一种抗体偶联药物连接子set0568的合成方法
技术领域
1.本发明涉及一种抗体偶联药物连接子的合成方法,尤其涉及一种set0568的合成方法,属于药物化学技术领域。
背景技术:
2.抗体药物偶联物(antibody drug conjugate,简称adc)是一类新型的抗肿瘤药物,其原理是将细胞毒素连接在抗体上,通过抗体对癌细胞表面特定抗原的识别,通过内吞作用进入癌细胞,从而将细胞毒素运输到靶点,达到靶向性治疗恶性肿瘤的目的。
3.adc与传统的小分子抗肿瘤药物相比,因能借助抗体的靶向识别性与毒素的高活性,故更具备特异性和有效性。
4.adc包括三个不同的组成部分,即抗体、连接子和细胞毒素。抗体实现靶向性,连接子保证adc在血液转运过程中的稳定性,而到达作用靶点后,毒素发挥对癌细胞的杀伤作用。根据作用机制的不同,适用于adc的毒素分为微管类抑制剂(microtubule inhibitors),dna损伤剂(dna damaging agents),rna聚合酶抑制剂(rna polymerase inhibitors)等。
5.目前,市场上销售和临床试验中的adc所采用的毒素主要为微管类抑制剂,主要包括dna拓扑异构酶i(topoisomerase i)抑制剂设计的化合物,比如dxd,以及基于美登素(maytansine-based)设计的化合物,比如dm1和dm4;dna拓扑异构酶ii(topoisomerase ii)抑制剂设计的化合物,比如一种高效的蒽环类新霉素代谢产物pnu-159682具有出色的细胞毒性,蒽环类药物是目前应用最广泛的化疗药物之一,特别是在治疗侵袭性非霍奇金淋巴瘤(nhl)方面非常有效。
6.连接子方面,主要应用的为不可裂解型,如缬氨酸-瓜氨酸(valine-citriline)和环己基甲酸(mcc),经过溶酶体水解后,药物仍然具有活性,并通过连接区与某个氨基酸残基结合在一起。
7.抗体药物偶联物的形成方式有多种。既可以通过抗体上的氨基或巯基和药物连接子进行化学反应偶联,也可以对抗体进行修饰,在抗体上引入特定功能基后,再和药物连接子进行化学反应偶联或酶催化反应偶联。
技术实现要素:
8.有鉴于此,本发明目的是为了克服现有技术的不足而提供一种用于抗体偶联药物的连接子set0568(mc-vc-pab-dmea-pnu159682)的合成方法,成功实现了set0568的高效合成,易于放大,便于商业化生产。
9.为达到上述目的,本发明采用的技术方案是:一种抗体偶联药物连接子set0568的合成方法,该合成方法包括以下步骤:
10.将化合物vc-pab(set0568-1)溶解在第一溶剂中,搅拌30min~60min,加入mc-osu搅拌1h-2h,加入第二溶剂进行后处理,得到(set0568-2)mc-vc-pab;
11.将化合物mc-vc-pab溶于第一溶剂中搅拌,内温降至15℃~20℃时加入dnpc,搅拌10min~20min,加入dipea,搅拌15h~18h,加入第二溶剂进行后处理,固体析出,抽滤,烘干,得到(set0568-3)mc-vc-pab-pnp;
12.将化合物(set0568-5)pnu-tbs溶于第三溶剂中,加入tbaf,降温至-5~0℃搅拌,反应20min-30min,分液萃取,干燥浓缩,得到(set0568-6)pnu-159682;
13.将pnu-159682与dnpc溶于第四溶剂,冰水搅拌降温0℃~5℃,加入三乙胺,保温0℃~5℃搅拌30min~60min,得到中间体(set0568-7)pnu-pnp,加入n,n
′‑
二甲基乙二胺,0~5℃左右搅拌1~2h,后处理,反应液浓缩,中压反相柱纯化,冻干,得到化合物(set0568-8)pnu-dmea;
14.在氮气保护下,将所述化合物pnu-dmea溶于第一溶剂中,加入mc-vc-pab-pnp,冰水浴降温至内温0~10℃,搅拌10~15min,加入dipea,继续进行冰水浴反应2~3h,后处理,冻干得化合物set0568。
15.本发明提供的抗体偶联药物的连接子set0568(mc-vc-pab-dmea-pnu159682)的合成方法合成的set0568为蒽环类衍生物,具有如下结构。
[0016][0017]
上述用于抗体偶联药物的毒素连接子分子set0568(mc-vc-pab-dmea-pnu159682)的合成方法,具有如下制备步骤:
[0018][0019]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备mc-vc-pab的步骤。
[0020]
优选地,制备mc-vc-pab时,vc-pab与mc-osu的混合摩尔比为1.0eq∶1.0eq-1.5eq。
[0021]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备mc-vc-pab-pnp的步骤。
[0022]
优选地,制备mc-vc-pab-pnp时,所述mc-vc-pab与所述dnpc的混合摩尔比为1.0eq∶1.5eq-2.5eq。
[0023]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备pnu-159682的步骤。
[0024]
优选地,制备pnu-159682时,所述pnu-tbs与所述tbaf的混合摩尔比为1.0eq∶1.0eq-1.5eq。
[0025]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备pnu-dmea的步骤。
[0026]
优选地,制备pnu-dmea时,所述pnu-159682、dnpc、三乙胺、n,n
′‑
二甲基乙二胺的混合摩尔比为1.0eq∶1.5eq-3.0eq∶2.0eq-3.5eq∶1.0eq-2.5eq。
[0027]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备set0568的步骤。
[0028]
优选地,制备set0568时,所述pnu-dmea、mc-vc-pab-pnp和dipea的混合摩尔比为1.0eq∶1.0-1.5eq∶2.0eq-3.0eq。比如可以为1.0eq∶1.0-1.4eq∶2.0eq-2.5eq,1.0eq∶1.0-1.3eq∶2.0eq-2.2eq;优选为1.0eq∶1.1eq∶2.0eq。
[0029]
优选地,第一溶剂为dmf或dma;第二溶剂为dcm、ea、pe、mtbe、乙醚中的一种或几种的混合溶剂;第三溶剂为dcm或thf;第四溶剂为dcm、thf或dmf。
[0030]
这里需要说明的是,第一溶剂,第二溶剂、第三溶剂和第四溶剂在每一步骤的使用中,还可以相同也可以不同,比如,制备mc-vc-pab时,将化合物vc-pab溶解在第一溶剂中,
制备mc-vc-pab-pnp时,将化合物mc-vc-pab溶于第一溶剂中,这两步中的第一溶剂可以相同也可以不同,可以均采用dmf或dma,也可以采用dmf溶解vc-pab,采用dma溶解mc-vc-pab。
[0031]
由于上述技术方案运用,本发明与现有技术相比具有下列优点:本发明的抗体偶联药物连接子set0568的合成方法新颖高效,易于放大和商业化生产。
[0032]
主要附图说明
[0033]
图1为化合物mc-vc-pab的hplc谱图。
[0034]
图2为化合物mc-vc-pab-pnp的hplc谱图。
[0035]
图3为化合物mc-vc-pab-pnp的lcms谱图。
[0036]
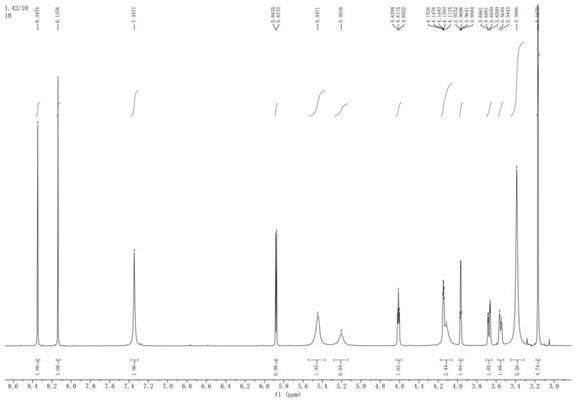
图4为化合物mc-vc-pab-pnp的hnmr谱图。
[0037]
图5为化合物set0568-7的lcms谱图。
[0038]
图6为化合物pnu-dmea的lcms谱图。
[0039]
图7为化合物mc-vc-pab-dmea-pnu159682的hplc谱图。
[0040]
图8为化合物mc-vc-pab-dmea-pnu159682的lcms谱图。
[0041]
图9为化合物mc-vc-pab-dmea-pnu159682的hnmr谱图。
[0042]
图10为对比例1的化合物的hplc谱图。
[0043]
图11为对比例1的set0568-4的hplc谱图。
具体实施方式
[0044]
本发明的抗体偶联药物连接子set0568的合成方法中包括制备set0568-2(mc-vc-pab)的步骤。具体按照以下方式进行:
[0045][0046]
在本发明的一具体实施方式中,制备mc-vc-pab时,vc-pab与mc-osu的混合摩尔比为1.0eq∶1.0-1.5eq。比如可以为1.0eq∶1.2eq。
[0047]
在本发明的一具体实施方式中,将化合物vc-pab溶解在第一溶剂中,搅拌30min~60min。比如可以搅拌33min、35min、38min、40min、45min、52min、55min、58min.
[0048]
在本发明的一具体实施方式中,制备mc-vc-pab时的后处理包括以下步骤:
[0049]
向滤液中加入dcm,搅拌30min~35min后直接抽滤,滤饼用dcm打浆30min~35min,再次抽滤,烘干,得黄色固体。
[0050]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备(set0568-3)mc-vc-pab-pnp的步骤。具体按照以下方式进行:
[0051][0052]
在本发明的一具体实施方式中,制备mc-vc-pab-pnp时,mc-vc-pab与dnpc的混合摩尔比为1.0eq∶1.5-2.5eq。比如可以为1.0eq∶1.0eq-1.3eq,1.0eq∶1.1eq-1.3eq,1.0eq∶1.1eq-1.2eq;优选为1.0eq:1.2eq。
[0053]
在本发明的一具体实施方式中,制备mc-vc-pab-pnp时,后处理包括以下步骤:
[0054]
反应液过滤,向滤液中加入dcm,搅拌30min~35min后,加入mtbe搅拌30min~35min后抽滤。滤饼用dcm搅拌20min~30min后加入mtbe搅拌30min后抽滤。滤饼烘干,得固体。
[0055]
制备mc-vc-pab-pnp时,所述mc-vc-pab与所述dnpc的混合摩尔比为1.0eq∶1.5eq-2.5eq。比如可以为1.0eq∶1.5eq-2.3eq,1.0eq∶1.8eq-2.3eq,1.0eq∶1.8eq-2.1eq;优选为1.0eq∶2.0eq。
[0056]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备(set0568-6)pnu-159682的步骤。具体按照以下方式进行:
[0057][0058]
在本发明的一具体实施方式中,制备pnu-159682时,所述pnu-tbs与所述tbaf的混合摩尔比为1.eq∶1.0-1.5eq。比如可以为1.0eq∶1.0eq-1.4eq,1.0eq∶1.1eq-1.4eq,1.0eq∶1.2eq-1.4eq;优选为1.0eq∶1.3eq。
[0059]
在本发明的一具体实施方式中,制备pnu-159682时,分液萃取包括以下步骤:
[0060]
加入水(比如20ml)和dcm(比如50ml),其中,水与dcm的体积比为20∶50,水相采用dcm(比如50ml)反萃取,合并有机相,完成萃取。
[0061]
在本发明的一具体实施方式中,制备pnu-159682时,干燥浓缩时,可以采用无水硫酸钠进行干燥。
[0062]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备pnu-dmea(set0568-8)的步骤。具体包括以下步骤:
[0063][0064]
在本发明的一具体实施方式中,制备pnu-dmea时,所述pnu-159682、dnpc、三乙胺、n,n
′‑
二甲基乙二胺的混合摩尔比为1.0eq∶1.5eq-3.0eq∶2.0eq-3.5eq∶1.0eq-2.5eq。比如可以为1.0eq∶1.8eq-2.5eq∶2.8eq-3.2eq∶1.2eq-1.8eq。优选为1.0eq∶2.0eq∶3.0eq∶1.5eq。
[0065]
在本发明的一具体实施方式中,制备pnu-dmea时,后处理采用旋蒸除去反应溶剂。然后粗品set0568-8中压反相制备纯化,120g反相柱,波长234nm,水/乙腈体系纯化,40%~50%乙腈比例。
[0066]
在本发明的抗体偶联药物连接子set0568的合成方法中包括制备set0568的步骤。
[0067][0068]
在本发明的一具体实施方式中,制备set0568时,所述pnu-dmea、mc-vc-pab-pnp和dipea的混合摩尔比为1.0eq∶1.0-1.5eq∶2.0-3.0eq。比如可以为1.0eq∶1.0-1.4eq∶2.0eq-2.5eq,1.0eq∶1.0-1.3eq∶2.0eq-2.2eq;优选为1.0eq∶1.1eq∶2.0eq。
[0069]
在本发明的一具体实施方式中,制备set0568时,后处理是中压反相制备纯化,波长234nm,流速50ml/min,0.05%nh4hco3水/乙腈,40~50%乙腈比例出产品。
[0070]
在本发明的一具体实施方式中,第一溶剂为dmf或dma;第二溶剂为dcm、ea、pe、mtbe、乙醚中的一种或几种的混合溶剂;第三溶剂为dcm或thf;第四溶剂为dcm、thf或dmf。
[0071]
下面将结合对本发明优选实施方案进行详细说明。
[0072]
实施例1
[0073]
化合物(set0568-2)mc-vc-pab的合成
[0074]
将set0568-1(vc-pab)131g(1.0eq),加入到2l三口瓶中,加入dmf1.5l,若搅拌30分钟-60分钟不澄清,布氏漏斗过滤,滤去不溶物,滤液转移至2l反应瓶中,加入mc-osu127.2g(1.2eq),搅拌一小时后,hplc监测反应完全。后处理:滤液转移至10l塑料桶中,加入dcm(6l),搅拌30~35分钟后直接抽滤,滤饼用dcm(2l)打浆30~35分钟后再次抽滤,烘干,得黄色固体135g。收率:68.2%。化合物(set0568-2)mc-vc-pab的hplc谱图如图1所示。
[0075]
化合物(set0568-3)mc-vc-pab-pnp的合成
[0076]
取set0568-2(mc-vc-pab,135g,1.0eq)溶于dmf中,内温降至15~20℃时加入dnpc(143.4g,2.0eq),搅拌15min,再加入dipea(3.1g,0.1eq),搅拌15~18h。取样hplc监控,无原料则反应完全。后处理:反应液过滤,将滤液转移至20l塑料桶中,加入dcm(5.8l),搅拌30~35min后,加入mtbe(3.9l)搅拌30~35min后抽滤。滤饼用1.5l dcm搅拌20~30min后加入
3l mtbe搅拌30min后抽滤。滤饼烘干,得固体147g,收率:84.5%,lcms:737.77,检测结果:738.31[m h] 。
[0077]
化合物(set0568-3)mc-vc-pab-pnp的hplc谱图(图2)、lcms谱图(图3)和hnmr谱图(图4)所示。
[0078]
化合物(set0568-6)pnu-159682的合成
[0079]
取化合物set0568-5500mg(pnu-tbs,1.0eq)于50ml单口瓶,加入thf5ml,降温至-5~0℃搅拌,取tbaf 0.86ml(1m in thf,1.3eq)滴加至反应瓶内,反应约20min,lcms检测原料反应完全,加入水20ml及dcm 50ml,水相50ml dcm反萃,合并有机相,无水硫酸钠干燥,抽滤,滤液浓缩得到(set0568-6)pnu-159682,直接用于下一步。
[0080]
化合物set0568-8pnu-dmae的合成
[0081]
将上一步得到的set0568-6(pnu-159682粗品,1.0eq),dnpc 400mg(2.0eq),加入dcm 10ml,冰水搅拌降温0~5℃左右,三乙胺/tea200mg(3.0eq)加入反应体系,保温0~5℃搅拌30~60min,lcms检测,原料转化完全,得到中间体set0568-7,lcms:806.73,检测结果807.52[m h] ,直接进行下一步。
[0082]
取n,n
′‑
二甲基乙二胺86.5mg(dmea,1.5eq)加入反应体系,0~5℃左右搅拌1~2h。hplc/lc-ms检测,原料转化完毕。后处理:旋蒸除去反应溶剂。粗品set0568-8中压反相制备纯化,120g反相柱,波长234nm,水/乙腈体系纯化,40~50%乙腈比例出产品。收集产品冻干,得到(set0568-8)pnu-dmea,231mg,hplc:94%,lcms:755.77,检测结果:756.81[m h] 。
[0083]
set0568-7的lcms谱图如图5所示。set0568-8的lcms谱图如图6所示。
[0084]
化合物(set0568)mc-vc-pab-dmea-pnu159682的合成
[0085]
在室温和氮气保护下,将205mg set0568-8(pnu-dmae,1.0eq)溶于2ml dmf中,加入set0568-3220mg(mc-vc-pab-pnp,1.1eq),再进行冰水浴降温至内温0~10℃,搅拌10~15min后加入dipea(2.0eq,70mg),继续进行冰水浴反应2~3h,反应完全。后处理:中压反相制备纯化,波长234nm,流速50ml/min,0.05%nh4hco3水/乙腈,40~50%乙腈比例出产品,收集产品,冻干得到set0568(mc-vc-pab-dmea-pnu159682)54mg,hplc:92.2%,lcms:1354.43,检测结果1355.31[m h] 。
[0086]
化合物mc-vc-pab-dmea-pnu159682的hplc谱图(图7)、lcms谱图(图8)和hnmr谱图(图9)。
[0087]1h nmr(400mhz,dmso)δ13.99(s,1h),13.23(s,1h),9.96(s,1h),8.05(d,j=7.3hz,1h),7.92-7.86(m,2h),7.79(d,j=8.6hz,1h),7.66-7.54(m,4h),7.27(s,2h),6.99(s,2h),5.97(t,j=5.7hz,1h),5.47(s,1h),5.39(s,2h),5.20(d,j=20.3hz,2h),5.06(d,j=17.7hz,1h),4.95(d,j=15.8hz,3h),4.58(d,j=1.7hz,1h),4.38(dd,j=13.3,7.9hz,1h),4.23(d,j=1.8hz,1h),4.21-4.14(m,2h),3.98(s,4h),3.92(s,1h),3.66(t,j=9.0hz,1h),3.50(d,j=11.7hz,1h),3.43-3.37(m,4h),3.31(s,3h),3.06-2.81(m,12h),2.69(dd,j=32.4,20.5hz,4h),2.35(d,j=14.3hz,1h),2.13(ddd,j=35.0,17.5,10.3hz,4h),1.96(dd,j=13.4,6.6hz,1h),1.68(s,3h),1.57-1.36(m,8h),1.19(dd,j=18.5,10.7hz,8h),0.83(dd,j=12.2,6.7hz,8h).
[0088]
对比例1
[0089]
本对比例提供了抗体偶联药物连接子set0568的合成方法,具体包括以下步骤:
[0090][0091]
化合物(set0568-4)mc-vc-pab-dmea的合成
[0092]
取set0568-3(mc-vc-pab-pnp,1.5g,1.0eq)溶于20ml dmf中,内温20~25℃时加入n,n
′‑
二甲基乙二胺168mg(dmea,1.01eq),搅拌15min,再加入1-羟基苯并三唑381mg(hobt,1.5eq),搅拌5~10min。向体系中加入dipea(2.0eq,480mg),室温搅拌,hplc检测无原料则反应完全,见hplc谱图(图10),体系已经有二聚体和set0568-3的副产物生成。后处理:反应液加入mtbe100ml,打浆搅拌30~60min,布氏漏斗过滤,set0568-4见hplc谱图(图11),hplc纯度只有54.2%,可以看出反应体系持续有大量的副产物生成,产品并不稳定,收率明显偏低。
[0093]
对比例2
[0094]
本对比例提供了抗体偶联药物连接子set0568的合成方法,具体包括以下步骤:
[0095][0096]
化合物set0568-4的合成收率很低,反应有很多副反应发生,如:水解开环,二聚体等。所以,我们改变合成策略,换用本对比例2,用保护基团保护的dmea(n,n
′‑
二甲基乙二胺)set0568-4a,再通过脱保护拿到高收率的set0568-4。
[0097]
在实际的合成实践中,boc保护的set0568-4(set0568-5)的反应转化率和收率很高,但是在脱保护阶段boc保护的set0568-4,尝试不同的脱保护试剂如:hcl/二氧六环、hcl/etoac、hcl/et2o、tfa、tsoh/thf-ch2c12、me3sii/chcl3或者ch3cn,收率都偏低,并且有很多副反应发生;
[0098]
将保护基团换成fmoc后,fmoc保护的set0568-4(set0568-5)的反应转化率和收率同样很高,但是在脱保护阶段fmoc保护的set0568-4,尝试不同的脱保护试剂如:二乙胺、二异丙胺、1,8-二偶氮杂双螺环[5.4.0]十一-7-烯(dbu)、哌啶、乙醇胺、环己胺、吗啡啉、吡咯烷酮2等反应有很多副反应发生如水解、断链等副反应,收率偏低。
[0099]
所以对比例2最终的总收率依然很低,难以放大生产和商业化。
[0100]
上述实施例和对比例说明,为了本发明的(set0568)mc-vc-pab-dmea-pnu159682和合成方法可以高效合成,易于放大,便于商业化生产。
[0101]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。