键中间体1(化学名称为4-羟基-1-甲基-7-苯氧基异喹咻-3-甲酸酯)。该合成路线原料易得,但是醚化反应易与氨基发生副反应,导致产品难以纯化,同时双氧水羟基化反应收率低,仅32%,因此该路线工业化生产还需改进和优化。
[0021][0022]
合成路线六
[0023]
浙江工业大学的中国专利cn111592490a以酪氨酸为起始原料,依次通过酯化、酰胺化、醚化、环化、芳构化和氧化重排得到罗沙司他关键中间体1(化学名称为4-羟基-1甲基-7-苯氧基异喹咻-3-甲酸甲酯),该合成路线与传统路线相比较,原料廉价,反应温和,但在醚化过程使用了2-异丁酰基环己酮,导致工业化成本较高,仍需改进;另外,脱氢芳构化和氧化上羟基属于危险工艺且收率低,使得产品成本高,工业化难度大。
[0024][0025]
合成路线七
[0026]
同时,国内山东百诺医药股份有限公司(cn111393365a)报道了一种罗沙司他的制备方式:从中间体1,经溴代、水解、酰胺化、酯水解,得到罗沙司他,该路线收率高,每步反应的收率大于83.20%,总收率高达80.82%,且后处理采用重结晶的方法,不需过柱纯化,适用于工业化生产。
[0027][0028]
合成路线八
[0029]
重庆三圣实业股份有限公司(cn111533691a)报道了从中间体1与四甲基甲烷二胺加成得到1-((二甲氨基)甲基)-4-羟基-7-苯氧异喹咻基-3-羧酸甲酯,再经还原,得到甲基化产物片段,再与甘氨酸对接,得到罗沙司他。该路线合成效率高,制备得到的罗沙司他的收率在85-90%,原料利用率高,罗沙司他的纯度在95%以上,满足工业化的生产要求。
[0030][0031]
合成路线九
[0032]
南京海润医药有限公司(cn112441976a)报道了从中间体1,合成得到1-((乙酸甲酯基)甲基)-4-羟基-7-苯氧异喹咻基-3-羧酸甲酯,再经zn-乙酸体系还原,得到1-甲基-4-羟基-7-苯氧异喹咻基-3-羧酸甲酯。该路线反应条件温和,无需高温高压,在常压下即可进行反应,并且收率不低于90%,纯度不低于99.7%,并已经工业化生产。
[0033][0034]
合成路线十
[0035]
安礼特(上海)医药科技有限公司(cn113121438a)报道了从中间体1在硫酸亚铁催化下,双氧水氧化,中间体1与乙醛缩合,得到甲基化片段,再与甘氨酸反应,得到罗沙司他。该路线原料易得,各步收率不低于90%,纯度不低于99.7%,适合工业化放大。
[0036]
[0037]
合成路线十一
[0038]
综上所述,罗沙司他的合成核心在于异喹啉-4号位置羟基的构建,和-1号位置甲基的构建,目前通常都是先构建-4号羟基,制备中间体4-羟基-7-苯氧基-3-异喹啉甲酸酯(式ⅵ)及其衍生物,进而再-1号位置甲基化,后经水解、酰胺化等反应制得罗沙司他。大量报道表明甲基化的构建目前已经很成熟,收率、品质均很理想,但在式ⅵ制备过程中,或因起始原料昂贵,没有商业化途径,或因合成路线长而收率低、或因使用价格昂贵的催化剂、或因为使用危险较大的试剂、或因为副反应多难以纯化等,这些因素导致了式ⅵ难以大规模工业化生产,因此只要解决式ⅵ的合成问题,就可以切实解决罗沙司他工业化生产问题,所以开发绿色、环保、收率高、成本低、选择性好的罗沙司他中间体ⅵ具有迫切的需要和广阔的前景。
[0039]
技术实现要素:
[0040]
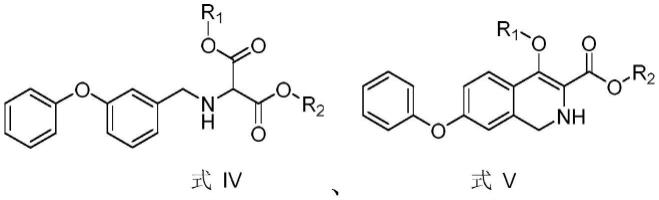
为了解决背景技术中提及的技术问题,本发明提供一种罗沙司他中间体(式ⅵ化合物)的制备方法,该方法原料易得、环保,路线新颖、合成线路短,制备得到的罗沙司他中间体的纯度好、收率高、成本低;同时,本发明还提供了在制备罗沙司他中间体(式ⅵ化合物)过程中的罗沙司他关键中间体(式ⅳ化合物和式
ⅴ
化合物)的具体结构式。
[0041]
本发明的目的可以通过以下技术方案实现:
[0042]
一种罗沙司他中间体的制备方法,合成路线如下所示:
[0043][0044]
其中,式
ⅷ
化合物、式ⅳ化合物、式
ⅴ
化合物、式ⅵ化合物中的r1、r2选自链状烷基、环状烷基或芳基,r1、r2可以是相同的基团,也可以是不同的基团;
[0045]
所述链状烷基为甲基、乙基、丙基,丁基、戊基、己基、庚基、辛基、壬基、癸基,优选为c1-c4烷基,更优选r1=r2为乙基;
[0046]
所述环状烷基为环丙基、环丁基、环戊基、环己基;
[0047]
所述芳基为未取代芳基或取代芳基,所述未取代芳基为苯基、苄基、萘基,所述取代芳基为甲苯基、二甲苯基、三甲苯基、三异丙基苯基、苯基乙基、苯基丙基、甲氧基苯基、氯苯基、硝基苯基;
[0048]
其中,式ⅲ化合物中的卤代基x为氯、溴、碘中的一种;
[0049]
包括以下步骤:
[0050]
s1、以间溴苯甲醛为原料,与苯酚进行取代反应,得到间苯氧基苯甲醛(式ⅰ);
[0051]
s2、间苯氧基苯甲醛经还原反应得到间苯氧基苄醇(式ⅱ);
[0052]
s3、间苯氧基苄醇经卤代,得到间苯氧基苄卤(式ⅲ);
[0053]
s4、间苯氧基苄卤与式
ⅷ
化合物发生取代反应,生成式ⅳ化合物;
[0054]
s5、式ⅳ化合物经关环,得到式
ⅴ
化合物;
[0055]
s6、式
ⅴ
化合物经水解、芳构化反应,得到式ⅵ化合物。
[0056]
进一步地,所述s1具体包括以下步骤:
[0057]
在氮气保护下,取间溴苯甲醛、苯酚、溶剂和碱催化剂混合均匀,高温下发生取代反应,得到间苯氧基苯甲醛。
[0058]
反应方程式如下所示:
[0059][0060]
进一步地,步骤s1中所述间溴苯甲醛与苯酚的摩尔比为1:1-1.4;所述溶剂为吡啶和喹啉按任意比混合,优选地,吡啶和喹啉的体积比为2-4:1;
[0061]
进一步地,所述碱催化剂为碳酸钾-氧化铜、碳酸钠-氧化铜、碳酸钾-氧化铁、碳酸钾-氧化锌、碳酸钠-氧化铁和碳酸钠-氧化锌中的任意一种,优选地,碱催化剂为碳酸钾和氧化铜按摩尔比1-1.5:1混合组成;
[0062]
进一步地,所述取代反应温度为150-190℃,优选地,反应温度为165-175℃;反应时间为12-48h,优选地,反应时间为26-30h。
[0063]
进一步地,步骤s1之后还包括间苯氧基苯甲醛的后处理,具体包括以下步骤:冷却,减压浓除溶剂,加入乙酸乙酯溶解,过滤,滤液分别使用1m hcl水溶液、氯化钠溶液洗涤,干燥,过滤,浓缩至干;
[0064]
进一步地,所述s2具体包括以下步骤:
[0065]
取间苯氧基苯甲醛加入反应溶剂中并降温至加料温度,再加入还原剂,低温反应,得到间苯氧基苄醇。
[0066]
反应方程式如下所示:
[0067]
[0068]
进一步地,步骤s2中所述反应溶剂为甲醇、乙醇或四氢呋喃,优选为甲醇;
[0069]
进一步地,所述间苯氧基苯甲醛与还原剂的摩尔比为1:1.1-1.5;
[0070]
进一步地,所述还原剂为硼氢化钠、氢化铝锂、硼氢化钾、硼氢化锂、硫代硼氢化钠、三仲丁基硼氢化锂中的一种,优选为硼氢化钠;
[0071]
进一步地,所述加料温度为0-30℃,优选为0-20℃;所述低温反应温度为0-50℃,优选为10-20℃;反应时间为1-24h,优选1-6h。
[0072]
进一步地,步骤s2之后还包括间苯氧基苄醇的后处理,具体包括以下步骤:加水淬灭,加入二氯甲烷萃取,干燥有机层,过滤,浓缩至干。
[0073]
进一步地,所述s3具体包括以下步骤:
[0074]
取间苯氧基苄醇加入反应溶剂中并降温至加料温度,滴加卤代试剂,低温反应,碳酸钠水溶液淬灭,调节ph,得到间苯氧基苄卤。
[0075]
反应方程式如下所示:
[0076][0077]
进一步地,步骤s3中所述反应溶剂为二氯甲烷;
[0078]
进一步地,所述间苯氧基苄醇与卤代试剂的摩尔比为2:1-1.4;
[0079]
进一步地,所述卤代试剂为ci2、br2、i2、soci2、pcl3、pcl5、pbr3、nbs中的一种,优选为pbr3;
[0080]
进一步地,所述加料温度为-20℃至10℃,优选为-10℃至0℃;所述低温反应的温度为0-20℃,优选为0-10℃;所述ph值为7-8。
[0081]
进一步地,步骤s3之后还包括间苯氧基苄卤的后处理,具体包括以下步骤:加入二氯甲烷萃取,干燥有机层,过滤,浓缩至干。
[0082]
进一步地,所述s4具体包括以下步骤:
[0083]
取间苯氧基苄卤、式
ⅷ
化合物、反应溶剂和碱,搅拌升温至反应温度,发生取代反应,监控至原料点消失,得到式ⅳ化合物。
[0084]
反应方程式如下所示:
[0085][0086]
进一步地,步骤s4中所述反应溶剂为二氯甲烷、乙腈或乙酸乙酯,优选为乙腈;
[0087]
进一步地,所述碱为碳酸钾或碳酸钠,优选为碳酸钠;
[0088]
进一步地,所述间苯氧基苄卤和式
ⅷ
化合物的摩尔比为1:1-2.5;所述反应温度为40-75℃,优选为50-60℃;反应时间为12-96h,优选为24-48h。
[0089]
进一步地,步骤s4之后还包括式ⅳ化合物的后处理,具体包括以下步骤:减压浓除溶剂,加入二氯甲烷溶解,水洗,干燥有机层,过滤,浓缩,过滤。
[0090]
进一步地,所述s5具体包括以下步骤:
[0091]
取式ⅳ化合物、反应溶剂、关环试剂,搅拌升温回流,反应监控至原料点消失,得到式
ⅴ
化合物。
[0092]
反应方程式如下所示:
[0093][0094]
进一步地,步骤s5中所述反应溶剂为n,n-二甲基甲酰胺、二甲基亚砜、甲苯、二甲苯、氯苯、二氯苯中的一种,优选为n,n-二甲基甲酰胺;
[0095]
进一步地,所述式ⅳ化合物与关环试剂的摩尔比为1:2-8,优选为1:3-6;
[0096]
进一步地,所述关环试剂为三氯氧磷或五氧化二磷,优选为三氯氧磷;
[0097]
进一步地,所述反应温度为150-160℃,优选为155-158℃;反应时间为1-48h,优选为1-12h。
[0098]
进一步地,步骤s5之后还包括式
ⅴ
化合物的后处理,具体包括以下步骤:减压浓除溶剂,收集固体,向固体中加入2m氢氧化钠溶液,搅拌,过滤,水洗,干燥。
[0099]
进一步地,所述s6具体包括以下步骤:
[0100]
取式
ⅴ
化合物、反应溶剂、酸性溶剂,搅拌升温回流,发生水解反应,降温后加入氧化剂,进行芳构化反应,得到式ⅵ化合物。
[0101]
反应方程式如下所示:
[0102][0103]
进一步地,步骤s6中所述反应溶剂为乙醇、甲醇、异丙醇、乙酸乙酯、氯仿、二氯甲烷、1,2-二氯乙烷、四氢呋喃、乙腈、2-甲基四氢呋喃中的一种,优选为乙醇;
[0104]
进一步地,所述式
ⅴ
化合物与酸性溶剂的摩尔比为1:1.1-2.5,优选为1:1.2-1.5;所述酸性溶剂为4m hcl溶液;
[0105]
进一步地,所述水解反应时间为1-24h,优选为1-5h;
[0106]
进一步地,所述氧化剂为二氧化锰、高锰酸钾、双氧水、过硫酸氢钾、叔丁基过氧化氢与碘化钾、叔丁基过氧化氢与碘化钠、六水合氯化铁,优选为二氧化锰;
[0107]
进一步地,所述式
ⅴ
化合物与氧化剂的摩尔比为1:2-8,优选为1:3-6;
[0108]
进一步地,所述芳构化反应温度为20-80℃,优选为30-50℃;反应时间为1-48h,优选为1-12h。
[0109]
进一步地,步骤s6之后还包括式ⅵ化合物的后处理,具体包括以下步骤:过滤,减压浓缩至干,加入乙腈,重结晶,过滤,干燥。
[0110]
进一步地,所述式ⅳ化合物和式
ⅴ
化合物为本发明的关键中间体。
[0111]
本发明的有益效果:
[0112]
本发明具体提出了一种罗沙司他中间体(式ⅵ化合物)的制备方法,是以间溴苯甲醛为原料,与苯酚取代,制备间苯氧基苯甲醛,经还原、卤代,得到间苯氧基苄卤,再与式
ⅷ
化合物发生取代反应,生成关键中间体(式ⅳ化合物),式ⅳ化合物经过成环反应,生成另一关键中间体(式
ⅴ
化合物),式
ⅴ
化合物再经水解和芳构化反应制得本发明的罗沙司他中间体(式ⅵ化合物)。本发明的合成线路新颖,所用的所有原料试剂易于获取或制备,且不使用高危险性和高污染性的试剂,安全环保,反应条件温和,操作方便可控,制备的罗沙司他中间体纯度好、收率高,成本优势明显,适合工业化生产,也为制备罗沙司他提供了新的思路。
具体实施方式
[0113]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0114]
实施例1
[0115]
一种罗沙司他中间体4-羟基-7-苯氧基-3-异喹啉甲酸乙酯的制备方法,包括以下步骤:
[0116][0117]
步骤s1、间苯氧基苯甲醛(式ⅰ)的合成
[0118]
氮气保护下,向圆底烧瓶中,依次加入吡啶(120ml)、喹啉(40ml)、碳酸钾(12.44g,90.0mmol)和氧化铜(7.16g,90.0mmol),搅拌均匀,加入苯酚(6.22g,66.0mol)和间溴苯甲醛(11.12g,60.0mmol)。升温至170℃,剧烈搅拌28h,冷却,减压浓除吡啶。加入乙酸乙酯(200ml)溶解,过滤,滤除不溶物,滤液使用1mhcl溶液、氯化钠水溶液依次洗涤,分取有机层,无水硫酸钠干燥,过滤,浓缩至干,得黄色油状物间苯氧基苯甲醛(10.82g,收率91%),直接用于下一步;
[0119]
反应方程式如下所示:
[0120][0121]
步骤s2、间苯氧基苄醇(式ⅱ)的合成
[0122]
向圆底烧瓶中,依次加入间苯氧基苯甲醛(10.0g,50.4mmol)、甲醇(50ml),在25-30℃下,搅拌15-20min。冷却至5-15℃,分批加入硼氢化钠(2.29g,60.5mmol)。控制温度10-15℃,搅拌2-3h。反应完成后,减压浓除溶剂,加水(50ml)淬灭,在25-30℃下,搅拌40-60min,使用二氯甲烷100mlx3萃取产物,合并有机层,无水硫酸钠干燥,浓缩至干,得到浅黄色油状物间苯氧基苄醇(9.49g,收率94%),直接用于下一步;
[0123]
反应方程式如下所示:
[0124][0125]
步骤s3、间苯氧基苄溴(式ⅲ)的合成
[0126]
向圆底烧瓶中,依次加入二氯甲烷(50ml)、间苯氧基苄醇(5.0g,25mmol),搅拌溶解,降温至-10℃~0℃,缓慢滴加pbr3(3.38g,12.5mmol),控温-5~0℃,滴完0-5℃反应1-2h,点板反应完(pe:ea=5:1),控温0-10℃,滴加10%碳酸钠溶液,调节ph至7-8,调毕,分取有机层,水层用二氯甲烷萃取,合并有机层,用水洗一次,干燥,浓缩得到液体间苯氧基苄溴5.99g,收率91%,纯度99.2%;
[0127]
反应方程式如下所示:
[0128]
步骤s4、n-4-苯氧基苄基-丙二酸二乙酯(式ⅳ)的合成
[0129]
向圆底烧瓶中,依次加入间苯氧基苄溴(13.16g,50mmol)、2.0eq氨基丙二酸二乙酯(17.52g,100mmol)、乙腈(130ml)和碳酸钠(13.25g,125mmol),升温至55℃,反应24-48h,点板监测原料反应完全(pe:ea=1:1),减压浓缩,浓除乙腈,加入二氯甲烷(150ml),水(100ml)洗涤有机层两次,无水硫酸钠干燥,过滤,浓缩,得到n-4-苯氧基苄基-丙二酸二乙酯,15.55g,收率87%,纯度97.5%;
[0130]
反应方程式如下所示:
[0131][0132]
步骤s5、4-烷氧基-7-苯氧基-1,2-二氢异喹啉甲酸乙酯(式
ⅴ
)的合成
[0133]
向圆底烧瓶依次加入n-4-苯氧基苄基-丙二酸二乙酯(13.22g,37mmol)、dmf(50ml)、三氯氧磷(28.37g,185mmol),升温至158℃,回流反应6h,冷却至室温,减压浓除溶剂,收集固体,向固体中加入2.0m的氢氧化钠300ml搅拌15min,过滤,滤饼经500ml水洗涤两次得湿品,干燥得类白色固体4-烷氧基-7-苯氧基-1,2-二氢异喹啉甲酸乙酯11.18g,收率89%,纯度99.4%;
48h,点板监测原料反应完全(pe:ea=1:1),减压浓缩,浓除乙腈,加入二氯甲烷(150ml),水(100ml)洗涤有机层两次,无水硫酸钠干燥,过滤,浓缩,得到n-4-苯氧基苄基-丙二酸二环丙酯16.21g,收率85%,纯度98.7%;
[0146]
反应方程式如下所示:
[0147][0148]
步骤s5、4-烷氧环丙基-7-苯氧基-1,2-二氢异喹啉甲酸环丙酯(式
ⅴ
)的合成
[0149]
向圆底烧瓶依次加入n-4-苯氧基苄基-丙二酸二环丙酯(14.11g,37mmol)、二甲苯(60ml)、五氧化磷(26.26g,185mmol),升温130-140℃,反应6h,冷却至室温,减压浓除溶剂,收集固体,向固体中加入2.0m的氢氧化钠300ml搅拌15min,过滤,滤饼经500ml水洗涤两次得湿品,干燥得类白色固体4-烷氧环丙基-7-苯氧基-1,2-二氢异喹啉甲酸环丙酯11.70g,收率87%,纯度97.0%;
[0150]
反应方程式如下所示:
[0151][0152]
步骤s6、4-羟基-7-苯氧基-3-异喹啉甲酸环丙酯(式ⅵ)的合成
[0153]
向圆底烧瓶依次加入乙醇(60ml)、4-烷氧环丙基-7-苯氧基-1,2-二氢异喹啉甲酸环丙酯(10.72g,29.5mmol),滴加4m hcl溶液(36.8g,35.4mmol),回流反应1-2h,降温至50℃以下,加入二氧化锰(9.56g,110mmol),30-50℃反应,tlc监测原料反应完全。过滤,减压浓除至基本无馏分,加入乙腈溶解,重结晶,过滤,干燥,得到类白色固体4-羟基-7-苯氧基-3-异喹啉甲酸环丙酯8.25g,收率87%,纯度98.1%.4-羟基-7-苯氧基-3-异喹啉甲酸乙酯分子式为c19h15no4,lc-ms测得分子离子峰m/z为322.1,[m h]
与理论值一致,元素分析:c,71.02;h,4.71;n,4.36;o,19.92。
[0154]
反应方程式如下所示:
[0155][0156]
实施例3
[0157]
一种罗沙司他中间体4-羟基-7-苯氧基-3-异喹啉甲酸环酚酯的制备方法,包括以
下步骤:
[0158][0159]
步骤s1、s2、s3与实施例1中的步骤s1、s2、s3均相同;
[0160]
步骤s4、n-4-苯氧基苄基-1,3-二苯氧基-2-氨基-丙二酸(式ⅳ)的合成
[0161]
向圆底烧瓶中,依次加入间苯氧基苄溴(13.16g,50mmol)、1,3-二苯氧基-2-氨基-丙二酸(27.13g,100mmol)、乙腈(150ml)和碳酸钾(17.28g,125mmol),升温至55℃,反应24-48h,点板监测原料反应完全(pe:ea=1:1),减压浓缩,浓除乙腈,加入二氯甲烷(150ml),水(100ml)洗涤有机层两次,无水硫酸钠干燥,过滤,浓缩,得到n-4-苯氧基苄基-1,3-二苯氧基-2-氨基-丙二酸19.73g,收率87%,纯度98.2%;
[0162]
反应方程式如下所示:
[0163][0164]
步骤s5、4,7-二苯氧基-1,2-二氢异喹啉甲酸酚酯(式
ⅴ
)的合成
[0165]
向圆底烧瓶依次加入n-4-苯氧基苄基-1,3-二苯氧基-2-氨基-丙二酸(16.78g,37mmol)、二甲苯(80ml)、三氯氧磷(28.37g,185mmol),升温130-140℃,反应8h,冷却至室温,减压浓除溶剂,收集固体,向固体中加入2.0m的氢氧化钠300ml搅拌20min,过滤,滤饼经500ml水洗涤两次得湿品,干燥得类白色固体4,7-二苯氧基-1,2-二氢异喹啉甲酸酚酯13.21g,收率82%,纯度99.2%;
[0166]
反应方程式如下所示:
[0167][0168]
步骤s6、4-羟基-7-苯氧基-3-异喹啉甲酸环酚酯(式vi)的合成
[0169]
向圆底烧瓶依次加入乙醇(60ml)、4,7-二苯氧基-1,2-二氢异喹啉甲酸酚酯(12.85g,29.5mmol),滴加4mhcl溶液(36.8g,35.4mmol),回流反应1-2h,降温至50℃以下,加入叔丁基过氧化氢(9.91g,110mmol),ki(1.3g,7.8mmol),30-50℃反应,tlc监测原料反应完全。过滤,减压浓除至基本无馏分,加入乙腈溶解,重结晶,过滤,干燥,得到类白色固体4-羟基-7-苯氧基-3-异喹啉甲酸环酚酯9.38g,收率89%,纯度99.1%。4-羟基-7-苯氧基-3-异喹啉甲酸环酚酯分子式为c22h15no4,lc-ms测得分子离子峰m/z为358.3,[m h]
与理论值一致,元素分析:c,73.94;h,4.23;n,3.92;o,17.91。
[0170]
反应方程式如下所示:
[0171][0172]
实施例4
[0173]
一种罗沙司他中间体4-羟基-7-苯氧基-3-异喹啉甲酸乙酯的制备方法,包括以下步骤:
[0174][0175]
步骤s1、s2与实施例1中的步骤s1、s2均相同;
[0176]
步骤s3、间苯氧基苄氯(式ⅲ)的合成
[0177]
向圆底烧瓶中,依次加入二氯甲烷(50ml)、间苯氧基苄醇(5.0g,25mmol),搅拌溶解,降温至-15℃~0℃,缓慢滴加pcl5(2.62g,12.5mmol),控温-5~0℃,滴完0-10℃反应1-2h,点板反应完(pe:ea=5:1),控温0-10℃,滴加10%碳酸钠溶液,调节ph至7-8,调毕,分取有机层,水层用二氯甲烷萃取,合并有机层,用水洗一次,干燥,浓缩得到液体间苯氧基苄氯4.92g,收率90%,纯度99.0%;
[0178]
反应方程式如下所示:
[0179][0180]
步骤s4、n-4-苯氧基苄基-1-甲基-2-乙基-2-氨基-丙二酸(式ⅳ)的合成
[0181]
向圆底烧瓶中,依次加入间苯氧基苄氯(10.93g,50mmol)、2.0eq1-甲基-3乙基-2氨基丙二酸(16.12g,100mmol)、二氯甲烷(130ml)和碳酸钾(17.28g,125mmol),升温至60℃,反应24-48h,点板监测原料反应完全(pe:ea=1:1),减压浓缩,浓除乙腈,加入二氯甲烷(150ml),水(100ml)洗涤有机层两次,无水硫酸钠干燥,过滤,浓缩,得到n-4-苯氧基苄基-1-甲基-2-乙基-2-氨基-丙二酸14.42g,收率84%,纯度98.7%;
[0182]
反应方程式如下所示:
[0183][0184]
步骤s5、4-烷氧甲基-7-苯氧基-1,2-二氢异喹啉甲酸乙酯(式
ⅴ
)的合成
[0185]
向圆底烧瓶依次加入n-4-苯氧基苄基-1-甲基-2-乙基-2-氨基-丙二酸(12.71g,37mmol)、氯苯(60ml)、五氧化磷(26.26g,185mmol),升温至回流,反应6h,冷却至室温,减压浓除溶剂,收集固体,向固体中加入2.0m的氢氧化钠300ml搅拌15min,过滤,滤饼经500ml水洗涤两次得湿品,干燥得类白色固体4-烷氧甲基-7-苯氧基-1,2-二氢异喹啉甲酸乙酯10.83g,收率90%,纯度99.0%;
[0186]
反应方程式如下所示:
[0187][0188]
步骤s6、4-羟基-7-苯氧基-3-异喹啉甲酸乙酯(式ⅵ)的合成
[0189]
向圆底烧瓶依次加入乙醇(60ml)、4-烷氧甲基-7-苯氧基-1,2-二氢异喹啉甲酸乙酯(9.60g,29.5mmol),滴加4mhcl溶液(36.8g,35.4mmol),回流反应1-2h,降温至50℃以下,加入30%双氧水(12.47g,110mmol),30-50℃反应,tlc监测原料反应完全。过滤,减压浓除
至基本无馏分,加入乙腈溶解,重结晶,过滤,干燥,得到类白色固体4-羟基-7-苯氧基-3-异喹啉甲酸乙酯8.12g,收率89%,纯度99.1%,4-羟基-7-苯氧基-3-异喹啉甲酸乙酯分子式为c18h15no4,lc-ms测得分子离子峰m/z为310.1,[m h]
与理论值一致,元素分析c,69.89;h,4.89;n,4.53;o,20.69。
[0190]
反应方程式如下所示:
[0191][0192]
在说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
[0193]
以上内容仅仅是对本发明所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离发明或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。