噻吩并吡啶酮衍生物在治疗常染色体显性多囊肾病adpkd中的用途
技术领域
1.本发明涉及噻吩并吡啶酮衍生物在治疗常染色体显性多囊肾病(adpkd)中的用途。
背景技术:
2.常染色体显性多囊肾病(adpkd)是最常见的单基因遗传性肾病,个体发病率为五百分之一到千分之一(p.igarashi等,j.am.soc.nephrol.,13(9),2384-2398,2002)。编码多囊蛋白(polycystin)-1和多囊蛋白-2的pkd1和pkd2基因中的突变分别造成85%和15%的adpkd病例。adpkd的特征在于是双侧肾大量囊肿的进行性发展和生长,导致许多异常,如急慢性疼痛、肾结石和尿路感染,其中最重要的是肾功能丧失。大约70%的adpkd患者在中位年龄58岁时进展为终末期肾病(esrd)(e.m.spithoven等,kidney int.,86(6),1244-1252,2014)。
3.对adpkd已经提出了许多疗法,针对驱动囊肿生长的各种信号传导通路。它们包括加压素受体抑制剂如托伐普坦(tolvaptan),生长抑素类似物如奥曲肽(octreotide)和兰瑞肽(lanreotide),普伐他汀(pravastatin)和酪氨酸激酶抑制剂如特伐替尼(tesevatinib)。其中,托伐普坦是fda批准的第一种药物治疗,用于减缓有快速进展adpkd风险的成人的肾功能衰减。然而,由于其潜在的肝细胞毒性,托伐普坦需要经常监测用其治疗的患者的肝功能。这些化合物靶向被认为涉及adpkd发病机制的关键过程(增殖或分泌)之一。
4.近期表明,参与囊液分泌的囊性纤维化跨膜传导调节蛋白(cftr)和可以驱动囊肿上皮细胞增殖的哺乳动物雷帕霉素靶蛋白(mtor)通路可能都受到amp活化蛋白激酶(ampk)的负调控。因此,ampk的激活剂可以同时靶向adpkd的这两个关键过程。在这种情况下,建议使用已知的ampk间接激活剂二甲双胍来同时抑制囊肿扩增的分泌和增殖成分,从而减缓肾囊肿生成(v.takiar等,pnas,第108卷,第6号,2462-2467,2011)。然而,已知二甲双胍作为副作用会引起乳酸性酸中毒。由于二甲双胍由肾脏清除,慢性肾病被认为是该并发症的潜在诱因。此外,担心二甲双胍在人肾脏中的治疗性ampk激活可能需要二甲双胍的高口服剂量,至少因为其生物利用度降低。
5.已经提出,除二甲双胍外,其它ampk激活剂也可以用于治疗adpkd。因此,wo2017/011917公开了许多由宽泛的化学式定义的噻吩并吡啶酮,但没有说明它们可以如何合成以及包括哪些具体化合物。
6.最近,水杨酸盐的前药二聚体和直接ampk激活剂—双水杨酯,被证明比二甲双胍更有效地减轻突变小鼠的囊性肾病严重程度(w.n.leohnard等,ebiomedicine,第47卷,436-445,2019)。双水杨酯通过与ampkβ1同种型的药物结合结构域直接相互作用来激活ampk。然而,尽管与二甲双胍相比,双水杨酯的生物利用度更好,但其提供治疗效果所需的口服剂量仍然高。
7.因此,仍然需要与为此目的所提出的化合物相比,将会以较低的剂量和/或更高的功效和/或减少的副作用对治疗adpkd有用的替代化合物。
8.发明人现已表明,特定的噻吩并吡啶酮衍生物可以用于以低于其它ampk激活剂的口服剂量治疗adpkd。这些化合物在wo2014/001554中被宽泛地公开为ampk激活剂,但迄今尚未有建议将它们用于治疗adpkd。它们已被证明是包含β1亚基在内的各种ampk同种型的直接激活剂。
技术实现要素:
9.本发明涉及一种式(i)所示的噻吩并吡啶酮衍生物,或其药学上可接受的盐和/或溶剂合物,或包含它们的药物组合物,在治疗adpkd中的用途:
[0010][0011]
其中:
[0012]
r1表示氢原子或卤素原子,
[0013]
r2表示茚满基或四氢化萘基基团,所述基团被一个或多个(例如2、3、4、5、6或7个)选自卤素原子、烷基基团、羟基、烷氧基基团、氨基、单或二烷基氨基基团、羧基基团、烷氧基羰基基团、单或二烷基氨基羰基基团、甲酰胺、氰基、烷基磺酰基和三氟甲基基团中的基团取代或不被取代,
[0014]
r3表示芳基基团,所述基团被一个或多个(例如2、3、4或5个)选自卤素原子、烷基基团、羟基、烷氧基基团、芳烷氧基基团、氨基、单或二烷基氨基基团、羧基基团、烷氧基羰基基团、单或二烷基氨基羰基基团、甲酰胺、氰基、烷基磺酰基和三氟甲基基团中的原子或基团取代或不被取代。
[0015]
本发明还涉及一种治疗adpkd的方法,所述方法包括向有此需要的对象施用有效量的如上所述的噻吩并吡啶酮衍生物、或包含有效量的如上所述的噻吩并吡啶酮衍生物和药学上可接受的支撑体的药物组合物。
[0016]
本发明还涉及如上所述的噻吩并吡啶酮衍生物或包含它的药物组合物在制造用于治疗adpkd的药物中的用途。
附图说明
[0017]
图1显示了与对照培养基相比较,在10μm、100μm和1mm二甲双胍或10μm、25μm或50μm pxl770存在下,由pimdck细胞形成的囊肿的体积。
[0018]
图2显示了在10μm、100μm和1mm二甲双胍或10μm、25μm或50μm pxl770存在下,由pimdck细胞形成的囊肿的照片。
[0019]
图3显示了评估本发明的各种噻吩并吡啶酮与类似化合物相比,对囊肿增大的功
效的体外实验结果。
[0020]
发明详述
[0021]
定义
[0022]
如本文所用的以下术语,除非另有明确说明,否则具有以下含义。
[0023]
术语“卤素原子”是指选自氟、氯、溴和碘原子的原子。
[0024]
术语“烷基基团”是指1至5个碳原子的直链或支链饱和链,例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基。优选地,烷基基团是1至3个碳原子的直链或支链饱和链,例如甲基、乙基、正丙基或异丙基基团。
[0025]
术语“芳基基团”是指c6-c18芳族基团,例如苯基或萘基基团,所述基团任选被一个或多个选自卤素原子、烷基基团、羟基(oh)、烷氧基基团、氨基(nh2)、单或二烷基氨基基团、羧基(cooh)、烷氧基羰基基团、单或二烷基氨基羰基基团、甲酰胺(conh2)、氰基(cn)、烷基磺酰基基团和三氟甲基(cf3)中的原子或基团取代。更具体地,所述芳基基团可以被氟、氯、溴原子、羟基、甲氧基、乙氧基、氨基、二甲基氨基、二乙基氨基、甲基、乙基、正丙基、正丁基、异丙基、仲丁基、异丁基、叔丁基、羧基、甲氧羰基、乙氧羰基、甲酰胺、二甲基氨基羰基、甲基氨基羰基、氰基、甲磺酰基或三氟甲基基团取代或不被取代。
[0026]
术语“芳烷基基团”是指如上定义的烷基基团,其氢原子被如上定义的芳基基团代替。芳烷基的实例是苄基基团。
[0027]
术语“烷氧基”基团是指通过氧原子与分子的其余部分连接的如上定义的烷基基团。在烷氧基基团之中可以提及的是甲氧基和乙氧基基团。
[0028]
术语“芳烷氧基”基团是指通过氧原子与分子的其余部分连接的如上定义的芳烷基基团。在芳烷氧基基团之中可以提及的是苄氧基基团。
[0029]
术语“烷基氨基基团”是指通过氮原子与分子的其余部分连接的如上定义的烷基基团。在烷基氨基之中,可以提及的是二甲基氨基和二乙基氨基基团。
[0030]
术语“烷氧基羰基基团”是指通过羰基基团与分子的其余部分连接的如上定义的烷氧基基团。
[0031]
术语“烷基氨基羰基基团”是指通过羰基基团与分子的其余部分连接的如上定义的烷基氨基基团。
[0032]
术语“烷基磺酰基”是指通过so2基团与分子的其余部分连接的如上定义的烷基。在烷基磺酰基基团之中,可以提及的是甲基磺酰基和乙基磺酰基基团。
[0033]
化合物的“溶剂合物”在本发明中是指惰性溶剂分子在化合物上由于它们相互的吸引力而形成的加合物。例如,溶剂合物是水合物或醇合物。
[0034]
本发明涉及式(i)的噻吩并吡啶酮衍生物,或其药学上可接受的盐和/或溶剂合物,或包含它们的药物组合物,用于治疗adpkd,
[0035][0036]
其中:
[0037]
r1表示氢原子或卤素原子,
[0038]
r2表示茚满基或四氢化萘基基团,所述基团被一个或多个(例如2、3、4、5、6或7个)选自卤素原子、烷基基团、羟基、烷氧基基团、氨基、单或二烷基氨基基团、羧基基团、烷氧基羰基基团、单或二烷基氨基羰基基团、甲酰胺、氰基、烷基磺酰基和三氟甲基基团中的基团取代或不被取代,
[0039]
r3表示芳基基团,所述基团被一个或多个(例如2、3、4或5个)选自卤素原子、烷基基团、羟基、烷氧基基团、芳烷氧基基团、氨基、单或二烷基氨基基团、羧基基团、烷氧基羰基基团、单或二烷基氨基羰基基团、甲酰胺、氰基、烷基磺酰基和三氟甲基基团中的原子或基团取代或不被取代。
[0040]
所述式(i)化合物的药学上可接受的盐的实例可以通过本领域通常已知的程序使所述式(i)的化合物与各种有机和无机碱反应而产生相应的碱加成盐来获得。这样的碱是,例如:碱金属氢氧化物,包括氢氧化钾、氢氧化钠和氢氧化锂;碱金属碳酸盐,包括碳酸钾和碳酸钠;碱土金属氢氧化物,例如氢氧化钡和氢氧化钙;碱土金属碳酸盐;碱金属醇盐,例如乙醇钾和丙醇钠;以及各种有机碱,例如哌啶、二乙醇胺和n-甲基谷氨酰胺。同样包括所述式(i)的化合物的铝盐。
[0041]
因此,所述式(i)的化合物的盐包括铝盐、铵盐、钙盐、铜盐、铁(iii)盐、铁(ii)盐、锂盐、镁盐、锰(iii)盐、锰(ii)盐、钾盐、钠盐和锌盐,但这并不意欲表示限制。在上述的盐中,优选的是单、二和三钠或钾盐,最优选钾盐。
[0042]
所述式(i)的化合物的任何药学上可接受的盐、或该化合物本身,可在本发明中以其溶剂合物之一的形式使用。化合物的“溶剂合物”在本发明中是指惰性溶剂分子在化合物上由于它们的相互吸引力而形成的加合物。因此,溶剂合物的性质取决于在所述碱与所述式(i)的化合物反应期间使用的溶剂。溶剂合物的实例包括醇溶剂合物,例如甲醇或乙醇溶剂合物,和水合物,包括一、二、三或四水合物,但这并不意欲表示限制。
[0043]
在一个特定的实施方式中,至少一个以下条件被满足,优选全部以下条件被满足:
[0044]
·
r1表示卤素原子,特别是氯原子,
[0045]
·
r2未被取代或被1或2个包含至少一个羟基基团的取代基取代,
[0046]
·
r2是四氢化萘基基团,
[0047]
·
r3表示未被取代或被1或2个取代基取代的苯基基团,
[0048]
·
所述式(i)的化合物是盐的形式,优选是钠盐或钾盐、更优选钾盐,
[0049]
·
所述式(i)的化合物是溶剂合物的形式,优选是水合物、更优选一水合物。
[0050]
更优选地,至少一个以下条件被满足,优选全部以下条件被满足:
[0051]
·
r1表示卤素原子,特别是氯原子,
[0052]
·
r2被1或2个包含至少一个羟基基团的取代基取代,
[0053]
·
r2是四氢化萘基基团,
[0054]
·
r3表示未被取代的苯基基团,
[0055]
·
所述式(i)的化合物是盐的形式,优选是钠盐或钾盐、更优选钾盐,
[0056]
·
所述式(i)的化合物是溶剂合物的形式,优选是水合物、更优选一水合物。
[0057]
在另一个实施方式中,至少一个以下条件被满足,优选全部以下条件被满足:
[0058]
·
r1表示卤素原子,特别是氯原子,
[0059]
·
r2被1或2个包含至少一个羟基基团的取代基取代,
[0060]
·
r2是茚满基基团,
[0061]
·
r3表示未被取代或被1或2个取代基取代的苯基基团。
[0062]
本发明的噻吩并吡啶酮衍生物的实例如下:
[0063]
2-氯-4-羟基-3-茚满-5-基-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮
[0064]
2-氯-5-(4-氟苯基)-4-羟基-3-茚满-5-基-7h-噻吩并[2,3-b]吡啶-6-酮
[0065]
2-氯-4-羟基-3-茚满-5-基-5-(3-甲氧基苯基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0066]
2-氯-4-羟基-3-茚满-5-基-5-(4-甲氧基苯基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0067]
3-(2-氯-4-羟基-3-茚满-5-基-6-氧代-7h-噻吩并[2,3-b]吡啶-5-基)苯甲腈
[0068]
2-氯-4-羟基-3-茚满-5-基-5-(3-甲基苯基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0069]
2-氯-5-(4-氟苯基)-4-羟基-3-(4-羟基茚满-5-基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0070]
2-氯-5-(3-氟苯基)-4-羟基-3-(4-羟基茚满-5-基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0071]
2-氯-4-羟基-3-(4-羟基茚满-5-基)-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮
[0072]
2-氯-5-(2-氟苯基)-4-羟基-3-(4-羟基茚满-5-基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0073]
2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮
[0074]
3-(2-氯-4-羟基-6-氧代-3-四氢化萘-6-基-7h-噻吩并[2,3-b]吡啶-5-基)苯甲腈
[0075]
2-氯-3-(5-氧四氢化萘-6-基)-5-苯基-噻吩并[2,3-b]-4,6-二羟基吡啶三钠
[0076]
2-氯-4-羟基-5-苯基-3-四氢化萘-6-基-7h-噻吩并[2,3-b]吡啶-6-酮
[0077]
2-氯-5-(4-氟苯基)-4-羟基-3-(5-羟基四氢化萘-6-基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0078]
2-氯-3-(5-氧四氢化萘-6-基)-6-氧代-5-苯基-7h-噻吩并[2,3-b]-4-羟基吡啶二钠
[0079]
2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-(3-甲基苯基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0080]
2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-(4-甲基苯基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0081]
2-氯-5-(3-氟苯基)-4-羟基-3-(5-羟基四氢化萘-6-基)-7h-噻吩并[2,3-b]吡
啶-6-酮
[0082]
2-氯-3-(5-羟基四氢化萘-6-基)-6-氧代-5-苯基-7h-噻吩并[2,3-b]-4-羟基吡啶钠
[0083]
2-氯-3-(5-羟基四氢化萘-6-基)-6-氧代-5-苯基-7h-噻吩并[2,3-b]-4-羟基吡啶钾。
[0084]
所述式(i)的化合物通常可以如wo 2014/001554中所公开的那样制备。
[0085]
这样的化合物的实例包括:
[0086]
·
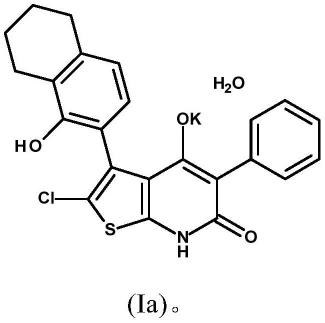
pxl770,其是2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮的一水合钾盐,对应于下面式(ia)的结构:
[0087][0088]
·
2-氯-5-(4-氟苯基)-4-羟基-3-茚满-5-基-7h-噻吩并[2,3-b]吡啶-6-酮,具有式(ib):
[0089][0090]
·
2-氯-4-羟基-3-茚满-5-基-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮,具有式(ic):
[0091][0092]
pxl770可根据包括以下步骤的方法制备:
[0093]
(a)使式(ii)的化合物与碳酸钾在包含水和选自乙酸正丁酯和异丙醇的溶剂的溶
液中反应:
[0094][0095]
(b)形成沉淀;和
[0096]
(c)回收步骤(b)中获得的沉淀,优选通过过滤回收。
[0097]
所述式(ii)的化合物及其制备方法已在专利申请wo 2014/001554中公开。
[0098]
或者,所述式(ii)的化合物可通过包括以下步骤的改进的方法获得:
[0099]
(a)在碱存在下,使6-乙酰基-5-羟基四氢化萘与亲电子苄基源、优选苄基溴反应;
[0100]
(b)在六甲基二硅氮烷和乙酸存在下,使步骤(a)中获得的化合物与氰基乙酸乙酯反应;
[0101]
(c)在碱存在下,使步骤(b)中获得的化合物与硫反应;
[0102]
(d)任选形成步骤(c)中获得的化合物的盐,优选盐酸盐;
[0103]
(e)使步骤(c)或(d)中获得的化合物与亲电子氯源、优选n-氯代琥珀酰亚胺反应;
[0104]
(f)使步骤(e)中获得的化合物与苯乙酰氯反应;
[0105]
(g)使步骤(f)中获得的化合物与碱反应;
[0106]
(h)使步骤(g)中获得的化合物与三溴化硼或三氯化硼、优选三氯化硼反应;和
[0107]
(i)任选回收步骤(h)中获得的化合物。
[0108]
通常,步骤(b)可以包括加热步骤(a)中获得的混合物、优选在接近所述混合物回流的温度下加热的子步骤(b1),随后是冷却所得混合物、例如在-15℃和35℃之间的温度下冷却的子步骤(b2)。表述“接近所述混合物回流”通常是指在步骤(a)中的溶剂体系(例如,水/异丙醇或水/乙酸正丁酯)沸点的90%至100%之间的温度。
[0109]
在所述加热子步骤和子步骤(b2)之间可以进行蒸馏步骤,优选在减压下进行。
[0110]
步骤(b)允许形成晶形沉淀,通过向步骤(b2)添加晶种可有利于或触发该形成。
[0111]
在一个优选的实施方式中,在步骤(c)中通过过滤回收所述沉淀。然后可相继用一种或多种溶剂,优选水、乙酸正丁酯和/或叔丁基甲基醚,对其进行洗涤。
[0112]
由此获得固体形式、例如粉末形式的式(ia)的化合物,即pxl770,通过衍射仪使用cu k(α)辐射测量,其具有以下xrpd(x射线粉末衍射)峰:
[0113][0114]
在以下描述中,用语“噻吩并吡啶酮衍生物”是指所述式(i)的化合物、或其药学上可接受的盐和/或溶剂合物之一。
[0115]
本发明的一个目的是一种治疗常染色体显性多囊肾病(adpkd)的方法,所述方法包括向有此需要的对象施用有效量的噻吩并吡啶酮衍生物或包含有效量的噻吩并吡啶酮衍生物以及药学上可接受的支撑体的药物组合物。
[0116]
本发明还涉及所述噻吩并吡啶酮衍生物或包含它的组合物在制造用于治疗常染色体显性多囊肾病(adpkd)的药物中的用途。
[0117]
根据本发明使用的药物组合物可通过任何常规方法制备。所述噻吩并吡啶酮衍生物可以与至少一种固体、液体和/或半液体赋形剂或佐剂一起、并且如果需要的话与一种或多种其它活性成分组合,转化为合适的剂型。
[0118]
术语“药学上可接受的支撑体”是指从药理学/毒理学角度为对象可接受、以及从物理/化学角度关于组成、配制、稳定性、对象接受度和生物利用度方面为制药化学家可接受的载体、佐剂或赋形剂。
[0119]
术语“载体”、“佐剂”或“赋形剂”是指添加到药物组合物中用作载体、佐剂和/或稀释剂以向对象递送治疗剂的任何物质,其本身不是治疗剂,而是为了改善治疗剂的处理或储存性质、或为了实现或便于将所述组合物的剂量单位形成为分立的制品。本发明的药物组合物,单独地或组合地,可以包含一种或几种选自分散剂、增溶剂、稳定剂、防腐剂等试剂或介质。
[0120]
术语“治疗”是指常染色体显性多囊肾病(adpkd)的治疗、预防(prevention)和预防(prophylaxis)。如本文所公开的术语“治疗”是指预防疾病或其至少一种症状。这也意指改善、预防至少一个与正在治疗的疾病相关的可测量的身体参数,该参数在对象中是可觉察的或觉察不出的。术语“治疗”还指抑制或减缓身体上疾病的进展,稳定生理上可觉察的
症状,例如稳定身体参数,或二者兼而有之。术语“治疗”还指延迟疾病或病症的发作。在一些特定的实施方式中,本发明的化合物作为预防措施施用。在本上下文中,“预防”是指降低与疾病相关的至少一种症状发生的风险。
[0121]
术语“治疗”可以包括用噻吩并吡啶酮衍生物或包含它的药物组合物预防或限制囊肿生长。如本文所用的“治疗”还涵盖了减小囊肿体积。因此,术语“治疗”包括与adpkd相关的症状的治疗。
[0122]
所述治疗包括将本发明的噻吩并吡啶酮衍生物或药物组合物施用于患有所宣布的病症的对象以治愈、延迟或减缓进展,从而改善患者的状况。
[0123]
在本发明的上下文内,术语“对象”是指哺乳动物,更特别是人类。本发明的待治疗对象可以基于与疾病相关的若干标准来适当地选择。在adpkd的情况下,所述治疗更特别适合于在pkd1和/或pkd2基因中具有至少一个突变的患者或有adpkd风险的患者。“有adpkd风险”的患者包括通过超声检查法检测,年龄在15-39岁且有三个或更多个单侧或双侧肾囊肿的患者、年龄在40-59岁且每个肾脏有两个或更多个囊肿的患者、和年龄在60岁或以上且每个肾脏有四个或更多个囊肿的患者。或者,通过磁共振成像观察,30岁或以下且有至少十个肾囊肿的患者通常被认为有adpkd风险。
[0124]
药物组合物可以按剂量单位形式施用,每剂量单位包含预定有效量的活性成分。
[0125]
药物组合物可以适于通过任何所需的合适方法施用,例如通过口服(包括口颊或舌下)、直肠、鼻、局部(包括口颊、舌下或经皮)、阴道或肠胃外(包括皮下、肌内、静脉内或皮内)方法。这样的组合物可以使用制药领域中已知的所有方法通过例如将活性成分与赋形剂或佐剂合并来制备。优选地,本发明的药物组合物适于口服施用。
[0126]
适于口服施用的药物组合物可以作为独立的单位例如胶囊或片剂;粉剂或颗粒剂;在水性或非水性液体中的溶液或悬浮液;可食用泡沫或泡沫食品;或乳液,例如水包油液体乳液或油包水液体乳液,进行施用。
[0127]
因此,例如,在以片剂或胶囊形式口服施用的情况下,活性成分组分可以与口服的无毒且药学上可接受的惰性赋形剂相结合。粉剂是通过将化合物粉碎至合适的细小尺寸并将其与以类似方式粉碎的药用赋形剂混合而制备的,所述赋形剂例如可食用碳水化合物,如淀粉或甘露糖醇。也可以存在调味剂、防腐剂、分散剂和染料。
[0128]
胶囊可通过制备如上所述的粉末混合物并用其填充成形的明胶壳来生产。在填充操作之前,可以向所述粉末混合物添加助流剂和润滑剂,例如高度分散的硅酸、滑石粉、硬脂酸镁、硬脂酸钙或固体形式的聚乙二醇。也可以添加崩解剂或增溶剂,例如琼脂、碳酸钙或碳酸钠,以改善胶囊服用后药物的利用度。
[0129]
另外,如果需要或必要,也可以将合适的粘合剂、润滑剂和崩解剂以及染料并入所述混合物中。合适的粘合剂包括淀粉、明胶、天然糖例如葡萄糖或β-乳糖、由玉米制成的甜味剂、天然和合成橡胶例如阿拉伯胶、黄蓍胶或藻酸钠、羧甲基纤维素、聚乙二醇、蜡等。这些剂型中所用的润滑剂包括油酸钠、硬脂酸钠、硬脂酸镁、苯甲酸钠、乙酸钠、氯化钠等。崩解剂包括但不限于淀粉、甲基纤维素、琼脂、膨润土、黄原胶等。片剂是例如如下所述配制的:制备粉末混合物,将所述混合物制粒或干压,添加润滑剂和崩解剂,以及压制整个混合物以产生片剂。粉末混合物是如下所述制备的:将以合适的方式粉碎的所述化合物与如上所述的稀释剂或碱混合,并任选与粘合剂例如羧甲基纤维素、藻酸盐、明胶或聚乙烯吡咯烷
酮、溶解延迟剂例如石蜡、吸收促进剂例如季盐、和/或吸收剂例如膨润土、高岭土或磷酸二钙混合。所述粉末混合物可以通过将其用粘合剂例如糖浆、淀粉糊、阿拉伯胶、或纤维素或聚合物材料溶液润湿并将其压过筛子来制粒。作为制粒的替代方法,所述粉末混合物可以通过压片机,产生形状不均匀的块,将其破碎以形成颗粒。所述颗粒可以通过添加硬脂酸、硬脂酸盐、滑石粉或矿物油进行润滑,以防粘在片剂铸模上。然后压制所述经润滑的混合物以产生片剂。本发明的化合物也可以与自由流动的惰性赋形剂合并,然后不进行所述制粒或干压步骤,直接压制以产生片剂。可以存在由虫胶密封层、糖或聚合物材料层和蜡光泽层组成的透明或不透明保护层。可以将染料添加到这些包衣中,以便能够区分不同的剂量单位。
[0130]
适于口服施用的药物组合物也可以通过固体或液体分散体的喷雾干燥来配制。
[0131]
口服的液体,例如溶液、糖浆和酏剂,可以制备成剂量单位的形式,使得给定的量包含预定量的化合物。糖浆可以通过将所述化合物溶解在具有合适调味剂的水溶液中来制备,而酏剂则使用无毒醇类介质来制备。悬浮液可以通过将化合物分散在无毒介质中来配制。也可以添加增溶剂和乳化剂例如乙氧基化异硬脂醇和聚氧乙烯山梨醇醚、防腐剂、风味添加剂例如薄荷油或天然甜味剂或糖精、或其它人造甜味剂等。
[0132]
如果需要,可以将用于口服施用的剂量单位制剂封装在微胶囊中。所述制剂也可以按延长或延迟释放的方式制备,例如通过在聚合物、蜡等中包被或包埋颗粒材料。
[0133]
根据本发明使用的噻吩并吡啶酮衍生物也可以按脂质体递送系统例如小单层脂囊、大单层脂囊和多层脂囊的形式施用。脂质体可以由各种磷脂例如胆固醇、硬脂胺或磷脂酰胆碱形成。
[0134]“有效量”是指如上定义的化合物预防、消除或减少所治疗的疾病在人类中的有害效应的量。应理解,本领域技术人员可根据患者、病理、施用方式等来调整施用剂量。例如,所述噻吩并吡啶酮衍生物可每天施用一次或两次,对人类患者的日剂量为0.5mg至300mg,优选20mg至1000mg,更优选60mg至500mg。它可以作为长期药物每周施用4、5、6或7天。
[0135]
在本发明的一个特定实施方式中,所述噻吩并吡啶酮衍生物以包含0.5mg至1500mg、优选20mg至1000mg、更优选60mg至500mg的噻吩并吡啶酮衍生物的剂量单位施用。
[0136]
本发明还将在以下实施例中更详细地描述,这些实施例并不意图限制本发明的范围,本发明的范围由权利要求书限定。
具体实施方式
[0137]
实施例1:pxl770的合成
[0138]
分析方法
[0139]
xrpd
[0140]
使用配备有cu(kα辐射)x射线管和pixcel检测器系统的panalytical xpert pro衍射仪进行x射线粉末衍射(xrpd)分析。样品以透射模式并夹在低密度聚乙烯薄膜之间进行分析。xrpd图谱使用highscore plus 2.2c软件进行分类、操作和索引。
[0141]
tg/dta
[0142]
在perkin elmer diamond热重/差温分析仪(tg/dta)上进行热重(tg)分析。校准标准是铟和锡。样品放在铝样品盘中,插入tg炉中并精确称重。样品在氮气流中以10℃/分
钟的速率从30℃加热到300℃。在分析样品之前,将炉温在30℃平衡。
[0143]
1a)1-(5-苄氧基四氢化萘-6-基)乙酮(1)的合成
[0144][0145]
将6-乙酰基-5-羟基四氢化萘(100g,1eq.)溶于乙腈(300ml)。添加k2co3(1.1eq.)和苄基溴(1.05eq.)后,加热该悬浮液(76℃)。48小时后,添加苄基溴(0.1eq)。总共74小时后,滤出固体并用乙腈(200ml)洗涤,并将合并的滤液蒸发。
[0146]
获得作为浆状物的化合物1:m=148.6g,定量产率,纯度96.6%a/a。
[0147]
1b)2-氨基-4-(5-苄氧基四氢化萘-6-基)噻吩-3-甲酸乙酯(2)的合成
[0148][0149]
将乙酸(70ml)加热至t=65℃。用10分钟添加hmds(1.5eq.)。然后,添加化合物1(69.5g,1eq.)和氰基乙酸乙酯(1.5eq.)在乙酸(140ml)中的溶液。将所得混合物在t=65℃下搅拌24小时。
[0150]
冷却至室温后,添加naoh水溶液(1m,140ml)和tbme(210ml)。进行层分离。有机层用naoh水溶液(1m,4
×
140ml)洗涤,直到水相的ph为碱性(ph=13)。将该有机层用hcl水溶液(1m,140ml)和h2o(2
×
140ml)洗涤。
[0151]
添加etoh(240ml)、nahco3(1.3eq.)和硫(1.0原子当量)。加热至回流180分钟后,将该反应混合物浓缩至210ml并与tbme(3
×
140ml)一起共蒸发。冷却至室温后,将该悬浮液过滤并用tbme(70ml)洗涤固体。合并的滤液浓缩至210ml,并在室温下滴加hcl的二烷溶液(1.1eq.)。接种后观察到沉淀。在室温下滴加庚烷(350ml)。搅拌14小时后,过滤该悬浮液。用庚烷(3
×
70ml)洗涤并干燥后,回收作为固体的化合物2。m=83.2g,产率71%,纯度93.7%a/a。
[0152]
1c)4-(5-苄氧基四氢化萘-6-基)-5-氯-2-[(2-苯基乙酰基)氨基]噻吩-3-甲酸乙酯(3)的合成
[0153][0154]
将化合物2(17.69g,1eq.)溶于二氯甲烷(140ml)。所得溶液用冰/水冷却。在搅拌下,添加n-氯代琥珀酰亚胺(1.05eq.)。该混合物在几分钟内变黑。1小时后,添加苯乙酰氯(1.25eq.)。
[0155]
在0℃下1小时和在室温下2小时后,将该混合物蒸发至约35ml,添加etoh(2
×
70ml),并再次蒸发。将该混合物用etoh(35ml)稀释并用冰/水冷却。产物沉淀。过滤出固体并用冷etoh(3x18ml)洗涤。
[0156]
获得作为固体的化合物3:m=20.99g,产率94.2%,纯度99.3%a/a。
[0157]
1d)3-(5-苄氧基四氢化萘-6-基)-2-氯-4-羟基-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮(4)的合成
[0158][0159]
将化合物3(19.88g,1eq.)溶于甲基四氢呋喃(120ml),并将反应混合物冷却至-16℃和-10℃之间的温度(nacl/冰)。分四份添加叔丁醇钾(5eq.)。然后,将该反应混合物升温至室温,并在室温下搅拌65分钟。在t=0-5℃(水/冰)下进行2n hcl(5eq.)的滴加,并剧烈搅拌所得混合物。用nacl
(水溶液)
(11%,1
×
50ml)和水(2
×
50ml)洗涤有机相。将有机相浓缩至~50%溶液。添加甲基四氢呋喃(80ml),将所得溶液浓缩至~50%溶液。添加tbme(100ml),并将所得溶液浓缩至~50%溶液(该步骤重复3次)。然后,添加tbme(25ml)、化合物4的晶种和正庚烷(20ml),所得溶液在室温下搅拌过夜。将该混合物浓缩至约50ml,过滤,用母液漂洗、用正庚烷(2
×
40ml)洗涤并干燥。获得作为粒状固体的化合物4。产率88%,纯度99.5%a/a。
[0160]
1e)2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮(i)的合成
[0161][0162]
将化合物4(15g,1eq.)溶于75ml二氯甲烷并冷却至t=-10℃/-15℃(用冰/nacl)。滴加bcl3(1.5eq.,溶液:1mol/l的二氯甲烷溶液)并将所得混合物在室温下搅拌15小时。用冰/水冷却所得混合物,并添加水(75ml)。剧烈搅拌所得混合物并用水/meoh(9:1v/v,5
×
45ml)提取有机相。浓缩有机相,用甲苯(3
×
90ml)进行溶剂交换并用甲苯稀释至达到90ml甲苯的最终体积。将所得混合物加热至回流并添加15ml甲醇。获得有少许粒子的淡褐色溶液。在t=40℃添加晶种,升温至t=52℃并冷却至室温。将所得混合物搅拌过夜,然后用冰/nacl(t=-10℃/-15℃)冷却100分钟。过滤出沉淀的产物,用甲苯/庚烷1:2v/v(15ml)和庚烷(15ml)洗涤并干燥。得到化合物(i)的晶体:产率87%,纯度99.0%a/a。
[0163]
1f)2-氯-4-羟基-3-(5-羟基四氢化萘-6-基)-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮的一水合钾盐(ia)的合成
[0164]
将化合物(i)悬浮在水/异丙醇混合物(1/1,每种溶剂各5份)中,然后添加0.50至0.55eq的碳酸钾。添加碳酸钾结束时ph为约12(ph试纸)。在50℃搅拌3小时后,该悬浮液变稠,ph为约8(ph试纸)。将温度升至80℃,直至获得溶液(10-15分钟)。如果需要,可以在所述过程的此刻进行澄清。添加7份水,然后将反应混合物冷却至40℃(观察到混浊溶液)。在40℃减压蒸馏(从180毫巴到40毫巴)溶剂,直到反应器中留下7份溶剂。此时可发生钾盐一水合物的结晶。添加4.2份水,并用化合物(i)接种该混合物(1至2%的晶种)。然后将该悬浮液在7小时内从40℃冷却至5℃(5℃/小时),并在5℃保持数小时。过滤该悬浮液。将滤饼用1.42份水洗涤两次。收集的固体在40℃真空干燥,得到所要求的化学纯度(即98% )的最低80%产率的化合物(ia)。
[0165]
实施例2:pxl770的表征
[0166]
a)化合物(ia)的x射线粉末衍射(xrpd)数据表明它由结晶材料组成。化合物(ia)的xrpd描述如表1所示。
[0167]
表1
[0168]
[0169][0170]
b)tg/dta分析显示,从30-100℃的初始重量损失为1.1%,随后由于结合水的损失,从117-160℃的重量损失较大,为3%。第二次重量损失伴随着大量吸热,4%的合并重量损失接近一水合物的理论重量损失(3.75%w/w)。该化合物在超过240℃时分解。
[0171]
实施例3:体外实验
[0172]
方法:
[0173]
将plmdck细胞在37℃、21%o2和5%co2下在改良mem培养基中培养,所述培养基含有“earl’s平衡盐溶液”、2mm l-谷氨酰胺、10%热休克灭活fcs、50iu/ml青霉素和50μg/ml链霉素。对于囊肿形成,将plmdck细胞胰蛋白酶化并溶解在i型胶原蛋白悬浮液内,然后转移到24孔板中。采用每个条件和单个实验4个孔。接下来,添加含有10μm毛喉素(对照;ctrl)或10μm毛喉素 给定浓度的二甲双胍或10μm毛喉素 给定浓度的pxl770的细胞培养基。所述实验进行5天。之后,使用带有zeiss axiocam 105彩色相机的zeiss primo vert显微镜(均为zeiss microscopy gmbh,jena,德国),在每个孔的4个不同区域(0、3、6和9点钟方向)拍摄照片。然后,用imagej(v.1.48)以盲法测量囊肿的直径。假设囊肿为球形,利用公式4/3πr3计算体积。计算每个条件和实验的所有囊肿的平均值。对照囊肿体积设置为100%。结果如图1所示。
[0174]
还拍摄了囊肿的照片,并在图2显示(比例尺:100μm)。
[0175]
结果:
[0176]
如图1和2所示,pxl770以低于二甲双胍的剂量显著抑制囊肿生长。
[0177]
实施例4:比较实验
[0178]
在本实施例中,在体外实验中比较了各种噻吩并吡啶酮化合物,即:
[0179]
pxl770:2-氯-3-(5-羟基四氢化萘-6-基)-6-氧代-5-苯基-7h-噻吩并[2,3-b]-4-羟基吡啶钾
[0180]
pxl700:2-氯-4-羟基-3-茚满-5-基-5-(3-吡啶基)-7h-噻吩并[2,3-b]吡啶-6-酮
[0181]
pxl702:2-氯-4-羟基-3-茚满-5-基-5-苯基-7h-噻吩并[2,3-b]吡啶-6-酮
[0182]
pxl695:2-氯-5-(4-氟苯基)-4-羟基-3-茚满-5-基-7h-噻吩并[2,3-b]吡啶-6-酮,
[0183]
pxl037:
[0184][0185]
为了研究这些化合物的效果,采用了使用包埋在3d培养基基质中的多囊肾病(pkd)患者来源的原代细胞(称为hupkd模型)进行的体外3d囊肿增大试验。囊肿的增大可以通过高内涵显微镜检术来可视化和量化。
[0186]
方法
[0187]
已用hupkd05细胞进行了3d hupkd囊肿增大试验。
[0188]
3d培养和化合物暴露:将hupkd05细胞(pkd1突变:c.5622g》ap.trp1874*)与primcyst凝胶(ocello bv)混合。使用cybio felix 96/250自动液体分配器(analyik jena ag)将15μl细胞-凝胶混合物移液至384孔板(greinerμclear,greiner bio-one b.v.)。将凝胶-细胞混合物以每孔450个囊肿的最终细胞密度铺板。37℃下凝胶聚合30分钟后,每孔添加33μl培养基(不含血清)。细胞在凝胶中生长24小时,然后将细胞与刺激囊肿增大的去氨加压素(ddavp)(tocris)以及以下分子pxl770、pxl037、pxl700、pxl695、pxl702共暴露48小时。
[0189]
表1:暴露条件概述
[0190][0191]
样品处理:48小时后,将培养物用4%甲醛(sigma aldrich)固定,同时用0.2%triton-x100(sigma aldrich)透化并在4℃避光下用0.25μm罗丹明-鬼笔环肽(sigma aldrich)和0.1%hoechst 33258(sigma aldrich)在1
×
pbs(sigma aldrich)中染色2天。固定和染色后,用1
×
pbs洗涤板,用greiner silverseal(greiner bio-one b.v.)密封并在成像前在4℃储存。
[0192]
成像和图像分析:使用带有4
×
nikon物镜的molecular devices imagexpress micro xls(molecular devices)进行成像。对于每个孔,为两个通道制作了在z方向上大约35张图像,在每个图像中捕获整个z平面。使用ominer
tm
软件(ocello bv)进行图像分析。利用对hoechst染色的细胞核和罗丹明-鬼笔环肽染色的细胞f-肌动蛋白的检测对囊肿进行分割。通过计算每个聚焦平面中每个物体的像素(px)面积来确定囊肿面积。将其取每孔的平均值,并在溶剂对照(0%)和仅有刺激剂(100%)之间进行归一化。使用graphpad prism进行统计,在graphpad prism 6(graphpad software,la jolla,ca)中作图。
[0193]
结果
[0194]
上述实验的结果示于图3。
[0195]
控制条件:在去氨加压素(ddavp)刺激和未刺激(溶剂对照,dmso)之间建立了显著性试验窗口(图1)(ddavp仅与dmso对比,p《0.0001)
[0196]
pxl770、pxl695、pxl700、pxl702和pxl037:
[0197]
pxl770、pxl695、pxl700、pxl702和pxl037以4个浓度进行测试:在ddavp(2.5μm)存在下10、5、2.5、0.5μm。
[0198]
pxl770剂量依赖性地减小囊肿面积,这种效果在5和10μm时与单独ddavp相比具有显著性,pxl770 5和10μm均达到50%减小(与单独ddavp相比,分别是p《0.01和p《0.05)。虽然图3中未显示,但应注意pxl770在25μm时提供了75%的囊肿面积减小(与单独ddavp相比,p《0.0001)。
[0199]
pxl695和pxl702剂量依赖性地减小囊肿面积。
[0200]
相反,与单独ddavp相比,pxl037和pxl700没有表现出显著的效果。
[0201]
结论
[0202]
在hupkd囊肿增大试验中评估了pxl770、pxl037、pxl770、pxl695和pxl702,以研究
它们在抑制去氨加压素(ddavp)诱导的囊肿增大中的效力。
[0203]
pxl770剂量依赖性地降低囊肿增大。5和10μm的pxl770抑制了几乎50%的诱导增大。
[0204]
用pxl695和pxl702实现了类似的效力和功效。
[0205]
pxl037和pxl700在高达10μm时仍对囊肿面积没有效果。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。